
Abstract
Given the shared risk factors for transmission, co-infection of hepatitis B virus (HBV) with hepatitis C virus (HCV) and/or human immunodeficiency virus (HIV) is quite common, and may lead to increases in morbidity and mortality. As such, HBV vaccine is recommended as the primary means to prevent HBV super-infection in HCV- and/or HIV-infected individuals. However, vaccine response (sero-conversion with a hepatitis B surface antibody titer >10 IU/L) in this setting is often blunted, with poor response rates to standard HBV vaccinations in virally infected individuals when compared with the healthy subjects. This phenomenon also occurs to other vaccines in adults, such as pneumococcal and influenza vaccines, in other immunocompromised hosts who are really at risk for opportunistic infections, such as individuals with hemodialysis, transplant, and malignancy. In this review, we summarize the underlying mechanisms involving vaccine failure in these conditions, focusing on immune exhaustion and immune senescence—two distinct signaling pathways regulating cell function and fate. We raise the possibility that blocking these negative signaling pathways might improve success rates of immunizations in the setting of chronic viral infection.
Similar content being viewed by others


Introduction
Super-infection with bacterial, fungal, or viral pathogens in individuals with chronic viral infections, such as human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV) is common. Other immunocompromised conditions, such as transplantation, hemodialysis, and malignancy, are also common clinical scenarios in which super-infections occur. As such, recommendations for immunization with available vaccines in adults have been generated to prevent the morbidity and mortality associated with super-infection in these hosts. Unfortunately, these patients often do not respond well to vaccinations. This review focuses on this problem, discussing the potential mechanisms underlying poor vaccine responses and possible approaches to increase the vaccine success rates in immunocompromised hosts.
Co-Infection of HBV with HIV or HCV Increases Liver-Related Morbidity and Mortality
Co-infection of HBV with HIV and/or HCV is common, given their shared risk factors for transmission (Bruguera et al. 1992; Cheruvu et al. 2007; Duberg et al. 2008; Filippini et al. 2007; Kellerman et al. 2003; Thio et al. 2002; Torbenson et al. 2004; Weber et al. 2006; Zarski et al. 1998). An estimated 60,000 people in the United States have chronic co-infection of HBV with HIV (Kellerman et al. 2003). Importantly, co-infection of HBV in HIV/HCV-infected patients significantly increases the liver-related morbidity and mortality (Duberg et al. 2008; Thio et al. 2002; Weber et al. 2006), underscoring the importance of prevention of HBV infection in people with HIV or HCV (Fig. 1).

a Risk of liver-related mortality in patients with HIV and or HBV infection in a Multi-center Cohort Study in Baltimore (Thio et al. 2002). The liver-related mortality rate was reported 1.1/1000 person years, and was higher in men with HIV-1 and HBsAg (14.2/1000) than in those with only HIV-1 infection (1.7/1000, p < 0.001) or only HBsAg (0.8/1000, p < 0.001). b Cause of death in individuals with HCV and or HBV infection in a nationwide community-based register study in Sweden (Duberg et al. 2008). The standardized mortality ratio, considering all mortality, is 8.5 for HCV/HBV, 5.8 for HCV, and 2.3 for HBV alone. Both studies concluded that super-infection with HBV in HIV- or HCV-infected patients will significantly increase the risk for liver-relaled mortality, underscoring the importance of prevention of HBV in individuals with HIV or HCV
In the Multi-center AIDS Cohort Study, HIV/HBV co-infected men were almost 19 times more likely to die of liver disease than those infected with HBV alone, and eight times more likely to die of liver disease than those infected with HIV-1 alone (Thio et al. 2002). In an observational study of more than 23,000 HIV-infected patients done in Switzerland, active HBV infection measured as hepatitis B surface antigen (HBsAg), hepatitis B e antigen (HBeAg), and HBV-DNA-positive was an independent predictor of liver-related death with an adjusted relative risk of 3.73 (Weber et al. 2006). In the general adult population in western countries such as Australia, US, and Italy, the risk of developing chronic hepatitis after contracting HBV is 5–10 %; this number, however, can be doubled or even tripled in HBV/HIV co-infected patients, not to mention in HBV/HIV/HCV-infected individuals (Bodsworth et al. 1991; Cheruvu et al. 2007; Filippini et al. 2007). However, there are considerable geographic differences in this rate, as for example in immigrants and refugees (Rossi et al. 2012). Additionally, co-infected patients often present with an elevated, sustained serum HBV DNA level, with less likelihood of spontaneous HBsAg and HBeAg seroconversion—the loss of HBsAg with development of an anti-HBsAg antibody and the loss of HBeAg with development of an anti-HBeAg antibody, respectively (Biggar et al. 1987; Bodsworth et al. 1989, 1991; Gilson et al. 1997; Lazizi et al. 1988). On the other hand, HBV and/or HCV infection can drive HIV infection with more rapid progression to AIDS, perhaps explained by an increased viral load in HIV-infected cells and a faster decline as well as lower nadir of CD4+ cell counts (Eskild et al. 1992; Horvath and Raffanti 1994). Additionally, activation factors such as Nef, Tat, Rantes, Mip-1 for HIV, and X-protein, nuclear factor (NF)-1, NF-3, AP-1 for HBV, are activated intracellularly as well as extracellularly and lead to enduring activation of cells. Co-infection of HBV in HCV patients leads to more severe liver disease as well, with higher rates and faster progression to liver cirrhosis and liver cancer (Bodsworth et al. 1991; Filippini et al. 2007).
HBV Vaccine Response is Blunted in HCV and/or HIV-Infected Individuals
HBV vaccine is the primary means to prevent HBV infection and liver-related diseases. Because co-infection increases liver-related mortality, it is highly recommended that HIV- and/or HCV-infected individuals who lack evidence of previous infection or immunity be immunized for HBV, with either Engerix® HBV, Fendrix® or HBVaxPro-40®, or Twinrix® HAV/HBV combination vaccine if the individual is non-immune to hepatitis A virus (HAV). Notably, the combination of HAV and HBsAg in Twinrix® induces a higher immune response against HBsAg, and a combined vaccine from a further manufacturer is not commercially available. However, compared with the general population, the response rates to HBV vaccine (defined as development of anti-HBsAg with titer >10 IU/L) in HIV and/or HCV-infected patients is significantly diminished (20–70 vs. 90–95 %) (Collier et al. 1988; Kao and Chen 2002; Keeffe 2005; Kramer et al. 2009; Shafran et al. 2007; Tsai et al. 2000). Recent studies suggest that the anti-HBsAg protective level of 10 IU/L is no longer sufficient and has been proposed to be elevated to around 100 IU/L (Allain and Canotti 2012; Stramer et al. 2011).
The efficacy of HBV vaccination varies depending on the immune status of the recipients. For example, HBV hypo-responsiveness is strongly correlated with aging, with seroconversion rates showing declines as early as age 35 and markedly waning over the ensuing decades (Fisman et al. 2002). Following a full vaccination series, 90 % of healthy adults and 95 % of infants and children have protective serum antibody concentrations (Kao and Chen 2002; Tsai et al. 2000). Antibody levels usually decline with time to below the protective level of 10 IU/L in up to 50 % of vaccinees after 15 years; but clinically significant breakthrough infections are rare. There is also a marked sex difference in response to HBV vaccine (Klein 2012; Klein et al. 2010).
In patients with HIV infection, the poor response rate is closely related to CD4+ cell counts. However, recent data show that even in the setting of CD4 counts >200 or 500 cells/mm3, seroconversion rates in HIV-infected patients are still much lower than age-matched healthy subjects (Shafran et al. 2007). In the case of HCV infection, vaccine response is related to the stage of liver disease at the time of vaccine initiation, with poor response in those with severe liver fibrosis and cirrhosis (Kramer et al. 2009). Nevertheless, our recent data suggest that even in the setting of relatively preserved hepatic function, seroconversion rates in HCV-infected individuals are still much lower than age-matched healthy subjects (53 vs. 94 %) (Moorman et al. 2011). Interestingly, HCV patients who receive Twinrix vaccination have a better response rate than those who receive HBV vaccine alone, suggesting that HAV antigen in the Twinrix might enhance the HBV vaccine response.
The phenomenon of poor response to immunizations in HIV/HCV-infected patients has also been found with other vaccines in addition to HBV, including HAV, influenza, or pneumococcal vaccines (Malaspina et al. 2005; Rodriguez-Barradas et al. 2003). Additionally, similar phenomena are also observed following routine adult immunizations in the setting of other immunosuppressive conditions, such as organ transplantation, cancer chemotherapy, and chronic renal failure, suggesting a universal or shared mechanism of vaccine nonresponse in immunocompromised hosts.
To improve the seroconversion of HBV immunization, several approaches—including different administration routes (subcutaneous vs. intradermal injection), higher doses of HBV vaccine (40 vs. 20 μg), and adding adjuvants (CPG 7909, levamisole, GM-CSF)—have been tried for nonresponders (Jacques et al. 2002; Kim et al. 2003; Nystrom et al. 2008; Rahman et al. 2000; Ramon et al. 1996). These approaches have led to varying degrees of improvement in healthy subjects, but have had limited success in virally infected individuals, in part due to a lack of information regarding cellular and molecular mechanisms that inhibit immune responses in this setting.
Possible Mechanisms Involved in HBV Vaccine Failure During Chronic Viral Infections
The reasons for vaccine nonresponse in 5–10 % of healthy subjects and 40–60 % of HCV or HIV-infected individuals remain poorly understood, although several factors are known to play a role, such as age, gender, smoking, obesity, and certain human leukocyte antigen (HLA) alleles (De Silvestri et al. 2001; Godkin et al. 2005; Lango-Warensjo et al. 1998). More specifically, nonresponsiveness to HBV vaccine in adults is strongly associated with the HLA-C4A locus. This association was also demonstrated in neonates who failed to mount a successful antibody response to challenge with HBV vaccine (De Silvestri et al. 2001). Additionally, HLA-DRB1*0301 has been associated with nonresponse to vaccination with HBV envelope proteins (HBsAg), with no altered susceptibility to viral persistence (Godkin et al. 2005). Interestingly, the amino acids that differ between the responders and the nonresponders are located in the peptide-binding groove of the HLA molecule, which seems to determine the response against HBsAg (Lango-Warensjo et al. 1998). These results suggest a role for HLA alleles to direct either a response or a nonresponse against HBsAg. Notably, some HLA class II genotypes were found to be identically shared by vaccine responders and nonresponders, indicating the influence of other factors in addition to the HLA system in the response to HBV vaccine. Additional causes besides HLA that may influence the HBV immune response include host genetic factors and cytokine genetic polymorphisms, amongst others (Macedo et al. 2010; Ryckman et al. 2010).
The mechanism for vaccine-induced immune responses is thought to be clonal activation and expansion of antigen-specific memory T and B lymphocytes upon encountering antigen. Given the fact that these nonresponders also have poor recall responses to tetanus toxoid or Candida, it has been suggested that HBV vaccine failure may be due to a defect in HBsAg-reactive T cells (Albarran et al. 2005; Bauer and Jilg 2006; Goncalves et al. 2004; Salazar et al. 1995), or in antigen-presenting cells (APCs) (Hohler et al. 2002; Verkade et al. 2007). Indeed, a recent study of patients with chronic renal disease who failed HBV vaccination revealed a more profound defect in monocyte-derived dendritic cell function, leading to diminished helper T cell and B cell responses (Verkade et al. 2007). Thus, it appears that defective APC and impaired helper T cell functions may underlie the dampened B cell response to vaccination, although this remains controversial (Desombere et al. 1995, 2005). Another study of HBV vaccine response in liver transplant recipients shows evidence of accumulation of HBsAg-specific regulatory T cells (Bauer et al. 2007), suggesting that broader immune modulations may play a pivotal role in vaccine response during chronic viral infections.
Immune Exhaustion in HBV Vaccine Failure During Chronic Viral Infections
It is well established that the immune system is precisely regulated by an intricate balance between positive and negative signals to ensure adequate responses against pathogens and yet prevent over-activation of lymphocytes and thus cause autoimmunity. The activation and proliferation of lymphocytes requires two signals: an antigen-specific signal (signal 1) and a co-stimulatory signal (signal 2) that is independent of the antigen receptor complex. While signal 1 determines the antigen-specific reaction, co-stimulatory signal 2 is pivotal in determining whether recognition of antigen by T-helper lymphocytes leads to full cell activation and proliferation or to cell exhaustion and apoptosis.
Besides positive stimulatory signaling, the immune system has developed negative feedback mechanisms to prevent unnecessary activation of immune responses. The recently described programmed death-1 (PD-1), suppressor of cytokine signaling-1 (SOCS-1) and T cell immunoglobulin domain protein-3 (Tim-3) inhibitory pathways represent such feedback mechanisms to maintain the balance between positive and the negative intracellular signals in T and B lymphocytes following antigenic encounter (Alexander 2002; Chen 2004; Egan et al. 2003; Hafler and Kuchroo 2008; Kuchroo et al. 2003; Nishimura and Honjo 2001; Okazaki and Honjo 2007; Sharpe and Freeman 2002). Compelling evidence is emerging for the involvement of these negative signaling molecules in antiviral immune evasion, autoimmune responses, and tumorigenesis (Barber et al. 2006; Boni et al. 2007; Chong et al. 2005; Dong et al. 2002; D’Souza et al. 2007; Golden-Mason et al. 2007; Iwai et al. 2003; Jin et al. 2010; Jones et al. 2008; Khoury and Sayegh 2004; McMahan et al. 2010; Morita et al. 2000; Vali et al. 2010), which raises the possibility that therapeutic strategies targeting these inhibitory pathways might be of clinical benefit.
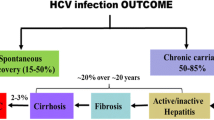
HCV infection is characterized by a high level of PD-1 and Tim-3 expressions on immune cells; up to 80 % of infected individuals develop persistent infection, and approximately 50 % of them fail to react appropriately to standard HBV vaccination. Inadequate HBV vaccine responses in chronic HCV infection provide an excellent model for examining the role of PD-1 and Tim-3 in regulation of immune response as a mechanism for vaccine non-responsiveness in immunocompromised hosts (Fig. 2). We have previously demonstrated that PD-1, SOCS-1, and Tim-3 are upregulated in APCs and T cells during chronic HCV infection, with dysregulation of immune responses contributing to persistent viral infection (Frazier et al. 2010; Ma et al. 2011; Moorman et al. 2009, 2012; Ni et al. 2010; Yao et al. 2007, 2008, 2010; Zhang et al. 2011a, b, 2012). Therefore, the PD-1 and Tim-3 pathways are both critical to terminating immune responses and are candidates for roles in vaccine nonresponsiveness. We have recently demonstrated PD-1 and Tim-3 up-regulation on monocytes and T cells from HCV-infected, HBV vaccine nonresponders compared with responders and observed a differential expression of IL-12/IL-23 in monocytes and IL-2/IL-17 in T cells (Moorman et al., unpublished data). Given the fact that the most common reason for vaccine failure in chronic viral infection is limited lymphocyte proliferative potential, a better understanding of this mechanism by which virus usurps host signaling machinery to modulate immune responses may open new avenues to enhance vaccine efficacy and immunotherapy.

A model for vaccine nonresponse in chronic viral infection. Multiple mechanisms are involved in the vaccine nonresponse during chronic viral infection; of note, Tim-3 and p16ink4a are only two negative signaling pathways of the mechanisms following HBsAg challenge. Persistent HIV/HCV-induced Tim-3 and/or p16ink4a expressions regulate DC, NK, Treg and T/B lymphocyte functions, resulting in a cytokine profile that contributes to blunted HBV vaccine responses in chronically virus-infected individuals. Therefore, defining the mechanisms and manipulating Tim-3/p16ink4a pathways may provide a novel therapy to restore HBV vaccine responses in the setting of chronic viral infection. This approach may apply to improve vaccine responses in other immunocompromised hosts
Immune Senescence in HBV Vaccine Failure During Chronic Viral Infections
In addition to inducing immune exhaustion that impairs essential functional activity, persistent viral infections can also lead to immune senescence, with accelerated premature aging due to telomere erosion or unrepaired DNA damage (Appay et al. 2007; Effros et al. 2008; Ferrando-Martínez et al. 2011; Voehringer et al. 2001). In the aging process, ataxia telangiectasia Rad3-related (ATR) and ataxia telangiectasia mutated (ATM) kinases are activated by double strand breaks in DNA or chromatin disruption (Sancar et al. 2004; Zou and Elledge 2003), which in turn, activate the DNA damage checkpoint (Bakkenist and Kastan 2003; Brown and Baltimore 2003). This senescence process seems to crosstalk with the cell exhaustion signal through a cascade of intracellular regulatory proteins, leading to cell cycle arrest and poor immune responses (Fig. 3). It has been well established that an age-related decline in immune responses in the elderly results in greater susceptibility to infection and reduced responses to vaccination. This decline in immune structure and function affects both innate and adaptive immune responses, and in parallel, the production of inflammatory mediators increases. The adaptive immune system depends on its proliferative capacity; however, the T cell repertoire, once established, is relatively robust to aging and only decompensates when stressed. Such stressors include chronic infections such as HCV and HIV, even when viral replication is controlled. Chronic immune activation in the presence of T cell exhaustion and DNA damage responses in these patients synergizes to develop an immune phenotype that is more characteristic of the elderly, with the declining ability of their immune system to respond to vaccines and to protect from infection (Le Saux et al. 2012).

A putative scheme suggesting that immune exhaustion and immune senescence signaling block over-activated helper T cell progression through distinct but cross-linked pathways. Immune exhaustion, mediated by inhibitory receptor signaling, such as PD-1/Tim-3, prevents PI3 K/Akt phosphorylation. This in turn lifts the block on forkhead box O (FOXO) transcription factors and activates p27kip1, causing G1-S phase transition. Immune senescence, on the other hand, activated by DNA or chromatin disruption to stimulate AT R and ATM kinases, which in turn, activate DNA damage checkpoint such as p53. p21cip1. p38 and p16ink4a. This causes G1 growth arrest by blocking the activations of cyclins and cyclin-dependent kinases (CDKs). thus induction of cell apoptosis. It is well known that the senescence-associated killer cell lectin-like receptor subfamily G member-1 (KLRG-1) also mediates its inhibitory signals by preventing Akt phosphorylation on Ser473, and this removes the block on p27kip1 transcription, enabling the G1-S phase transition. Interplay of immune exhaustion and senescence with regard to vaccine responses would be an interesting topic of research
Accumulating evidence suggests that cells of the immune system may have a limited lifespan in vivo following repeated antigenic stimulation. In this context, persistent activation during chronic HIV and/or HCV infection may lead to an exhaustion as well as senescence of immune resources. This may occur at two levels: clonal (virus-specific suppression) and global (general immune suppression). Some virus-specific T lymphocytes start expressing senescence markers (CD57, p16ink4a, KLRG-1, loss of CD28) soon after primary infection. Persistently activated, virus-specific T cell clones may eventually reach stages of senescence and disappear through cell apoptosis, resulting in the loss of antigen-specific CD4+ and/or CD8+ T cell populations important to controlling viral replication. In addition, HIV infection is characterized by the accumulation of highly differentiated CD8+CD28− T cells over time. Along with the decline of T cell renewal capacities, this may reflect a general aging of the lymphocyte population. Similar observations have been found in non-infected elderly individuals, suggesting that premature immune senescence occurs in the setting of chronic viral infections as a result of persistent immune stimulation. Accelerated immunosenescence in the setting of HIV/HCV diseases results in an aging state that diminishes the ability of the immune system to contain virus while at the same time facilitating viral replication and spread. Clinically, these changes result in a lower capacity to respond to new infections or vaccines as well as an increased frequency of age-associated end-organ disease (e.g. cardiovascular complications, cancer, and neurologic disease) that is associated with increased morbidity and mortality.
Essential features of immune senescence include reduced number and function of APCs in blood; reduced natural killer cell cytotoxicity; and decreased naive T and B cells with an increase in terminally differentiated lymphocytes. In particular, an accumulation of late differentiated effector/memory T cells contributes to a decline in the capacity of the adaptive immune system to respond to novel antigens. Consequently, vaccine responsiveness is compromised in the elderly, especially frail patients, as well as virally infected individuals. Indeed, we have recently found a significantly increased CD8+CD28− T cell accumulation in HCV-infected, HBV vaccine nonresponders versus responders (unpublished data). In the future, the development and use of markers of immunosenescence to identify patients who may have impaired responses to vaccination, as well as the use of end-points other than antibody titers to assess vaccine efficacy, may help to reduce morbidity and mortality due to chronic viral infections.
Because of the effect of aging on APC function, Treg-mediated immune suppression, reduced proliferative capacity of T cells, and other diminished immune responses, the efficacy of vaccines often wanes with advanced age. Strikingly, chronic HIV/HCV infections compress the aging process, accelerating comorbidities and frailty. A recent study demonstrated that young HIV-infected patients with less than 4 years of infection have early immune exhaustion leading to premature aging and senescence that is comparable to the elderly, suggesting virus-induced premature immune senescence associated with high rates of immune exhaustion following short-term infection (Ferrando-Martínez et al. 2011). We have also explored the role of HCV-mediated immune exhaustion and immune senescence in HBV vaccine responses during chronic HCV infection. We found that HCV-infected individuals exhibit higher expressions of both exhaustion and senescence markers—including PD-1/Tim-3 and KLRG-1/p16ink4a—in APC or helper T cells; this is associated with impaired cellular functions that are more significant in HBV vaccine non-responders compared with responders (unpublished data). Additionally, we have previously demonstrated that HCV arrests cell cycle progression through stabilization of p27kip1—an inhibitor of cell cycle regulatory proteins CDK and cyclin D/E (Yao et al. 2003). These findings have led to an intersection of the fields of virus-mediated immune exhaustion and immune senescence with regard to vaccine responses (Fig. 3). The mechanisms behind how HIV/HCV infection induces immune exhaustion and immune senescence, and whether these two distinct pathways interact each other during immune responses, have yet to be clearly defined but are essential for developing specific strategies to improve vaccine responses in the setting of viral infection.
Conclusion
Immune exhaustion and immune senescence are two distinct signaling pathways that coordinately regulate cell function and fate. Further investigation into their roles in vaccine responses during chronic viral infections is critical to identifying high priority topics and gaps for future research. Although there has been substantial progress in identifying the mechanisms that regulate both processes separately, it is unclear how these processes interrelate and whether blocking pathways that maintain either the exhausted or the senescent state or both can boost vaccine responses, especially in chronically virus-infected individuals. Answers to the questions posed are likely to help prioritize and balance strategies to slow the progression of persistent viral infections, to address co-morbidities and drug toxicity, and to enhance our understanding of the underlying mechanisms for HIV/HCV infections and their associated poor vaccine responses. Investigation in this area has the potential to improve vaccine development and advise more effective strategies to reduce the risk of vaccine-preventable illness in chronically virus-infected individuals. Additionally, novel approaches, such as viral vectors for antigen delivery, DNA-based vaccines and innovative adjuvants, and in particular Toll-like receptor agonists, may help to achieve optimal vaccine protection against chronic infectious diseases. Such studies will lead to novel guidelines for improving the vaccine response in immunocompromised hosts who are at high risk for infection, ultimately initiating steps to reduce clinical morbidity and mortality.
Abbreviations
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- HBsAg:
-
Hepatitis B surface antigen
- HBeAg:
-
Hepatitis B e antigen
- HAV:
-
Hepatitis A virus
- HLA:
-
Human leukocyte antigen
- APCs:
-
Antigen-presenting cells
- PD-1:
-
Programmed death-1
- SOCS-1:
-
Suppressor of cytokine signaling-1
- Tim-3:
-
T cell immunoglobulin domain protein-3
- IL-2:
-
Interleukin-2
- Treg:
-
Regulatory T cells
- KLRG-1:
-
Killer cell lectin like receptor G-1
- ATR and ATM:
-
Ataxia telangiectasia Rad3-related and ataxia telangiectasia mutated kinases
References
Albarran B, Goncalves B, Salmen B et al (2005) Profiles of NK, NKT cell activation and cytokine production following vaccination against hepatitis B. APMIS 113:526–535
Alexander WS (2002) Suppressors of cytokine signaling (SOCS) in the immune system. Nat Rev Immunol 2:410–416
Allain JP, Canotti D (2012) Hepatitis B virus in transfusion medicine: still a problem? Biologicals 40:180–186
Appay V, Almeida JR, Sauce D et al (2007) Accelerated immune senescence and HIV-1 infection. Exp Gerontol 42:432–437
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499–506
Barber DL, Wherry EJ, Asopust DM et al (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687
Bauer T, Jilg W (2006) Hepatitis B surface antigen-specific T and B cell memory in individuals who had lost protective antibodies after hepatitis B vaccination. Vaccine 24:572–577
Bauer T, Gunther M, Bienzie I et al (2007) Vaccination against hepatitis B in liver transplant receipients: pilot analysis of cellular immune response shows evidence of HBsAg-specific regulatory T cells. Liver Transpl 13:434–442
Biggar R, Goedert J, Hoofnagle J (1987) Accelerated loss of antibody to hepatitis B surface antigen among immunodeficient homosexual men infected with HIV. N Engl J Med 316:630–631
Bodsworth N, Donovan B, Nightingale B (1989) The effect of concurrent human immunodeficiency virus infection on chronic hepatitis B. a study of 150 homosexual men. J Infect Dis 160:577–582
Bodsworth NJ, Cooper DA, Donovan B (1991) The influence of human immunodeficiency virus type 1 infection on the development of the hepatitis B virus carrier state. J Infect Dis 163:1138–1140
Boni C, Fisicaro P, Valdatta C et al (2007) Charaterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol 81:4215–4225
Brown EJ, Baltimore D (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 17:615–628
Bruguera M, Cremades M, Salinas R et al (1992) Impaired response to recombinant hepatitis B vaccine in HIV-infected persons. J Clin Gastroenterol 14:27–30
Chen L (2004) Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol 4:336–347
Cheruvu S, Marks K, Talal AH (2007) Understanding and management of hepatitis B/HIV and hepatitis B/hepatitis C virus coinfection. Clin Liver Dis 11:917–943
Chong MM, Metcalf D, Jamieson E et al (2005) Suppressor of cytokine signaling-1 in T cells and macrophages is critical for preventing lethal inflammation. Blood 106:1668–1675
Collier A, Corey L, Murphy V et al (1988) Antibody to human immunodeficiency virus (HIV) and suboptimal response to hepatitis B vaccination. Ann Intern Med 109:101–105
D’Souza M, Fontenot AP, Mack DG et al (2007) Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J Immunol 179:1979–1987
De Silvestri A, Pasi A, Martinetti M et al (2001) Family study of non-responsiveness to hepatitis B vaccine confirms the importance of HLA class III C4A locus. Genes Immun 2:367–372
Desombere I, Hauser P, Rossau R et al (1995) Non responders to hepatitis B vaccine can present evelope particles to T lymphocytes. J Immunol 154(15):520–529
Desombere I, Cao P, Gijbels Y et al (2005) Non-responsiveness to hepatitis B surface antigen vaccines is not caused by defective antigen presentation or a lack of B7 co-stimulation. Clin Exp Immunol 140:126–137
Dong H, Strome SE, Salomao DR et al (2002) Tumor-associated B7–H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8:793–800
Duberg AS, Torner A, Daviosdottir L et al (2008) Cause of death in individuals with chronic HBV and/or HCV infection, a nationwide community-based register study. J Viral Hepat 15:538–550
Effros RB, Fletcher CV, Gebo K et al (2008) Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis 47:542–553
Egan PJ, Lawlor KE, Alexander WS et al (2003) Suppressor of cytokine signaling-1 regulates acute inflammatory arthritis and T cell activation. J Clin Invest 111:915–924
Eskild A, Magnus P, Petersen G et al (1992) Hepatitis B antibodies in HIV-infected homosexual men are associated with more rapid progression to AIDS. AIDS 6:571–574
Ferrando-Martínez S, Ruiz-Mateos E, Romero-Sánchez MC et al (2011) HIV infection-related premature immunosenescence: high rates of immune exhaustion after short time infection. Curr HIV Res 9:289–294
Filippini P, Coppola N, Pisapia R et al (2007) Virological and clinical aspects of HBV-HCV coinfection in HIV positive patients. J Med Virol 79:1679–1685
Fisman DN, Agrawal D, Leder K (2002) The effect of age on immunologic response to recombinant hepatitis B vaccine: a meta-analysis. Clin Infect Dis 35:1368–1375
Frazier AD, Zhang CL, Ni L et al (2010) Program death-1 pathway affects suppressor of cytokine signaling-1 expression in T cells during hepatitis C infection. Viral Immunol 23:487–495
Gilson RJ, Hawkins AE, Beecham MR et al (1997) Interactions between HIV and hepatitis B virus in homosexual men: effects on the natural history of infection. AIDS 11:597–606
Godkin A, Davenport M, Hill AV (2005) Molecular analysis of HLA class II associations with hepatitis B virus clearance and vaccine nonresponsiveness. Hepatology 41:1383–1390
Golden-Mason L, Palmer B, Klarquist J et al (2007) Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol 81:9249–9258
Goncalves L, Albarran B, Salmen S et al (2004) The nonresponse to hepatitis B vaccination is associated with impaired lymphocyte activation. Virology 326:20–28
Hafler DA, Kuchroo V (2008) TIMs: central regulators of immune responses. J Exp Med 205:2699–2701
Hohler T, Stradmann-Bellinghausen B, Starke R et al (2002) C4A deficiency and nonresponse to hepatitis B vaccination. J Hepatol 37:387–392
Horvath J, Raffanti S (1994) Clinical aspects of the interactions between human immunodeficiency virus and the hepatotropic viruses. Clin Infect Dis 18:339–410
Iwai Y, Terawaya S, Ikegawa M et al (2003) PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med 198:39–50
Jacques P, Moens G, Desombere I et al (2002) The immunogenicity and reacogenicity profile of a candidate hepatitis B vaccine in an adult vaccine non-responder population. Vaccine 20:3644–3649
Jin HT, Anderson AC, Tan WG et al (2010) Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA 107:14733–14738
Jones RB, Ndhlovu LC, Barbour JD et al (2008) Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med 205:2763–2779
Kao JH, Chen DS (2002) Global control of hepatitis B virus infection. Lancet Infect Dis 2:395–403
Keeffe EB (2005) Acute hepatitis A and B in patients with chronic liver disease: prevention through vaccination. Am J Med 118((Suppl 10A)):21S–27S
Kellerman SE, Hanson DL, McNaghten AD et al (2003) Prevalence of chronic hepatitis B and incidence of acute hepatitis B infection in human immunodeficiency virus-infected subjects. J Infect Dis 188:571–577
Khoury SJ, Sayegh MH (2004) The role of the new negative T cell costimulatory pathways in regulating autoimmunity. Immunity 20:529–538
Kim MJ, Nafziger AN, Harro CD et al (2003) Revaccination of healthy nonreasponders with hepatitis B vaccine and prediction of seroprotection response. Vaccine 21:1174–1179
Klein SL (2012) Sex influences immune responses to viruses, and efficacy of prophylaxis and treatments for viral diseases. Bio Essays 34:1050–1059
Klein SL, Jedlicka A, Pekosz A (2010) The Xs and Y of immune responses to viral vaccines. Lancet Infect Dis 10:338–349
Kramer ES, Hofmann C, Smith PG et al (2009) Response to hepatitis A and B vaccine alone or in combination in patients with chronic hepatitis C virus and advanced fibrosis. Dig Dis Sci 54:2016–2025
Kuchroo VK, Umetsu DT, DeKruyff RH et al (2003) The TIM gene family: emerging roles in immunity and disease. Nat Rev Immunol 3:454–462
Lango-Warensjo A, Cardell K, Lindblom B (1998) Haplotypes comprising subtypes of the DQB1*06 allele direct the antibody response with hepatitis B surface antigen. Tissue Antigens 52:374–380
Lazizi Y, Grangeot-Keros L, Delfraissy J et al (1988) Reappearance of hepatitis B virus in immune patients infected with the human immunodeficiency virus type 1. J Infect Dis 158:666–667
Le Saux S, Weyand CM, Goronzy JJ (2012) Mechanisms of immune senescence: lessons from models of accelerated immune aging. Ann NY Acad Sci 1247:69–82
Ma CJ, Ni L, Zhang Y et al (2011) PD-1 negatively regulates IL-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages during chronic hepatitis C infection. Immunology 132:421–431
Macedo LC, Isolani AP, Visentainer JE et al (2010) Association of cytokine genetic polymorphrisms with the humoral immune response to recombinant vaccine against HBV in infants. J Med Virol 82:929–933
Malaspina A, Moir S, Orsega SM et al (2005) Compromised B cell responses to influenza vaccination in HIV-infected individuals. J Infect Dis 191:1442–1450
McMahan RH, Golden-Mason L, Nishimura MI et al (2010) Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest 120:4546–4557
Moorman JP, Dong ZP, Ni L et al (2009) Abnormal B lymphocyte activation associated with TALL-1 overexpression and SOCS-1 deregulation in chronic HCV infection. Immunology 128:227–235
Moorman JP, Zhang CL, Ni L et al (2011) Impaired hepatitis B vaccine responses during chronic hepatitis C infection: involvement of the PD-1 pathway in regulating CD4+ T cell responses. Vaccine 29:3169–3176
Moorman JP, Wang JM, Zhang Y et al (2012) Tim-3 controls regulatory and effector T cell balance during HCV Infection. J Immunol 189:755–766
Morita Y, Naka T, Kawazoe Y et al (2000) Signals transducers and activators of transcription (STAT)-indueced STAT inhibitor (SS-1)/suppressor of cytokine signaling-1 (SOCS-1) suppreses tumor necrosis factor α-induced cell death in fibroblasts. Proc Natl Acad Sci USA 97:5405–5410
Ni L, Ma CJ, Zhang Y et al (2010) PD-1 modulates Regulatory T cells and suppresses T cell responses in HCV-associated Lymphoma. Immunol Cell Biol 89:535–539
Nishimura H, Honjo T (2001) PD-1: an inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol 22:265–268
Nystrom J, Cardell K, Bjornsdottir TB et al (2008) Improved cell mediated immune responses after successful re-vaccination of non-responders to the hepatitis B virus surface antigen (HBsAg) vaccine using the combined hepatitis A and B vaccine. Vaccine 26:5967–5972
Okazaki T, Honjo T (2007) PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 19:813–824
Rahman F, Dahmen A, Herzog-Hauff S et al (2000) Cellular and humoral immune responses induced by intradermal or intramuscular vaccination with a major hepatitis B surface antigen. Hepatology 31:521–527
Ramon JM, Bou R, Oromi J (1996) Low-dose intramuscular revaccination against hepatitis B. Vaccine 14:1647–1650
Rodriguez-Barradas MC, Alexandraki I, Nazir T et al (2003) Response of human immunodeficiency virus-infected patients receiving highly active antiretroviral therapy to vaccination with 23-valent pneumococcal polysaccharide vaccine. Clin Infect Dis 37:438–447
Rossi C, Shrier I, Marshall L et al (2012) Seroprevalence of chronic hepatitis B virus infection and prior immunity in immigrants and refugees: a systematic review and meta-analysis. PLoS One 7:e44611
Ryckman KK, Fielding K, Hill AV et al (2010) Host genetic factors and vaccine-induced immunity to HBV infection: haplotype analysis. PLoS One 5:e12273
Salazar M, Deulofeut H, Granja C et al (1995) Normal HBsAg presentation and T cell defect in the immune response of nonresponders. Immunogenetics 41:366–374
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K et al (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85
Shafran SD, Mashinter LD, Lindemulder A et al (2007) Poor efficacy of intradermal administration of recombinant hepatitis B virus immunization in HIV-infected individuals who fail to respond to intramuscular administration of hepatitis B virus vaccine. HIV Med 8:295–299
Sharpe AH, Freeman GJ (2002) The B7-CD28 superfamily. Nat Rev Immunol 2:116–126
Stramer SL, Wend U, Candotti D et al (2011) Nucleic acid testing to detect HBV infection in blood donors. N Engl J Med 364:236–247
Thio CL, Seaberg EC, Skolasky RJ et al (2002) HIV-1, hepatitis B virus, and risk for liver-related mortality in the Multicenter Cohort Study (MACS). Lancet 360:1921–1926
Torbenson M, Kannangai R, Astemborski J et al (2004) High prevalence of occult hepatitis B in Baltimore injection drug users. Hepatology 39:51–57
Tsai IJ, Chang MH, Chen HL et al (2000) Immunogenicity and reactogenicity of the combined hepatitis A and B vaccine in young adults. Vaccine 19:437–441
Vali B, Jones RB, Sakhdari A et al (2010) HCV-specific T cells in HCV/HIV co-infection show elevated frequencies of dual Tim-3/PD-1 expression that correlate with liver disease progression. Eur J Immunol 40:2493–2505
Verkade MA, van Druningen CJ, de Op Hoek CT et al (2007) Decreased antigen-specific T cell proliferation by moDC among hepatitis B vaccine non-responders on haemodialysis. Clin Exp Med 7:65–71
Voehringer D, Blaser C, Brawand P et al (2001) Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol 167:4838–4843
Weber R, Sabin CA, Friis-Moller N et al (2006) Liver-related deaths in persons infected with the human immunodeficiency virus: the D:A:D Study. Arch Intern Med 166:1632–1641
Yao ZQ, Eisen-Vandervelde A, Ray S et al (2003) HCV core/gC1qR interaction arrests T cell cycle progression through stabilization of the cell cycle inhibitor p27kip1. Virology 314:271–282
Yao ZQ, King E, Prayther D et al (2007) T cell dysfunction by hepatitis C virus core protein involves PD-1/PDL-1 signaling. Viral Immunol 20:276–287
Yao ZQ, Prayther D, Trabu C et al (2008) Differential regulation of SOCS-1 signaling in T and B lymphocytes by HCV core protein. Immunology 125:197–207
Yao ZQ, Ni L, Zhang Y et al (2010) Differential regulation of T and B lymphocytes by PD-1 and SOCS-1 signaling in hepatitis C virus-associated non-Hodgkin’s lymphoma. Immunol Invest 40:243–264
Zarski JP, Bohn B, Bastie A et al (1998) Characteristics of patients with dual infection by hepatitis B and C viruses. J Hepatol 28:27–33
Zhang Y, Ma CJ, Ni L et al (2011a) Crosstalk between PD-1 and SOCS-1 in HCV core-mediated IL-12 suppression. J Immunol 186:3093–3103
Zhang Y, Ma CJ, Wu XY et al (2011b) Tim-3 negatively regulates IL-12 production by monocytes in HCV infection. PLoS One 6:e19664
Zhang Y, Cheng J, Ma CJ et al (2012) Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. J Leukoc Biol 91:189–196
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300:1542–1548
Acknowledgments
This work was supported by an NIH NIDDK grant to ZQY/JPM (R01DK093526). The authors disclose no conflicts of interest. They would like to appreciate the supporting staff from the Hepatitis (HCV/HIV) Program at James H. Quillen VAMC. This publication is the result of work supported with resources and the use of facilities at the James H. Quillen Veterans Affairs Medical Center. The contents in this publication do not represent the views of the Department of Veterans Affairs or the United States Government.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Yao, Z.Q., Moorman, J.P. Immune Exhaustion and Immune Senescence: Two Distinct Pathways for HBV Vaccine Failure During HCV and/or HIV Infection. Arch. Immunol. Ther. Exp. 61, 193–201 (2013). https://doi.org/10.1007/s00005-013-0219-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-013-0219-0