
Abstract.
Hutchinson-Gilford progeria (HGPS) is a premature aging syndrome associated with LMNA mutations. Progeria cells bearing the G608G LMNA mutation are characterized by accumulation of a mutated lamin A precursor (progerin), nuclear dysmorphism and chromatin disorganization. In cultured HGPS fibroblasts, we found worsening of the cellular phenotype with patient age, mainly consisting of increased nuclear-shape abnormalities, progerin accumulation and heterochromatin loss. Moreover, transcript distribution was altered in HGPS nuclei, as determined by different techniques. In the attempt to improve the cellular phenotype, we applied treatment with drugs either affecting protein farnesylation or chromatin arrangement. Our results show that the combined treatment with mevinolin and the histone deacetylase inhibitor trichostatin A dramatically lowers progerin levels, leading to rescue of heterochromatin organization and reorganization of transcripts in HGPS fibroblasts. These results suggest that morpho-functional defects of HGPS nuclei are directly related to progerin accumulation and can be rectified by drug treatment.
Similar content being viewed by others


Hutchinson-Gilford progeria syndrome (HGPS, OMIM 176670) is a rare pre-mature aging disorder clinically characterized by postnatal growth retardation, midface hypoplasia, premature atherosclerosis, absence of subcutaneous fat, alopecia and generalized osteodysplasia with osteolysis and pathologic fractures [1–3]. Death occurs in the teenage usually due to coronary artery disease [2]. HGPS is caused by sporadic mutations in LMNA (OMIM 150330) coding for alternatively spliced lamins A and C. At least five different mutations within the LMNA gene have been found in patients with HGPS [3–5]. However, most HGPS-described cases harbor an identical C-to-T transition at position 1824 of the coding sequence in the context of a CpG dinucleotide [2]. This mutation results in a silent polymorphism at codon 608 within exon 11 (G608G), which causes the activation of a cryptic donor-splicing site, resulting in deletion of 150 bp in the mRNA [5]. A truncated pre-lamin A isoform is therefore translated which has been recently referred to as progerin [4, 5]. Progerin lacks 50 amino acids including the endoprotease cleavage site present at the C terminus of the wild-type lamin A precursor [2]. Therefore, the HGPS pathogenesis is likely to arise from accumulation of the mutated 67 kDa isoform of farnesylated pre-lamin A [2]. Nuclear envelope-nuclear lamina abnormalities and loss of peripheral heterochromatin have been described in HGPS fibroblasts [6]. We observed these defects in the three cell lines examined in this study and showed that they are more represented in cell lines from older patients, concomitant with the increased appearance of progerin in those cells. In HGPS nuclei, transcript distribution was altered, while transcriptional activity was slightly decreased, despite the massive chromatin decondensation. In an attempt to destabilize the farnesylated protein, we treated HGPS cells with a farnesyl-transferase inhibitor (mevinolin). We observed a slight decrease in the amount of progerin and rescue of heterochromatin organization in a low percentage of mevinolin-treated cells. In a second step, we attempted to restore heterochromatin areas in HGPS nuclei using drugs known to modify chromatin organization. Unexpectedly, rescue of heterochromatin organization and transcript distribution was obtained in a high percentage of HGPS fibroblasts receiving combined treatment with mevinolin and trichostatin A. This treatment also dramatically lowered the progerin level.
Materials and methods
Cell cultures. Skin fibroblast cultures from three HGPS patients carrying a G608G lamin A/C mutation were obtained from the Progeria Research Foundation, while control cultures were obtained in our laboratory from skin biopsies of unaffected controls (age 2 to 50) following written consent. Fibroblast cultures were established and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (FCS) and antibiotics (penicillin, 50 units/ml; streptomycin, 50 mg/ml; amphotericin B, 250 µg/ml).
The three HGPS cell lines were: HGADFN001 (age 9 years, passage 13), HGADFN003 (age 2 years, passage 13), HGADFN127 (age 3 years, passage 7). The experiments were performed at these passage numbers, except when stated otherwise.
Drug treatments. To induce defarnesylation of progerin, 25 µM mevinolin (Sigma) was added to the culture medium for 18 h. The following chromatin-modifying agents were used: (i) 5-azadeoxycytidine (Sigma), (ii) anacardic acid (Sigma), (iii) trichostatin A (TSA) (Sigma). 5-Azadeoxycytidine was applied to cultured fibroblasts for 18 h following mevinolin treatment. This drug inhibits DNA methylation and, at the dosage here employed (10 µM for 18 h), elicits constitutive heterochromatin decondensation [7]. The inhibitor of histone acetyl transferases, anacardic acid, was applied to cultured fibroblasts for 18 h at 9 µM. The histone deacetylase (HDAC) inhibitor TSA (Sigma) [8] was applied to cell cultures as follows. Samples were pretreated or not with mevinolin for 18 h, thereafter culture medium was replaced and 1.5 µM TSA was added for 24 h [8].
Antibodies. Antibodies employed for Western blot (WB) analysis or immunofluorescence (IF) labeling were: anti-lamin A/C monoclonal (Novocastra Laboratories), diluted 1:50 for IF analysis; anti-lamin A/C goat polyclonal (Santa Cruz, SC-6215), diluted 1:100 for IF analysis and 1:100 for WB analysis; anti-pre-lamin A goat polyclonal (Santa Cruz, SC-6214) [9] diluted 1:100 for WB analysis; anti-emerin mouse monoclonal (Novocastra Laboratories) diluted 1:50 for IF analysis and 1:100 for WB analysis; anti-trimethylated-H3 histone (lysine 9) (tri-H3K9), mono-methylated (lysine 9) H3 histone (mono-H3K9) and dimethylated-H3 histone (lysine 9) (di-H3K9) (Abcam) diluted 1:30 for IF analysis; anti-BrdU monoclonal antibody (BD-Biosciences) diluted 1:1000 for IF analysis; anti-lamin A 2H10 monoclonal antibody [10] diluted 1:400 for IF analysis.
In situ transcription assay. Living fibroblasts were treated according to published protocols [10]. Briefly, cells were permeabilized in a buffer containing 20 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 0.5 mM EGTA, 25% glycerol, 10 µg/ml digitonin, 1 mM PMSF, RNasin (20 U/ml). The transcription assay was performed for 5 min in a buffer containing 50 mM Tris-HCl, pH 7.4, 100 mM KCl, 10 mM MgCl2, 0.5 mM EGTA, 25% glycerol, 2 mM ATP, 0.5 mM CTP, 0.5 mM GTP, 0.5 mM BrUTP, 1 mM PMSF and RNasin (20 U/ml). Alpha-amanitin (10 µg/ml) (Sigma) was added to the transcription buffer to obtain negative controls (not shown). Cells were washed in PBS and fixed in 2% paraformaldehyde at room temperature. Incorporated nucleotides were revealed by immunofluorescence labeling with anti-BrdU antibody.
Western blot. Western blot analysis of cellular lysates from human fibroblasts was done as follows. Cells were lysed in buffer containing 10 mM Tris-HCl, pH 7.4, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM NaVO3, 150 M NaCl, 1 mM EDTA, 1 mM PMSF, 1 µM aprotinin, leupeptin, pepstatin. Proteins were loaded in Laemmli sample buffer and subjected to SDS-PAGE followed by immunochemical reactions. Immunoblotted bands were detected by enhanced chemiluminescence (Amersham).
Immunofluorescence. Human fibroblasts grown on coverslips were fixed in paraformaldheyde 4% in PBS at 4 °C and post-fixed in methanol at −20 °C for 7 min. Samples were incubated with PBS containing 4% BSA to saturate non-specific binding. Incubation with primary antibodies was performed overnight at 4 °C, while secondary antibodies were applied for 1 h at room temperature. Slides were mounted with an anti-fade reagent in glycerol and observed with a Nikon E 600 fluorescence microscope equipped with a digital camera. Pictures were elaborated with Photoshop-6 software.
Electron microscopy. Cell pellets from control and HGPS fibroblasts (subjected or not to drug treatment) were fixed with 2.5% glutaraldehyde-0.1 M phosphate buffer pH 7.3 for 1 h at room temperature. After post-fixation with 1% osmium tetroxide (OsO4) in veronal buffer for 1 h, pellets were dehydrated in an ethanol series, infiltrated with propylene oxide and embedded in Epon resin. Ultrathin sections (60 nm thick) were stained with uranyl acetate and lead citrate (10 min each) and were observed at 0° tilt angle with a Philips EM 400 transmission electron microscope, operated at 100 kV. At least 200 nuclei per sample were observed.
Regressive EDTA staining was carried out according to the procedure of Bernhard [11]. Briefly, ultrathin sections of Epon-embedded control or HGPS fibroblasts (fixed with 2.5% glutaraldehyde in phosphate buffer pH 7.3, avoiding OsO4 post-fixation) were floated for 10 min on 5% aqueous uranyl acetate at room temperature, washed with distilled water and treated with 0.2 M EDTA for 60 min. Sections were washed with distilled water and contrasted for 10 min with standard lead citrate at room temperature.
Results
Progerin accumulation in HGPS fibroblasts correlates with nuclear defects. Progerin was detected in the three examined cell lines by Western blot analysis (fig. 1A). An increased proportion of progerin relative to mature lamin A was determined in the cell line HGADFN001 from the older patient (fig. 1A). The lamin A/C level was reduced in HGADFN001 fibroblasts (fig. 1A). Emerin expression was not affected in HGPS cells (fig. 1A).

Nuclear defects in HGPS fibroblasts. (A) Western blot analysis of lamins in HGPS fibroblasts. Lamin A and C were expressed at normal levels in the cell lines HGADFN003 (passage 13) and HGADFN127 (passage 7), while both lamin A and C levels were decreased in the cell line HGADFN001 (passage 13). Progerin (67 kDa) was detected in all examined cellular lysates, with an increased proportion relative to mature lamin A in the older patient cell line HGADFN001. The emerin level was not altered in HGPS cells. Molecular-weight markers are reported in kDa. The age of each patient is indicated (y, years), the number of cell passages is indicated in the lower row (p, passages). (B) One control cell line and three HGPS cell lines (HGADFN003, HGADFN127 and HGADFN001; see Materials and methods for details) were examined by IF analysis using anti-lamin A/C (green) and anti-emerin antibodies (red). The nuclear shape was altered in HGPS fibroblasts and the nuclear envelope invaginations are evident. Emerin mostly colocalized with lamin A/C. Nuclear shape abnormalities increased in the older cell line. (C) Ultrastructural analysis of control and HGPS nuclei (HGADFN003, HGADFN127 and HGADFN001; see Materials and methods for details). Nuclear invaginations are evident in 50% of examined nuclei from HGADFN127 cells and their frequency increased in HGADFN003 and HGADADFN001 cells. Heterochromatin loss was observed in all HGPS cell lines and heterochromatin areas were completely absent from HGADFN0001 nuclei. Nuclear invaginations including the outer nuclear membrane (arrowheads) or selectively involving the inner nuclear membrane and the lamina (including the perinuclear cisterna, arrows) are shown. (D) Quantitation of misshapen nuclei shown in B. Nuclei showing at least five invaginations were considered nuclei with invaginations (left columns). Nuclei showing an increase in the major diameter of at least 20% of control nuclei were considered enlarged nuclei (right columns). One thousand nuclei were scored per cell line (see legend for details). Triplicate staining experiments (lamin A/C-emerin staining) were examined. Mean values including standard errors of the mean were calculated and reported as percentage of counted nuclei.
Nuclear shape was dramatically altered in HGPS fibroblasts, with enlarged nuclei characterized by invaginations and blebs (fig. 1B). Double-immunofluorescence labeling of HGPS fibroblasts was performed using anti-lamin A/C and anti-emerin antibodies. Lamin A/C and emerin mostly co-localized in progeria nuclei (fig. 1B). Worsening of nuclear envelope defects with patient age was evident (compare HGADFN003 and HGADFN001 nuclei; fig. 1B).
Electron microscopy analysis of HGPS nuclei confirmed the worsening of nuclear defects with patient age (fig. 1C). Focal loss of peripheral heterochromatin was observed in most nuclei, while heterochromatin areas became almost undetectable in the older cell line (fig. 1C). Nuclear envelope invaginations were observed (fig. 1C). In some instances, nuclear invaginations also contained the outer nuclear membrane (fig. 1C; arrowheads), but more often were devoid of the outer nuclear membrane and included the perinuclear cisterna (fig. 1C; arrows).
Methylation of H3 histone is altered in HGPS nuclei. The methylation pattern of H3 histone was investigated in HGPS cells. Figure 2 shows immunofluorescence labeling of mono-, di- and tri-H3K9 in HGADFN127 fibroblasts. Mono-H3K9 staining was reduced in HGPS nuclei and was moderately detectable in 10–20% of nuclei (fig. 2A). The labeling pattern of di-H3K9 was comparable to controls (fig. 2B), while a dramatic alteration of tri-H3K9 was observed in HGPS cells. Twenty percent of HGADFN127 nuclei were negative for tri-H3K9 staining (fig. 2C). Even more striking alterations of labeling patterns were observed in the other HGPS cell lines (fig. 2C, graph). These results show that both facultative heterochromatin methylation (detected by mono-H3K9 labeling) and constitutive heterochromatin methylation (revealed by tri-H3K9 staining) [12] are altered in HGPS.

Immunofluorescence labeling of methylated H3 histone species in control and HGPS fibroblasts. H3K9 labeling was performed by specific antibodies and revealed by FITC-conjugated secondary antibody (green); lamin A/C was labeled by polyclonal antibody and revealed by Cy-3-conjugated secondary antibody (red); DNA was counterstained with DAPI; mono- H3K9 labeling (A); di- H3K9 labeling (B); tri-H3K9 labeling (C). A representative experiment performed in HGADFN127 fibroblasts is shown in this picture. Quantitation of H3K9 staining shown in each panel is reported on the right. Bars show the mean percentage of nuclei with reduced fluorescence intensity. Nuclei with reduced fluorescence intensity were considered the nuclei with less than 50% of the mean fluorescence intensity measured in control nuclei (Lucia Image Version 4.61 software was used to measure fluorescence intensity per area). Mean values of triplicate counting performed in each patient cell line (see legend for details) including standard errors of the mean were calculated.
Progerin accumulation is reduced by mevinolin treatment. In the attempt to destabilize progerin, we used mevinolin to obtain defarnesylation. Figure 3A shows the WB analysis of progerin in HGPS fibroblasts subjected to mevinolin treatment. Control fibroblasts were also treated with the farnesyltransferase inhibitor and subjected to Western blot analysis using anti-lamin A/C antibody (fig. 3) and anti-pre-lamin A antibody (not shown). Beside the expected increase in wild-type prelamin A caused by mevinolin in all the examined cell lines, a decrease in the amount of progerin was observed in HGPS lysates (fig. 3A). The lamin A level was slightly reduced by mevinolin treatment, while amounts of lamin C and emerin were not affected (fig. 3A).

WB analysis of lamins in drug-treated HGPS fibroblasts. Control and HGPS fibroblasts were subjected to drug treatment, lysed and protein separation was obtained by SDS-PAGE. An immunoblot was performed using anti-lamin A/C or anti-emerin antibodies. (A) Untreated control fibroblasts show an almost undetectable amount of pre-lamin A (lane 1). Untreated HGPS fibroblasts show a low amount of wild-type pre-lamin A and a high amount of progerin (lane 3). Increased pre-lamin A levels were observed after mevinolin addition in all examined cell lines (lanes 2 and 4). The amount of progerin was reduced in mevinolintreated HGPS cells (lane 4). (B) Further treatment with TSA led to a slight reduction in wild-type pre-lamin A levels (lanes 5, 8), while the progerin level was strikingly lowered (lane 8). TSA treatment (in the absence of mevinolin) slightly increased the lamin A/C level both in control (lane 6) and HGPS cells (lane 9), but minimally affected the progerin level (lane 9). The Wild-type prelamin A was reduced by addition of 5-azadeoxycytidine (5-AC) to mevinolin-treated control (lane 7) and HGPS cells (lane 10). The amount of progerin was not affected by 5-AC (lane 10). The lamin A/C level was minimally affected by 5-AC (lane 7 and 10). Emerin was not altered by drug treatments in control or HGPS cell lines. Actin bands are labeled as a loading control. A representative experiment is shown of three different experiments performed in each control and HGPS cell line. (C) Densitometric analysis of pre-lamin A and progerin immunoblotted bands shown in A and B. Pre-lamin A values are reported as the percentage of pre-lamin A amount detected in mevinolin-treated cells; progerin values are reported as the percentage of progerin amount detected in untreated HGPS cells. Values are the mean ± standard error of the mean of three independent experiments performed in HGADFN001 cells.
Progerin accumulation is abolished by mevinolin/TSA treatment. In the attempt to investigate heterochromatin dynamics in HGPS nuclei, we treated HGPS fibroblasts with a set of drugs capable of interfering with chromatin organization. The effect of each treatment on progerin accumulation was evaluated by WB analysis (fig. 3B). Treatment of progeria fibroblasts with 5-azadeoxycytidine slightly increased the amount of progerin (not shown). TSA treatment of HGPS fibroblasts slightly affected the progerin level (fig. 3B, lane 9). Addition of each chromatin-modifying drug to mevinolin-treated cells elicited the following effects. 5-Azadeoxycytidine addition lowered wild-type pre-lamin A levels both in control and HGPS cells (fig. 3B, lane 7 and 10). Dramatic lowering of the progerin level was observed in HGPS cells sequentially treated with mevinolin and TSA (fig. 3B, lane 8). A moderate reduction in wild-type pre-lamin A was also observed in these cells, while lamin A and C levels were slightly affected by TSA addition (fig. 3B, lane 8). Anacardic acid elicited effects on progerin and mature lamin A expression, which, however, were not consistent and require further investigation (not shown). We performed a statistical evaluation of wild-type pre-lamin A and progerin densitometric values referred to immunoblotted bands (fig. 3C). A significant decrease in progerin accumulation is clearly obtained by mevinolin/TSA administration (fig. 3C). The overall evaluation of graphs shows that each drug treatment elicited very different effects on wild-type compared to mutated lamin A precursor (fig. 3C).
Heterochromatin organization in HGPS nuclei is improved by mevinolin/TSA treatment. By ultrastructural analysis, heterochromatin areas were not detected in untreated HGADFN001 fibroblasts (fig. 4A), while they were observed at the nuclear periphery in 5% of mevinolin-treated HGADFN001 fibroblasts (fig. 4B). However, mevinolin treatment of HGPS fibroblasts apparently failed to improve nuclear shape abnormalities (not shown). TSA alone did not cause detectable changes in chromatin organization in control or in HGPS cells (fig. 4C). In all the examined HGPS cell lines, administration of mevinolin plus TSA elicited reorganization of peripheral heterochromatin (fig. 4D). Nuclear shape was also improved after the combined treatment with mevinolin and TSA (fig. 4D).

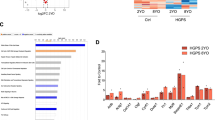
Heterochromatin organization in mevinolin/TSA-treated HGPS fibroblasts. (A–D) Experiments are representative of three independent analyses performed in each cell line, and the pictures were obtained by transmission electron microscopy. (A) Heterochromatin areas are evident at the nuclear periphery of untreated control nuclei, except at the nuclear pores (*). In untreated HGADFN001 nuclei, peripheral heterochromatin is absent. (B) Mevinolin treatment elicited formation of heterochromatin areas both in control and HGPS cells. The percentage of nuclei showing heterochromatin areas was increased by 5% in HGADFN001 nuclei. (C) TSA treatment did not significantly change the number of nuclei with heterochromatin areas either in control or in HGPS cells. (D) Mevinolin plus TSA treatment slightly affected heterochromatin organization in control fibroblasts, while it raised to 40% the percentage of HGADFN001 nuclei showing heterochromatin areas. (E) Double-IF staining of tri-H3K9 and lamin A/C in HGADFN001 fibroblast nuclei. Anti-tri-H3K9 antibody was revealed by FITC-conjugated secondary antibody (green); anti-lamin A/C antibody was revealed by Cy3-conjugated secondary antibody (red). Tri-H3K9 labeled discrete foci in control nuclei (control), while labeling was lost in 36% of HGPS nuclei (HGPS). Mevinolin treatment (mevinolin) did not affect the number of tri-H3K9-positive control cells, while slightly increasing the number of tri-H3K9-positive HGPS cells. TSA treatment (TSA) affected the distribution of tri-H3K9 foci in control cells, while it was not effective in HGPS cells. In HGPS nuclei, complete recovery of tri-H3K9 labeling was obtained after mevinolin + TSA treatment (mevinolin/TSA). (F) Quantitation of the electron microscopy analysis shown in panels A-D. The percentage of nuclei showing at least four heterochromatin areas was reported for control and HGADFN001 fibroblasts. (G) Quantitation of tri-H3K9 positive nuclei in HGADFN001 fibroblasts before and after each drug treatment (shown in E). Mean values of triplicate experiments including standard errors of the mean were calculated. One thousand nuclei per sample were counted.
To support the reported findings, we checked the distribution of tri-H3K9 before and after mevinolin/TSA treatment. Tri-H3K9 stained discrete sites in control nuclei, but labeling was not detected in most HGPS nuclei (fig. 2C, 4E). Tri-H3K9 staining was increased in a minor, not statistically significant percentage of mevinolin-treated HGPS cells (fig. 4E). Histone staining was restored by mevinolin/TSA treatment of each cell line (fig. 4E). Statistical analysis of the experiments shown in figure 4A–D is reported in figure 4F. The percentage of nuclei showing at least four heterochromatin areas rose to 40% in HGADFN001 cells after mevinolin/TSA treatment (fig. 4F). Figure 4G shows the statistical analysis of the experiments shown in Figure 4E. The percentage of HGPS nuclei showing tri-H3K9 staining is increased to almost 100% by mevinolin/TSA treatment (fig. 4G).
Ribonucleoprotein levels are reduced in HGPS cells and restored by mevinolin/TSA treatment. We sought to determine if the increased availability of dispersed chromatin could affect the transcriptional activity of HGPS nuclei. In particular, we wished to determine if dispersed chromatin areas observed by electron microscopy corresponded to domains of increased transcript accumulation. An altered distribution of mRNA transcripts was revealed by an in situ transcription assay. Control nuclei showed a uniform staining of BrU all over the nucleus (fig. 5A). The amount of incorporated BrU was reduced in enlarged HGPS nuclei showing major nuclear lamina defects and BrU-containing transcripts were distributed in the nucleus with a non-uniform pattern (fig. 5A). After treatment with mevinolin and TSA, BrU incorporation was comparable to controls in most HGPS nuclei, including some enlarged nuclei (fig. 5A). Uniform BrU staining was observed in HGPS nuclei after mevinolin/TSA treatment (fig. 5A). Reverse EDTA staining of control nuclei allowed us to identify ribonucleoproteins distributed to discrete clusters throughout the nuclei except at the nuclear periphery, where bleached chromatin was evident (fig. 5B, control). In mevinolin/TSA-treated control nuclei, a uniform distribution of transcripts all over the nucleus was observed (fig. 5B, mevinolin/TSA control). Ribonucleoprotein staining was dramatically reduced in 36% of HGPS nuclei and a faint gray staining was observed throughout the nucleus, including the nuclear periphery (fig. 5B, untreated HGPS). Bleached chromatin areas at the nuclear lamina were absent, while an irregular thickening of the lamina was revealed (fig. 5B, arrows). Mevinolin/TSA treatment of HGPS cells restored the ribonucleoprotein distribution (fig. 5B, mevinolin/TSA HGPS) and improved nuclear lamina organization leading to rescue of heterochromatin areas (arrowheads). An altered distribution of intranuclear lamin A associated with splicing factor compartments was observed in all HGPS cell lines (fig. 5C). Mevinolin/TSA treatment restored the intranuclear lamin A labeling pattern in HGPS nuclei (fig. 5C).

Transcript distribution was altered in HGPS nuclei. (A) In situ transcription assay showing uniform BrU staining in control nuclei (91% out of 200 counted nuclei) and reduced transcriptional activity and nonuniform BrU distribution in enlarged HGADF001 nuclei (41% out of 200 counted nuclei). Uniform BrU staining was observed in HGPS nuclei after mevinolin/TSA treatment (83% out of 200 counted nuclei) (right column). RNA transcripts incorporating BrU were labeled by anti-BrU antibody and revealed by Cy-3-conjugated secondary antibody (red). The nuclear lamina was labeled by anti-lamin A/C antibody and revealed by FITC-conjugated secondary antibody (green). The merged images (merge) underscore the different BrU distribution in control and HGPS nuclei. Images were obtained by fluorescence microscopy. (B) EDTA-reverse staining of a control and an HGADFN001 nucleus. In the control nucleus, clusters of electron-dense ribonucleoproteins were observed at the nuclear interior, while peripheral bleached heterochromatin was detected as unstained areas (arrowhead) (96% out of 200 observed nuclei); EDTA staining was observed throughout the nucleus in mevinolin/TSA-treated controls (74% out of 200 observed nuclei). In untreated HGPS nuclei, a diffuse gray staining was observed and electron-dense ribonucleoproteins were reduced (36% out of 200 observed nuclei) (the irregular thickness of the nuclear lamina was indicated by arrows); in mevinolin/TSA-treated HGPS nuclei ribonucleoprotein staining was restored and bleached heterochromatin areas were detected at the nuclear periphery (arrowheads) (87% out of 200 observed nuclei). Different patterns of ribonucleoprotein staining were seen within the rectangle areas; images were taken by transmission electron microscopy. (C) The labeling pattern of intranuclear lamin A labeled by the 2H10 antibody corresponded to a speckled distribution (control) (95% out of 200 observed nuclei); mevinolin/TSA treatment did not affect 2H10 labeling in the majority of control nuclei (76% out of 200 observed nuclei); lamin A 2H10 stained in irregularly distributed foci at the nuclear interior of HGADFN001 cells (HGPS, untreated) (53% out of 200 observed nuclei); a staining pattern comparable to control nuclei was obtained by treating HGPS cells with mevinolin/TSA (HGPS, mevinolin/TSA) (81% out of 200 observed nuclei). A representative experiment was shown of triplicate experiments performed for each HGPS cell line.
Discussion
The results reported in this study show that progerin accumulation, nuclear envelope alterations and chromatin disorganization in HGPS correlate with patient age and are associated with defects of transcript accumulation and distribution. We demonstrate that both nuclear-shape abnormalities and heterochromatin organization may be rescued by treating HGPS fibroblasts with a farnesyl-transferase inhibitor, followed by a chromatin-modifying drug. Reversal of the nuclear defects is related to lowering of the progerin level in drug-treated samples and also elicits recovery of the ribonucleoprotein staining pattern.
Worsening of the HGPS cellular phenotype with patient age is clearly shown by the formation of nuclear introflections, blebs and enlargement of nuclei, as well as by progressive loss of peripheral heterochromatin. These aspects are related to the increased proportion of progerin relative to mature lamin A found in HGPS cells from older patients. On the other hand, a combination of factors is likely to cause the progressive worsening of nuclear defects observed in HGPS. In our hands, fibroblasts from the 3-year-old patient are in fact less affected than those from the 2-year-old patient (which are at a higher passage number). We suggest that both passage number [6] and patient age contribute to worsening of the cellular phenotype. However, we cannot rule out the possibility of an involvement of the genetic background in the different severity of the cellular defects observed in HGPS cells from different patients.
Although we observe a slight reduction in mature lamin A in the older patient cell line, literature data [13, 14] and our results obtained in the present study strongly suggest that the severe defects of HGPS cells are due to progerin accumulation. Here we show that removal of progerin from HGPS fibroblasts can be obtained by drug treatment and is associated with an important improvement in the cellular phenotype. As expected, wild-type pre-lamin A also accumulates in mevinolin/TSA-treated cells. This could be expected to elicit a toxic effect, as wild-type prelamin A has been shown to be toxic for transgenic mice [9, 13, 15] and humans [16–18]. However, it is worth noting that, after mevinolin treatment, a non-farnesylated lamin A precursor accumulates. This protein elicits very different effects on heterochromatin organization and on the whole cellular phenotype in comparison to farnesylated pre-lamin A [G. Lattanzi et al., unpublished results], as suggested by Fong et al. [13].
Results obtained by drug treatments further confirm that the amount of progerin accumulated in each HGPS cell culture directly determines the severity of the cellular phenotype. In fact, a slight reduction of progerin level by mevinolin corresponds to recovery of heterochromatin organization in a minor (though significant) proportion of cells. On the other hand, recovery of the heterochromatin arrangement along with improvement of nuclear-shape abnormalities is observed in a high percentage of HGPS cells after progerin removal. It should be noted that TSA was effective in reducing the progerin level only in mevinolin-pretreated cells, indicating that defarnesylation of the mutated protein allows destabilization. This aspect is a crucial point in the understanding of HGPS pathogenesis. Recently published papers [19–22] show that farnesyl-transferase inhibitors cause relocalization of progerin at the nuclear interior. This effect appears to improve nuclear shape [19–22]. These and our results suggest that defarnesylation of progerin represents the first step toward an improvement of the cellular phenotype. Release of the mutated protein from one nuclear region to another appears to be the first effect of progerin defarnesylation [19–22]. The subsequent reduction in progerin level we obtained by TSA addition more likely affects newly established protein interactions (see below). The major role of progerin in the HGPS pathogenetic mechanism is also supported by other recently published data [14] showing that recovery of the nuclear phenotype is obtained in HGPS fibroblasts by correction of the aberrant LMNA splicing leading to reduced progerin transcription [14].
Increased HDAC activity and increased levels of HDACs bound to unphosphorylated pRB (the protein product of the retinoblastoma gene) have been found in senescent cells [8]. TSA, which inhibits HDAC activity, has been suggested to reverse the cellular phenotype from senescent to proliferating by activating the cyclin E promoter [8]. We cannot completely rule out the possibility that pre-lamin A (and/or progerin) might be involved in the protein complex including pRB, HDACs and the E2F transcription factor to affect the stability of the protein complex itself. Lamin A does in fact interact with pRB and E2F in the protein complex anchored by the laminaassociated polypeptide 2 alpha (LAP2a) [23]. However, the reason why TSA does not elicit the expected decondensation effect on chromatin in HGPS nuclei is not obvious. We hypothesize either an altered distribution of chromatin-regulating factors including HDACs, or a mislocalization of mutated lamin A precursor affecting the composition of chromatin-regulating complexes in HGPS. Both these situations might explain the unexpected effects produced by inhibition of HDACs.
Our results suggest that mevinolin/TSA treatment leads to progerin degradation rather than to impairment of progerin synthesis. The finding that mevinolin /TSA treatment reduces the progerin level, while wild-type pre-lamin A is not or only minimally affected suggests that there is not a down-regulation of pre-lamin A expression. Moreover, the amount of progerin is dramatically reduced only in cells pretreated with mevinolin. This strongly suggests that defarnesylation of progerin is necessary to obtain protein loss. It appears unlikely that TSA might down-regulate progerin synthesis only after mevinolin treatment. A destabilization mechanism appears more acceptable. Our preliminary results show that defarnesylation of pre-lamin A allows formation of protein-protein interactions (G. Lattanzi et al., unpublished results). We propose that the reduction in progerin level obtained by TSA addition more likely affects newly established protein interactions.
Massive chromatin decondensation observed in HGPS fibroblasts does not correspond to increased transcriptional activity. Instead, regressive staining of nuclei allowed us to show that the pool of ribonucleoproteins is reduced in HGPS nuclei. In agreement with this finding, the distribution of incorporated BrU was altered in enlarged HGPS nuclei, as determined by an in situ transcription assay. The reduced ribonucleoprotein staining that characterizes progeria nuclei is suggestive of an aberrant mechanism affecting transcript accumulation, possibly due to the disproportion between open chromatin areas and the available pool of the transcriptional machinery. This hypothesis is in agreement with the observation that intranuclear lamin A associated with splicing factor compartments is mislocalized in HGPS cells. Both the ribonucleoprotein staining pattern and the rate of BrU incorporation, as well as the localization of intranuclear lamin A sites were improved by mevinolin/TSA treatment of HGPS cells, indicating that reorganization of heterochromatin domains corresponds to functional recovery.
The reported data argue for a major role of progerin in chromatin organization and dynamics. Our results also suggest a role for wild-type pre-lamin A in heterochromatin dynamics. For example, we observed a dramatic decrease in pre-lamin A accumulation in cells treated with the chromatin demethylating agent 5-azadeoxycitidine. Interestingly, a role for pre-lamin A in the organization of chromatin domains has been suggested [24, 25] and a DNA-binding domain in the lamin A carboxy-terminus sequence has been determined [26]. Moreover, heterochromatin defects feature in all the laminopathies described so far [6, 10, 27–30]. Evidence of pre-lamin A involvement in chromatin dynamics will be presented in another study [G. Lattanzi et al., unpublished results].
References
De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I. et al. (2003) Lamin A truncation in Hutchinson-Gilford progeria. Science. 300: 2055
Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L. et al. (2003) Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 423: 293–298
Cao H. and Hegele R.A. (2003) LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). J. Hum. Genet. 48: 271–274
D'Apice M.R., Tenconi R., Mammi I., Ende J. van den and Novelli G. (2004) Paternal origin of LMNA mutations in Hutchinson-Gilford progeria. Clin. Genet. 65: 52–54
Csoka A.B., Cao H., Sammak P.J., Constantinescu D., Schatten G. and Hegele R. A. (2004) Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J. Med. Genet. 41: 304–308
Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B. et al. (2004) Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 101: 8963–8968
Haaf T. and Schmid M. (2000) Experimental condensation inhibition in constitutive and facultative heterochromatin of mammalian chromosomes. Cytogenet. Cell. Genet. 91: 113–123
Bandyopadhyay D., Okan N.A., Bales E., Nascimento L., Cole P.A. and Medrano EE.(2002) Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res. 62: 6231–6239
Pendas A.M., Zhou Z., Cadinanos J., Freije J.M., Wang J., Hultenby K. et al. (2002) Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 31: 94–99
Capanni C., Cenni V., Mattioli E., Sabatelli P., Ognibene A., Columbaro M. et al. (2003) Failure of lamin A/C to functionally assemble in R482L mutated familial partial lipodystrophy fibroblasts: altered intermolecular interaction with emerin and implications for gene transcription. Exp. Cell Res. 291: 122–134
Bernhard W. (1969) A new staining procedure for electron microscopical cytology. J. Ultrastruct. Res. 27: 250–265
Rice J. C., Briggs S.D., Ueberheide B., Barber C. M., Shabanowitz J., Hunt, D. F. et al. (2003) Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 12: 1591–1598
Fong L. G., Ng J. K., Meta M., Cote N., Yang S. H., Stewart C. L. et al. (2004) Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 101: 18111–18116
Scaffidi P. and Misteli T.(2005) Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 11: 440–445
Bergo M. O., Gavino B., Ross J., Schmidt W. K., Hong C., Kendall L. V. et. al. (2002) Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 99: 13049–13054
Agarwal A. K., Fryns J. P., Auchus R. J. and Garg A. (2003) Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 12: 1995–2001
Liu B., Wang J., Chan K. M., Tjia W. M., Deng W., Guan X. et. al (2005) Genomic instability in laminopathy-based premature aging. Nat. Med. 11: 780–785
Capanni C., Mattioli E., Columbaro M., Lucarelli E., Parnaik V. K., Novelli G. et al. (2005) Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 14: 1489–1502
Glynn M. W. and Glover T.W. (2005) Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum. Mol. Genet. 14: 1489–1502
Capell B. C., Erdos M. R., Madigan J. P., Fiordalisi J. J., Varga R., Conneely K. N. et al. (2005) Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 102: 12879–12884
Toth J. I., Yang S. H., Qiao X., Beigneux A. P., Gelb M. H., Moulson C. L. et al.(2005) Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA 102: 12873–12878
Yang S. H., Bergo M. O., Toth J. I., Qiao X., Hu Y., Sandoval S. et al. (2005) Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA 102: 10291–10296
Markiewicz E., Dechat T., Foisner R., Quinlan R. A. and Hutchison C. J. (2002) Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol. Biol. Cell. 13: 4401–4413
Maraldi N. M., Squarzoni S., Sabatelli P., Capanni C., Mattioli E., Ognibene A. et al. (2005) Laminopathies: involvement of structural nuclear proteins in the pathogenesis of an increasing number of human diseases. J. Cell. Physiol. 203: 319–332
Sasseville A. M. and Raymond Y. (1995) Lamin A precursor is localized to intranuclear foci. J. Cell. Sci. 108: 273–285
Stierlé V. V., Couprie J., Ostlund C., Krimm I., Zinn-Justin S., Hossenlopp P. et al. (2003) The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry 42: 4819–482
Filesi I., Gullotta F., Lattanzi G., d'Apice M.R., Capanni C., Nardone A.M. et al. (2005) Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy, Physiol Genomics Jul 26; [Epub ahead of print].
Ognibene A., Sabatelli P., Petrini S., Squarzoni S., Riccio M., Santi S. et al.(1999) Nuclear changes in a case of X-linked Emery-Dreifuss muscular dystrophy. Muscle Nerve 22: 864–869
Sullivan T., Escalante-Alcalde D., Bhatt H., Anver M., Bhat N., Nagashima K. et al. (1999) Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 147: 913–920
Sabatelli P., Lattanzi G., Ognibene A., Columbaro M., Capanni C., Merlini L. et al. (2001) Nuclear alterations in autosomaldominant Emery-Dreifuss muscular dystrophy. Muscle Nerve 24: 826–829
Acknowledgement
The authors are grateful to HGPS patients and their families who donated skin samples, without which this study would not have been possible. We would also like to express our gratitude to the entire Progeria Research Foundation cell and tissue bank (Peabody, Mass.), especially Dr. L. Gordon, Dr. S. Campbell and Mrs. R. Zahr for their cooperation. We thank R. Foisner for carefully reading the manuscript and for helpful discussion. The technical support of P. Sabatelli, A. Valmori and S. Grasso is gratefully acknowledged. This work was supported by grants from the Italian Health Ministry (P.F. No. 2003/123), from the Italian Ministry for University and Research (FIRB Project No. RBNE01JJ45_005) and Cofin 2004, Italy, by a ‘Telethon’ grant to G. N. and by a grant from ‘Fondazione Carisbo,’ Italy. V. K. P. was supported by the Council of Scientific and Industrial Research, India.
Author information
Authors and Affiliations
Corresponding author
Additional information
Received 16 July 2005; received after revision 7 September 2005; accepted 21 September 2005
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Columbaro, M., Capanni, C., Mattioli, E. et al. Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell. Mol. Life Sci. 62, 2669 (2005). https://doi.org/10.1007/s00018-005-5318-6
Published:
DOI: https://doi.org/10.1007/s00018-005-5318-6