
Abstract.
Polycystic kidney diseases (PKDs) represent a large group of progressive renal disorders characterized by the development of renal cysts leading to end-stage renal disease. Enormous strides have been made in understanding the pathogenesis of PKDs and the development of new therapies. Studies of autosomal dominant and recessive polycystic kidney diseases converge on molecular mechanisms of cystogenesis, including ciliary abnormalities and intracellular calcium dysregulation, ultimately leading to increased proliferation, apoptosis and dedifferentiation. Here we review the pathobiology of PKD, highlighting recent progress in elucidating common molecular pathways of cystogenesis. We discuss available models and challenges for therapeutic discovery as well as summarize the results from preclinical experimental treatments targeting key disease-specific pathways.
Similar content being viewed by others


Introduction
Polycystic kidney diseases (PKDs) are a large family of disorders characterized by the formation and growth of renal cysts often leading to end-stage renal disease (ESRD). The most common inherited PKDs can be transmitted as autosomal dominant or autosomal recessive traits. Autosomal dominant polycystic kidney disease (ADPKD) occurs with an incidence of 1 : 1000 and is characterized by the development of fluid-filled cysts in the kidneys, liver, pancreas and other organs, accounting for up to 10%ofESRDcases [1]. ADPKD is a systemic disorder with cardiovascular manifestations including cardiac valve abnormalities, cardiac hypertrophy and intracranial aneurisms. Intestinal diverticuli and abdominal wall hernia are also common. Complications include hypertension, abdominal pain, hematuria and urinary tract infections [2, 3]. The disease manifests in adulthood, with ∼50% of patients developing renal failure by age 60 [4, 5].
Autosomal recessive polycystic kidney disease (ARPKD) is a less frequent childhood disease with an incidence of 1 : 20 000. ARPKD is characterized by cystic kidneys and congenital hepatic fibrosis with a high level of mortality in affected newborns. Most cases manifest in utero or at birth with renal enlargement and biliary dysgenesis. A smaller number of patients display later onset of disease with portal hypertension or cholangitis [6]. There are several other less common inherited recessive cystic diseases including nephronophthisis (NPHP), Bardet-Biedl syndrome (BBS), Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS) that will not be discussed here and are described in other excellent reports [7–10].This review will mainly focus on recent advances in understanding the molecular pathogenesis of PKDs and development of novel therapeutic options.
Molecular genetics of PKDs
ADPKD. ADPKD is genetically and phenotypically heterogeneous. Approximately 85% of all ADPKD cases are caused by mutations in the PKD1 gene, encoding polycystin-1, which maps to human chromosome 16p13.3 [11 – 14]. The remaining 15%can be attributed to the PKD2 gene, encoding polycystin-2, which maps to 4q21 [15, 16]. Clinical presentations of the PKD1 and PKD2 mutations are very similar, although the disease phenotype in the latter is significantly milder. Typically, individuals with PKD2 display later mean age at diagnosis, hypertension and ESRD [17, 18]. Heterogeneity is also seen within families in age of onset, rate of cystic disease progression and extrarenal manifestations, suggesting the role of genetic and environmental factors [19].
The PKD1 genomic region of 53 kb is organized into 46 exons encoding a ∼14 kb mRNA. Sequences related to the PKD1 locus are duplicated several times proximally on chromosome 16 [20]. The PKD1 genomic region contains a number of unusual structural features, including high GC content and multiple simple repeats. Most interesting is the 2.5-kb polypyrimidine tract located within intron 21 [21]. Such lengthy polypyrimidine elements may interfere with replication, transcription or RNA processing. Because of the complex structure of the PKD1 gene, comprehensive mutation screening is difficult. Approximately 270 different PKD1 mutations have been described [19]. Most mutations are predicted to produce truncated protein and are unique to a single family, although missense mutations have also been identified.
The PKD2 gene has 15 exons encoding ∼5 kb mRNA [16]. Nearly 70 different mutations have been described [19]. While genotype/phenotype correlations in thePKD1gene suggest that mutations in the 5′ portion of the gene are associated with a more severe phenotype, no obvious correlations have been found in the PKD2 gene [22].
The combination of the unique unstable structural elements in the PKD1 gene with intrafamilial heterogeneity and the focal nature of cyst formation has led to the “two-hit hypothesis” for cyst formation, which suggests that cysts form in cells with an inherited mutation in one allele and a somatic mutation occurring in the other allele. This hypothesis received experimental support through identification of somatic mutations in a subset of kidney cysts [23, 24]. A similar mechanism was also found to play a role in PKD2 renal and hepatic cystogenesis [25]. Further studies suggest, however, that other mechanisms may be involved. Cystogenesis in humans and mice occurs in trans-heterozygotes with PKD1 and PKD2 mutations [26, 27]. In addition, lowering of Pkd1 expression is sufficient to cause PKD in mice [28]. It is possible that reduced expression of the normal PKD1 allele below a critical level due to genetic or environmental factors may lead to cyst formation in the kidneys of ADPKD patients [28]. On the other hand, over-expression of polycystin-1 in transgenic animals also results in cyst formation [29, 30]. These data suggest that abnormal levels of polycystin-1 expression can trigger pathogenic mechanisms leading to cyst formation.
ARPKD. Genetic data have shown that mutations in a single gene, PKHD1, located on chromosome 6p21 (encoding fibrocystin/polyductin) cause ARPKD [31, 32]. The PKHD1 gene spans a region of ∼500 kb, consists of 67 exons and encodes a large ∼16 kb mRNA. Over 300 disease-causing mutations have been identified [33, 34]. Genotype/phenotype correlations show that individuals with two truncating mutations die in the perinatal period, while one or two missense changes are associated with milder disease [33]. This finding was further supported by a study of neonatal survivors in which no two truncating mutations were identified, suggesting that missense mutations are required for survival of newborns [35]. The majority of PKHD1 mutations are unique to a single family, with no obvious hot spots identified [35].
PKD proteins
Polycystin-1. Polycystin-1, the product of the PKD1 gene, is a large transmembrane protein with an estimated molecular weight of ∼500 kD (Fig. 1) [12, 13]. The large extracellular N-terminal region contains several specific motifs including leucine-rich repeats (LRRs), C-type lectin domain, LDL-A region, multiple Ig-like domains (or PKD domains), REJ domain and GPS domain. It has 11 transmembrane domains, with a PLAT domain located in the first cytoplasmic loop and a small cytoplasmic tail with a G-protein-binding motif and coiled-coil region (Fig. 1). The 16 Ig-like domains are segmented such that the first Ig-like domain is localized between the LRRs and the C-type lectin domain, while the remaining 15 Iglike domains are clustered together between LDL-A and REJ domains. This Ig-like domain cluster forms strong homophilic interactions that are important for cell-cell adhesion [36, 37]. Polycystin is likely a multifunctional protein with important roles in cell-cell/matrix adhesion and ciliary functions [10, 38]. Polycystin-1 undergoes partial cleavage at the GPS domain such that N-terminal and C-terminal polypeptides remain non-covalently linked [39, 40]. It is subsequently cleaved at the second site, which releases its C-terminal tail [41]. The cytoplasmic tail of polycystin-1 enters the nucleus and regulates cell signaling events. This signaling function of polycystin-1 is regulated by polycystin-2 and may possibly be initiated by mechanical stimuli [41].

The structure of polycystin-1, polycystin-2 and fibrocystin (LRR, leucine-rich repeats; WSC, cell wall integrity and stress response component 1; PKD (Ig-like), Ig-like domains; LDL, low density lipoprotein domain; REJ, receptor for egg jelly; GPS, proteolytic G protein-coupled receptor proteolytic site; PLAT, lipoxygenase domain; EF, EF hand domain; TIG, immunoglobulin-like domains; TMEM2, homology with TMEM2 protein). Polycystin-1 and fibrocystin undergo cleavage at sites shown by the arrows.
Polycystin-2. Polycystin-2 is a protein of ∼110 kD with six transmembrane domains and cytoplasmic N- and C-terminal domains (Fig. 1) [16]. Polycystin-2 (or TRPP2) is thought to be a new member of the transient receptor potential (TRP) family of ion channels. It was shown to be a cation channel with some selectivity for Ca2+ [42] and functions in multiple subcellular locations including plasma membrane [43, 44], endoplasmic reticulum [45] and the primary cilia [46].
Polycystin-1 and -2 can function together as a complex as well as independently in a variety of subcellular compartments. Direct interaction between the cytoplasmic tails of the polycystins has been shown using yeast two-hybrid assay [47, 48].
Fibrocystin/polyductin. Fibrocystin is a large receptorlike membrane-associated protein of ∼450 kD with a single transmembrane domain, large extracellular N-terminal region and small cytoplasmic C-terminal tail. Its extracellular portion consists of several TIG domains (immunoglobulin-like) and a short cytoplasmic C-terminus with putative phosphorylation sites (Fig. 1) [49, 50]. Based on similarities with other TIG-containing proteins such as the hepatocyte growth factor receptor and the plexins, fibrocystin was suggested to function as a receptor or a ligand, since secreted forms may be generated from alternatively spliced transcripts [49]. Fibrocystin is expressed in cortical and medullary collecting ducts of the kidney as well as biliary and pancreatic ducts in a pattern consistent with the histologic features seen in ARPKD [50].
Similar to polycystins, fibrocystin is expressed in multiple subcellular locations including the basolateral membrane, cytoplasm and cilia. It has been shown that fibrocystin can undergo Notch-like processing with release of the ectodomain from primary cilia [51]. An independent study also showed cleavage of the ectodomain of fibrocystin as well as generation of a cytoplasmic fragment that translocates to the nucleus [52]. Such proteolytic cleavage can be elicited by stimulation of intracellular Ca2+ release or protein kinase C activation. Fibrocystin may form a complex with polycystin-2 to regulate calcium responses in kidney epithelia, but its exact role in normal and cystic epithelia is not known [53].
Cellular and molecular pathogenesis of PKDs
Proliferation, apoptosis and fluid secretion in cysts. The formation and growth of cysts in PKD is accompanied by increased proliferation and apoptosis of cyst-lining epithelia, loss of epithelial polarity and de-differentiation, dysregulation of cell/matrix interactions and transformation of the absorptive epithelial phenotype to a secretory phenotype [54 – 56]. Epidermal growth factor (EGF), transforming growth factor (TGF) alpha and EGF receptor (EGFR) play important roles in promoting cystic epithelial proliferation. In human PKDs and several animal models, EGFR is overexpressed and mislocalized to the apical membranes of cystic epithelial cells [57]. Overexpression of TGF alpha in transgenic animals leads to renal cyst formation [58]. Apoptosis is also essential for cystogenesis: deletion of the anti-apoptotic Bcl-2 and AP-2β genes and overexpression of the pro-apoptotic gene c-myc in mice results in renal cyst formation [59]. Cystic fluid is derived from glomerular filtrate in the early stages of ADPKD, when cysts are still attached to the parent tubule [60]. Cysts separate from the tubule of origin when they reach ∼200 μm in diameter and continue to expand through a transepithelial chloride secretion mechanism mediated by cAMP [61]. Chloride enters cells via the basolateral Na-K-Cl cotransporter and accumulates in the cytoplasm. A chloride channel in the apical membrane, CFTR, mediates movement of chloride into the cystic lumen. Chloride secretion drives sodium into the cystic cavity through paracellular mechanisms; this causes movement of water through aguaporins [61]. In contrast to ADPKD, cysts in ARPKD do not separate from affected collecting ducts. Therefore, proliferation but not transepithelial secretion is a major component causing cystic kidney volume enlargement in ARPKD.
Impaired cell-cell/matrix adhesion. The overlapping expression and localization patterns of polycystin-1 and -2 support their role as a complex in regulating multiple processes in tubular epithelia [62]. Both proteins are found in basolateral membranes and the primary cilium, where they may act together to regulate cellular adhesion and Ca2+ signaling. On the other hand, polycystin-2 is mainly expressed in endoplasmic reticulum, where it functions as a Ca2+ release channel [45]. In addition, polycystin-1 is highly expressed during development, with significant down-regulation of its expression in adult tissues. In contrast, expression of polycystin-2 seems to persist into adult life [62].
Experimental evidence from several groups has established an important role for polycystins in epithelial cell morphogenesis, including differentiation and maturation in vivo [63, 64]. Studies using in vitro models of tubulogenesis and cystogenesis based on MDCK cells demonstrated that expression of polycystin-1 at cell-cell junctions at controlled levels is critical for proper tubular differentiation [65]. It has been shown that polycystin-1 is directly involved in intercellular adhesion via formation of strong homophilic interactions of its PKD (Ig-like) domains as shown in Fig. 2 [36]. A direct role for Ig-like domains in cell-cell adhesion was demonstrated by specific perturbation of intercellular adhesion using antibodies against Ig-like domains in cell cultures [36, 37]. Polycystin-1 was localized to the cell-cell adhesion complexes with adherens junctions and desmosomal junctions in epithelial cells of different origin [65 – 67]. Because alterations in polycystin-1-mediated adhesion may cause the abnormal epithelial cell phenotype observed in ADPKD cells, including dedifferentiation and loss of epithelial polarity, several studies examined cell-cell adhesion junctions in primary cells derived from ADPKD kidneys [37, 68, 69]. As shown in Fig. 2, abnormal adherens and desmosomal junctions were found in ADPKD: intracellular junctions were devoid of desmosomal cadherins and associated proteins, which were sequestered to the cytoplasmic pools, and adherens junctions appeared disrupted, accompanied by a great reduction of Ecadherin expression and partial compensatory expression of N-cadherin [68].

Polycystin-1 in cell-cell/matrix adhesion in normal and ADPKD epithelia. (a) Polycystin-1 (PC-1) mediates cell-cell adhesion through homophilic interactions of its Ig-like domains. It localizes with desmosomal junctions (DJs) and adherens junctions (AJs). The components of DJs are shown: desmoplakin (Dp), desmogleins (Dsg), desmocollins (Dsc) and plakoglobin (Pg) [121]. AJs consist of E-cadherin (E-cad) as well as α, β and γ-catenins (catenins). The AJ complex provides a linkage to the actin cytoskeleton, and DJ link together intermediate filaments (IF) of epithelial cells. In ADPKD epithelial cells, dysfunctional PC-1 (crossed red lines) and desmosomal proteins are lost from cell-cell contacts and remain in intracellular vesicles. In addition, E-cadherin expression is reduced, resulting in compensatory expression of N-cadherin (N-cad). Thus, AJs and DJs are disrupted in ADPKD cells, while tight junctions (TJ) remain intact. (b) Expression of PC-1 in cell-matrix focal adhesion contacts. In normal epithelial cells, PC-1 is found in a complex with talin (TAL), paxillin (PAX), vinculin (VINC), focal adhesion kinase (FAK), c-src (SRC), p130-cas (CAS), nephrocystin (NPH1) and tensin (TEN). InADPKD cells, expression of FAK is lost from the focal adhesion complex.
Interestingly, co-immunoprecipitation studies in ADPKD cells using an anti-polycystin-1 antibody showed that E-cadherin was lost from the complex, while beta-catenin remained associated with polycystin-1. Moreover, beta-catenin was still expressed at the plasma membrane despite the loss of E-cadherin, suggesting the presence of an alternative cadherin [68]. N-cadherin, but not K-cadherin, was overexpressed in ADPKD cells and formed a complex with beta-catenin. In normal kidney, E-cadherin is expressed in distal segments of the nephron, while Ncadherin is expressed in proximal tubules [70]. In the ADPKD kidney, N-cadherin is markedly increased in cysts of distal origin [68]. However, the expression of N-cadherin in place of E-cadherin in ADPKD cells was not sufficient to maintain epithelial cell-cell adhesion [37, 69].
Polycystin-1 was shown to be indispensable in cellmatrix interactions (Fig. 2) [54, 71]. Abnormalities in the basement-membrane composition and expression of matrix metalloproteases and their inhibitors were identified in PKD kidneys. Interestingly, the inactivation of tensin and insertional mutation in laminin alpha five result in cystogenesis [72, 73]. It has been shown that polycystin-1 interacts with focal adhesion complex molecules including α1β2 integrin, vinculin, paxillin, p130-cas, talin and focal adhesion kinase (FAK) [38]. In ADPKD cells, the focal adhesion complex is disrupted due to loss of FAK (Fig. 2).
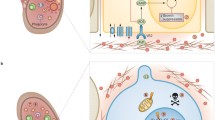
PKD as a ciliopathy. Polycystins, fibrocystin and numerous other proteins or cystoproteins such as nephrocystin-1, -3, -4, -5, inversin, ALMS1, OFD1, BBS1-8, cystin, polaris and Nek8, which cause PKD in humans and animals, were recently discovered in a distinct subcellular compartment, the primary cilium [10, 74, 75]. Cilia are microtubule-containing organelles that project from the surface of most eukaryotic cells [76]. The primary (non-motile) cilia are known to transduce sensory stimuli such as concentrations of growth factors, osmolarity and fluid flow [76]. They primary cilia are formed in fully differentiated cells during interphase and grow out of the basal bodies (Fig. 3). The ciliary basal body is formed by the centrosomes, more specifically by the mother centriole that moves to the membrane where the axoneme, the structural core, is formed [77]. Construction and maintenance of the axoneme requires a bidirectional intraflagellar transport system (IFT) to deliver structural components from the cell body to the tip of the cilium.
It was recently discovered that defects in ciliary structure or function underlie multiple human diseases with diverse phenotypes, including retinal degeneration, neural tube defects, obesity, polydactyly and PKD. Initial linkage between cilia and PKD came from mating behavior studies in Caenorhabditis elegans [78]. Mutations in the lov-1 and PKD-2 genes of C. elegans, which are closely related to human polycystins, were associated with mechanosensation defects of ciliated sensory neurons. Direct evidence linking defects in ciliogenesis and PKD came from the study of renal tissue-specific inactivation of KIF3A, a subunit of kinesin-II essential for cilia formation [79]: inactivation of KIF3A in renal tubular epithelial cells resulted in development of PKD. Several studies have shown that defects in ciliary assembly are associated with PKD. It was demonstrated that the primary cilia in the cystic kidney of mice with a mutation in the Tg737 gene (homologous to the IFT gene of Chlamydomonas, IFT88) are shorter than normal [80]. On the other hand, the primary cilia of cystic epithelial cells in jck mice were found to be significantly longer than the cilia in wild-type mice [75]; this study suggested that ciliary protein Nek8, which is mutated in the jck mice, may play a role in control of ciliary length. Taken together, these data established a link between ciliary dysfunction and PKD.
Cilia and mechanosensation in PKD. A number of studies support a role for the primary cilium as a mechanosensor in kidney tubular epithelia. Praetorius et al. reported that the primary cilia of renal epithelial MDCK cells can serve as a flow sensor [81]. Bending the cilium resulted in intracellular Ca2+ influx followed by calcium release from IP3-sensitive stores. The calcium signal was spread as a wave through gap junctions of cells [81]. The authors concluded that the primary cilium in MDCK cells is mechanically sensitive and responds to flow by increasing intracellular calcium. A subsequent study demonstrated that polycystins can serve as flow-sensitive ciliary mechanosensors in kidney epithelia [46]. More specifically, ciliary polycystin-1 and polycystin-2 function together with ryanodine receptors to mediate mechanotransduction into the intracellular Ca2+ signaling response (Fig. 3). The influx of Ca2+ across the plasma membrane constitutes the initial response to mechanical stimulation, and downstream signaling is mediated through intracellular Ca2+ release [46]. It is possible that polycystin-1 functions as a sensor of ciliary bending, while polycystin-2 transduces themechanical signal into a calcium response. Changes in intracellular Ca2+ concentration are known to regulate multiple cellular functions including gene expression, cell cycle, differentiation and apoptosis. Cells with mutated polycystin-1 fail to respond to the fluid flow: it was shown that in ADPKD-derived cells, the ciliary mechanosensation of fluid-flow shear stress by poly-cystins is absent [46].

Ciliary functions of cystoproteins: mechanosensation and cell cycle control. Polycystin-1 (PC-1), polycystin-2 (PC-2) and other cystoproteins (not shown) are expressed in primary cilia, basal bodies or centrosomes. The primary cilium forms in fully differentiated cells in the G0 phase of the cell cycle. A cilium protrudes from the basal body (centrosome) formed by two centrioles, a mother centriole (blue cylinder) and a daughter centriole (orange cylinder). As cells enter the cell cycle, the cilium is resorbed, and the centrosome can act as a mitotic spindle pole organizer. Flow-induced bending of cilia in renal epithelial cells triggers Ca2+ influx mediated by PC-2 in association with PC-1.The role of ciliary proteins in cell cycle control is supported by several findings: (1) In the normal state, PC-2 sequesters Id2 [proproliferative helix-loop-helix (HLH) protein] in the cytoplasm, preventing it from entering the nucleus. In PKD, when PC-1 or PC-2 are inactivated, Id2 translocates to the nucleus and interacts with E-protein, blocking its ability to induce growth-suppressive genes and resulting in increased activity of cdk2 and activation of cell proliferation; (2) PC-1 can activate the JAK/STAT signaling pathway, resulting in the induction of p21waf1 and cell cycle arrest in G0 and G1; (3) The intraflagellar transport component IFT88/polaris is capable of controlling G1/S transition of the cell cycle. IFT88/polaris remains associated with the centrosome throughout the cell cycle, where it forms a complex with Che-1, thus modulating its binding to Rb protein.
Cilia and cell cycle. There is an intimate link between cilia and the cell cycle. The basal bodies/centrosomes of the cilia act as organizers of the mitotic spindle poles during cell division, directly connecting ciliogenesis with cell cycle regulation, and cilia are resorbed when cells enter the cell cycle (Fig. 3). As we discussed previously, a number of cystoproteins causing PKD in humans and animals are expressed, at least partially, in cilia or the basal body of the cilia. Polycystins and other cystoproteins may play an important role in connecting the mechanosensory function of the cilia to the centrosome and thus influence cell cycle control. Disruption of cystoproteins associated with cilia or basal bodies could, therefore, lead to dysregulation of the cell cycle and proliferation, resulting in cystic disease.
Several lines of evidence support this hypothesis: Overexpression of exogenous polycystin-1 in cultured cells resulted in growth arrest in G0/G1 phase of the cell cycle (Fig. 3) [82]. Further analysis has shown that polycystin-1 expression inhibits Cdk2 and induces p21waf1. Polycystin-1 activates the JAK-STAT pathway, thereby up-regulating p21waf1 and inducing cell cycle arrest in G0/G1. Functional polycystin-2 was shown to be essential for this process [82]. Other cystoproteins localized to cilia have also been recently shown to directly regulate the cell cycle. For example, the IFT88/polaris protein, a component of the IFT, was shown to be tightly associated with the centrosome during cell cycle transitions [83]. Similar to polycystin-1, overexpression of polaris resulted in cell cycle arrest at the G0/G1 stage (Fig. 3). On the other hand, down-regulation of polaris mRNA promoted progression of the cell cycle. Polycystin-2 has been shown to participate in cell cycle regulation as well: it can associate with the helix-loop-helix protein Id2 and block its translocation to the nucleus, preventing proliferation [84]. The translocation of Id2 in cells with mutated polycystins is associated with downregulation of p21 expression, leading to an increase in CDK2 activity and cell cycle progression (Fig. 3). Thus, evidence of a direct link between cystoproteins, ciliary dysfunction and cell cycle dysregulation continues to accumulate.
Other signaling pathways affected in PKD
As we discussed previously, PKDs in humans and animals can be caused by different cystogenes and can be distinguished by the patterns of inheritance and clinical manifestation. On the other hand, cyst formation in these disorders is clearly characterized by common features at the cellular and molecular levels. Common cellular abnormalities of cystic epithelia include increased proliferation, apoptosis, loss of epithelial polarity accompanied by missorting of proteins and intracystic fluid secretion. Ciliary dysfunction may also constitute one of the common features for cystic diseases, since most known cystoproteins are associated with cilia/centrosome. Recent evidence suggests that cystoproteins may physically or genetically interact. For example, polycystin-2 was found to directly interact with fibrocystin to regulate calcium responses in the kidneys [53]. Genetic interaction between murine genes encoding polycystin-1 and fibrocystin was shown, suggesting that polycystin-1 and fibrocystin function in the same molecular pathway to maintain tubular integrity [85]. In addition, genetic interaction was also identified between cystogenes encoding polycystin-1 and Nek8 (mutated in the jck mouse model of PKD) (Natoli et al., unpublished observations). Therefore, elucidation of common pathogenic mechanisms involved in PKDs is an important step toward developing new therapies for effective treatment of cystic diseases.
cAMP-activated pathways. Studies using in vitro-cultured cells from ADPKD cysts and animal models of PKD have revealed an important role for cAMP-activated pathways. cAMP was shown to block proliferation of normal kidney epithelial cells through inhibition of the Ras/Raf/MEK/ERK pathway [86]. In sharp contrast, cAMP was able to induce proliferation of ADPKD-derived cystic epithelial cells through activation of the B-Raf/MEK/ERK pathway. This switch from conventional cAMP-induced inhibition of growth to the cAMP-induced stimulation of growth in cystic epithelia was mediated by decreased intracellular calcium levels [87]. Mutated polycystins disrupted Ca2+ signaling, which led to abnormal cAMP-stimulated proliferation of cystic cells. Cystic kidneys were shown to accumulate high levels of cAMP inmultiple models of PKD, making it plausible that activated cAMP pathways lead to both the increased proliferation and fluid secretion seen in cystic epithelia [86, 88, 89].
Wnt signaling in PKD. Activated Wnt signaling was initially implicated in PKD through studies demonstrating that overexpression of the cytoplasmic tail of polycystin-1 stabilizes endogenous beta-catenin and stimulates TCF-dependent gene transcription in vitro [90]. Microinjection of the polycystin-1 cytoplasmic tail induced dorsalization in zebrafish, suggesting that polycystin-1 is involved in modulating Wnt signaling during renal development [90]. Other studies supported this finding by connecting dysregulation of the beta-catenin pathway with PKD. Overexpression of beta-catenin in transgenic animals or inactivation of APC resulted in the development of cystic kidneys [91, 92]. Canonical beta-catenin-dependent Wnt signaling is required for kidney development, while constitutive beta-catenin signaling in maturing tubules leads to formation of cysts. Flow-induced signaling switches the Wnt pathway to a non-canonical beta-catenin-independent pathway through increased expression of inversin, a protein mutated in nephronophthisis type II [93]. This, in turn, results in reduced levels of the cytoplasmic dishevelled protein that activates destruction of the beta-catenin complex.
mTOR activation. The PKD1 gene is located in close proximity to the TSC2 gene, which encodes the tuberin protein responsible for the tuberous sclerosis complex (TSC), in a tail-to-tail orientation. Patients with contiguous PKD1-TSC2 gene syndrome develop a severe PKD. These observations suggested that polycystin-1 and tuberin may function in a common molecular pathway. A functional link between polycystin-1 and tuberin was established by a study showing that tuberin is required for membrane trafficking of polycystin-1 [94]. Polycystin-1 appeared to form a complex with tuberin and regulate the activity of mTOR, an important regulator of protein synthesis and cellular differentiation [95]. The mutations in polycystin-1 may therefore lead to persistent activation of mTOR in PKD. In fact, increased mTOR activity was identified in epithelial cells lining ADPKD cysts. Activation of mTOR was also found in animal models of recessive PKD such as the bpk and orpk-rescue, suggesting that mutations in polycystin-1 are not critical form mTOR activation. It is possible that signaling through other membrane receptors could lead to stimulation of mTOR pathway, which is likely to be a late stage of the cystogenesis cascade.
Therapeutic approaches for the treatment of PKD: preclinical and clinical testing
Preclinical PKD animal models. Greater understanding of the molecular mechanisms of cyst formation and growth has led to the discovery of novel potential therapeutic targets. Animal models of PKD have been critical in supporting studies of disease pathogenesis and in testing potential therapies. The availability of well-characterized and disease-relevant models is instrumental for successful development of viable therapies. Several preclinical PKD models have been described, some with spontaneous mutations and others generated through chemical/genetic manipulations [96]. Mutations in these models occurred in different genes, including Pkd1 and Pkd2. No existing model is ideal for preclinical testing in PKD; however, each represents a facet of human pathobiology (Table 1). In general, an ideal PKD model should carry a mutation in a gene orthologous to the human disease-carrying gene.The development of the disease in such orthologous models should reproduce human PKD relative to the severity, segmental origin of kidney cysts, extrarenal manifestations and known cellular and molecular abnormalities including increased proliferation, apoptosis, cAMP, protein missorting as well as activation of known signaling pathways such as Wnt, b-Raf/MEK/ERK and mTOR. Numerous attempts to produce such models have not yet been successful. A dozen different mouse models with targeted disruption of the Pkd1 gene have been created, including animals with conditional gene inactivation (reviewed in [96]). Mice with heterozygous Pkd1 and Pkd2 mutations develop a very mild and heterogeneous kidney cystic disease late in life and therefore are not suitable for therapeutic testing. Homozygous animals develop renal and pancreatic cysts at embryonic day 15.5 and die during embryogenesis. Mice with conditional inactivation of the Pkd1 gene develop kidney and liver cysts, but disease is too mild to be informative in preclinical testing [97]. An interesting mouse model with a mutation in the Pkd2 gene (Pkd2−/WS25) carrying an unstable allele combined with a Pkd2-null allele has been described [98]. Although these animals develop kidney and liver cystic disease that mimics human ADPKD, the model has not been widely used for therapeutic testing due to a very high degree of phenotypic heterogeneity in combination with a slow course of progression with no measurable loss of renal function as late as 4–6 months of age. The Pkd2−/WS25 model should be most useful as a secondary confirmatory model rather than the primary model for proof of concept therapeutic testing [96]. Several spontaneous mouse models of PKD, such as cpk, bpk and orpk, resemble human recessive disease with respect to cyst localization and the rapid rate of disease progression (Table 1). Cystic disease in the pcy mouse and Han:SPRD rat is slowly progressive with cyst formation similar to ADPKD. The jck mouse model is characterized by the development of cysts in multiple nephron segments and, despite the autosomal recessive mode of inheritance, resembles human ADPKD phenotypically and can be used for relatively rapid therapeutic testing [75]. Since no single model fully recapitulates all aspects of human PKD pathogenesis, it is important to test a potential therapy in more than one PKD model to determine its utility for clinical testing.
EGF receptor tyrosine kinase inhibitors. An important role for epidermal growth factor (EGF) and its receptor (EGFR) in contributing to cystic epithelial proliferation has been demonstrated. In human PKD and multiple animal models, EGFR is overexpressed and mislocalized to the apical membrane in cystic epithelial cells [57]. Transforming growth factor alpha (TGF alpha), a member of EGF family, also signals through EGFR. The role of TGF alpha in cystogenesis was addressed by renal expression of a TGF alpha transgene in the mouse [58]. Overexpression of TGF alpha in transgenic animals resulted in formation of renal cysts. Preclinical studies showed that inhibition of EGFR tyrosine kinase activity significantly reduced cystogenesis in mouse models of PKD, including bpk and orpk, as well as in the Han:SPRD rat model [99, 100], all models characterized by overexpression and mislocalization of EGFR to the apical membrane. However, no therapeutic effect was detected in the PCK rat; interestingly, although the PCK rat is an orthologous model of ARPKD, EGFR was not found to be mislocated to the apical membrane of cystic cells, which may explain the lack of therapeutic efficacy in this model [101].
Inhibition of apoptosis. Cystogenesis is characterized by enhanced apoptosis. Growth of cysts in 3-D collagen cultures of epithelial cells in vitro proceeds with increased apoptosis [102]. Moreover, deletion of the anti-apoptotic Bcl-2 and AP-2 beta genes as well as overexpression of pro-apoptotic c-myc in mice leads to renal cyst formation [103, 104]. To test the effect of inhibition of apoptosis in PKD, a caspase inhibitor was tested in the Han:SPRD rat model [105]. This study showed inhibition of PKD progression and attenuation of the loss of renal function.
Modulation of cAMP signaling. Multiple studies have shown the importance of cAMP-activated pathways in PKD. The critical role of the cAMP-activated B-Raf/MEK/ERK pathway was confirmed by the successful preclinical testing of a MEK inhibitor [106]: oral administration the MAP/ERK kinase inhibitor PD184352 to pcy mice significantly decreased kidney weight, serum creatinine levels and water intake and significantly increased urine osmolality. Thus, inhibitors of the B-Raf/MEK/ERK pathway may prove to be promising agents for PKD therapy.
Studies were conducted to specifically target the cystogenic cAMP and vasopressin pathways using vasopressin V2 receptor (VPV2R) inhibitors, and efficacy was shown in four different PKD animal models [107]. Treatment with Tolvaptan (human VPV2R antagonist, Otsuka Pharmaceutical) resulted in significant lowering of cAMP levels in PCK rat kidneys and inhibition of renal cystogenesis and fibrosis. Tolvaptan has entered human clinical trials in ADPKD, and phase II results have shown that it is well tolerated, with thirst and polyuria as side effects [88, 108]. It is important to note that effective reduction of plasma vasopressin levels through increased water intake decreased VPV2R expression and slowed PKD progression in PCK rats [109].
As somatostatin is capable of inhibiting cAMP-stimulated fluid secretion, it constitutes an alternative approach to the targeting of abnormal cAMP signaling in cysts. In a randomized placebo-controlled human ADPKD trial, the benefit of a 6-month treatment with long-acting somatostatin (octreotide-LAR, 40 mg intramuscularly every 28 days) was assessed [110]. This study indicated that somatostatin therapy is safe andmay slow disease progression. Several clinical trials with somatostatin are now being implemented.
mTOR inhibition. Treatment of the bpk and orpk-rescue mouse models with the mTOR inhibitor rapamycin showed effective inhibition of PKD [95]. This effect was mediated by selective induction of apoptosis in cystic cells. Similar results were previously observed in a separate study with rapamycin performed in Han:SPRD rats [111]. Treatment of human ADPKD transplant recipient patients with rapamycin resulted in a significant reduction in polycystic kidney volumes [95]. Several clinical trials are currently underway to test the efficacy of rapamycin and everolimus in ADPKD patients.
Cyclin-dependent kinase (CDK) inhibition. An important role of ciliary dysfunction in PKD pathology is highlighted by findings of the disrupted connection between cilia and the cell cycle. This dysregulation between cilia and cell cycle progression seems to occur early in cystogenesis and represents an attractive therapeutic target. The CDK inhibitor roscovitine was tested in cystic assay in vitro and in the preclinical PKD mouse models jck and cpk [89]. Roscovitine (Seliciclib, CYC202) is a potent and remarkably selective inhibitor of CDK2/cyclin E, CDK7/cyclin H, CDK9/cyclin T1 and CDK5/p35-p25 [112, 113]. Robust inhibition of PKD was shown in the jck mouse model of slowly progressive disease as well as in the cpk mouse model of aggressive PKD. Pulse treatment resulted in a persistent long-lasting effect such that continuous daily administration of the drug was not required to achieve efficacy. Roscovitine was equally effective against cysts formed in different segments of the nephron, a desirable feature for a potential drug against ADPKD, where cysts are formed in multiple parts of the nephron. The effect of roscovitine at the molecular level was mediated through cell cycle blockade, transcriptional inhibition and apoptotic arrest [89]. Thus, the apparent therapeutic benefit of CDK inhibition for PKD may be due to integrative effects on several key aspects of disease pathology. Seliciclib, an orally bioavailable compound, is currently in clinical trials as a cancer drug candidate [114]. Preliminary clinical testing showed that Seliciclib can be administered safely without major side effects. Known adverse events include transient elevations in serum creatinine, transient hypokalemia and reversible elevations in liver enzymes [114].
Stimulation of polycystin-2-mediated Ca2+release. The deregulation of Ca2+ signaling in cystic cells is believed to be mediated by polycystin-2 in complex with polycystin-1. The first example of a therapeutic strategy to promote an increase in cytosolic Ca2+ in PKD has been demonstrated by treatment with triptolide, an active diterpene used in traditional Chinese medicine [115]. Triptolide treatment of Pkd1-null homozygous embryos via maternal administration of the drug resulted in arrest of cellular proliferation and inhibition of cyst formation through restoration of Ca2+ signaling.
Integrated approach for drug discovery. The majority — if not all — of the drugs successfully tested in animal models so far were not specifically designed to treat PKD but rather were originally developed for other indications. Some of these drugs may require further optimization to achieve efficacy/toxicity profiles that are more appropriate for the PKD patient population. Because multiple optimized analogues would need to be tested, direct assessment of their efficacy in animal models is not feasible. The availability of high-throughput in vitro screening assays that are disease-relevant, robust, rapid and reproducible is crucial. In addition, such assays can be directly used for discovery of PKD-specific compounds that antagonize cyst development. It has been shown that MDCK cysts grown in a 3-D collagen matrix in vitro adequately reflect several aspects of cystogenesis in vivo and can be successfully used for drug testing (Fig. 4) [65, 116, 117]; the cystic assay can be used directly for high-throughput screening of a compound library or as a drug optimization assay. Recently it was shown that an embryonic cystic kidney organ culture assay can be a very valuable testing platform for rapid assessment of efficacy as well as preliminary testing for organ toxicity before entering into time- and material-consuming trials in animals ([118] and T. Natoli, unpublished results) (Fig. 4). With the availability of such a screening cascade and successful candidates already emerging from preclinical testing, the field is poised to suggest viable therapeutic options for the treatment of PKD.

Drug discovery platforms for anti-cystic therapeutic agents. High-throughput drug screening (HTS) of a small compound library can be performed using an in vitro assay of cystogenesis. Identified hits (active drugs) can then be rapidly assayed for preliminary efficacy and organ toxicity using a readily available cystic kidney organ culture assay that utilizes Pkd1−/− animals. Effective compounds can be selected for in vivo efficacy testing in animal models of PKD. Further optimization of compounds using medicinal chemistry and SAR (structure-activity relationship) can proceed through the same screening platforms. The discovery process can also be initiated through “proof of concept” in vivo testing (drug candidate validation), followed by the drug optimization cycle.
Assessment of therapeutic efficacy in clinical trials. Several potential therapies, including tolvaptan, sirolimus, everolimus and octreotide (as discussed above), are being tested in ongoing ADPKD clinical trials. In addition, a large clinical trial (HALT-PKD) has been implemented to address the benefit of inhibition of the renin-angiotensin system [119]; this trial will test whether using ACE inhibitors and angiotensin receptor blockers in combination is beneficial in comparison with ACE inhibitors alone. The main challenge in ADPKD clinical trials is the primary outcome measure. Because disease develops slowly over several decades, renal function begins to decline late in the course of the disease when significant and likely irreversible damage to the kidney occurs. More beneficial early intervention trials require availability of reliable endpoints predictive of the subsequent renal function decline. Results of the CRISP (Consortium for Radiologic Imaging Studies of PKD) study have demonstrated that measurement of kidney volume by magnetic resonance imaging (MRI) can be a reliable indicator of clinical outcome [120].
Conclusions
Recent advances in our fundamental understanding of the molecular pathogenesis of PKD have allowed selection of disease-relevant targets for therapeutic testing. Several successful studies in animal models that are currently being translated into human clinical trials strengthen the need to further explore disease-specific therapeutic targets. A rapidly growing number of well-characterized in vitro and in vivo models of cystic disease open opportunities for systematic drug discovery efforts. The availability of magnetic resonance imaging techniques to accurately measure PKD progression and, subsequently, the effect of therapeutic agents in a short period of time greatly enhances options for clinical trial design. It is likely that future treatment for PKD will require a combination therapy integrating several key pathways of cystogenesis. The future of the treatment options for PKD is a bright one.
References
Friedman, J. (1983) Cystic diseases of the kidney. In: Principles and practice of medical genetics. pp. 1002–1010, Emery, A. and Rimoin, D. (eds. ), Churchill Livingston, Edinburgh, UK.
Gabow, P. A. (1993) Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 329: 332–342.
Franz, K. A. and Reubi, F. C. (1983) Rate of functional deterioration in polycystic kidney disease. Kidney Int. 23: 526–529.
Churchill, D. N., Bear, J. C., Morgan, J., Payne, R. H., McManamon, P. J. and Gault, M. H. (1984) Prognosis of adult onset polycystic kidney disease re-evaluated. Kidney Int. 26: 190–193.
Parfrey, P. S., Bear, J. C., Morgan, J., Cramer, B. C., McManamon, P. J., Gault, M. H., Churchill, D. N., Singh, M., Hewitt, R., Somlo, S. and Reeders, S. (1990) The diagnosis and prognosis of autosomal dominant polycystic kidney disease. N. Engl. J. Med. 323: 1085–1090.
Zerres, K., Rudnik-Schoneborn, S., Steinkamm, C., Becker, J. and Mucher, G. (1998) Autosomal recessive polycystic kidney disease. J. Mol. Med. 76: 303–309.
Baala, L., Audollent, S., Martinovic, J., Ozilou, C., Babron, M. C., Sivanandamoorthy, S., Saunier, S., Salomon, R., Gonzales, M., Rattenberry, E., Esculpavit, C., Toutain, A. et al. (2007) Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 81: 170–179.
Delous, M., Baala, L., Salomon, R., Laclef, C., Vierkotten, J., Tory, K., Golzio, C., Lacoste, T., Besse, L., Ozilou, C., Moutkine, I., Hellman, N. E. et al. (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 39: 875–881.
Baala, L., Romano, S., Khaddour, R., Saunier, S., Smith, U. M., Audollent, S., Ozilou, C., Faivre, L., Laurent, N., Foliguet, B., Munnich, A., Lyonnet, S. et al. (2007) The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 80: 186–194.
Hildebrandt, F. and Otto, E. (2005) Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 6: 928–940.
Reeders, S. T., Breuning, M. H., Davies, K. E., Nicholls, R. D., Jarman, A. P., Higgs, D. R., Pearson, P. L. and Weatherall, D. J. (1985) A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature 317: 542–544.
Hughes, J., Ward, C. J., Peral, B., Aspinwall, R., Clark, K., San Millan, J. L., Gamble, V. and Harris, P. C. (1995) The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 10: 151–160.
Burn, T. C., Connors, T. D., Dackowski, W. R., Petry, L. R., Van Raay, T. J., Millholland, J. M., Venet, M., Miller, G., Hakim, R. M., Landes, G. M., Klinger, K.W., Qian, F. et al. (1995) Analysis of the genomic sequence for the autosomal dominant polycystic kidney disease (PKD1) gene predicts the presence of a leucine-rich repeat. The American PKD1 Consortium (APKD1 Consortium). Hum. Mol. Genet. 4: 575–582.
(1995) Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium. Cell 81: 289–298.
Kimberling, W. J., Kumar, S., Gabow, P. A., Kenyon, J. B., Connolly, C. J. and Somlo, S. (1993) Autosomal dominant polycystic kidney disease: localization of the second gene to chromosome 4q13–q23. Genomics 18: 467–472.
Mochizuki, T., Wu, G., Hayashi, T., Xenophontos, S. L., Veldhuisen, B., Saris, J. J., Reynolds, D. M., Cai, Y., Gabow, P. A., Pierides, A., Kimberling, W. J., Breuning, M. H. et al. (1996) PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272: 1339–1342.
Torra, R., Badenas, C., Darnell, A., Nicolau, C., Volpini, V., Revert, L. and Estivill, X. (1996) Linkage, clinical features, and prognosis of autosomal dominant polycystic kidney disease types 1 and 2. J. Am. Soc. Nephrol. 7: 2142–2151.
Wright, G. D., Hughes, A. E., Larkin, K. A., Doherty, C. C. and Nevin, N. C. (1993) Genetic linkage analysis, clinical features and prognosis of autosomal dominant polycystic kidney disease in Northern Ireland. Q. J. Med. 86: 459–463.
Rossetti, S., Consugar, M. B., Chapman, A. B., Torres, V. E., Guay-Woodford, L. M., Grantham, J. J., Bennett, W. M., Meyers, C. M., Walker, D. L., Bae, K., Zhang, Q. J., Thompson, P. A. et al. (2007) Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 18: 2143–2160.
(1994) The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell 77: 881–894.
Van Raay, T. J., Burn, T. C., Connors, T. D., Petry, L. R., Germino, G. G., Klinger, K.W. and Landes, G. M. (1996) A 2.5 kb polypyrimidine tract in the PKD1 gene contains at least 23 H- DNA-forming sequences. Microb. Comp. Genomics 1: 317–327.
Rossetti, S., Chauveau, D., Kubly, V., Slezak, J. M., Saggar-Malik, A. K., Pei, Y., Ong, A. C., Stewart, F., Watson, M. L., Bergstralh, E. J., Winearls, C. G., Torres, V. E. and Harris, P. C. (2003) Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet 361: 2196–2201.
Qian, F., Watnick, T. J., Onuchic, L. F. and Germino, G. G. (1996) The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 87: 979–987.
Badenas, C., Torra, R., Perez-Oller, L., Mallolas, J., Talbot-Wright, R., Torregrosa, V. and Darnell, A. (2000) Loss of heterozygosity in renal and hepatic epithelial cystic cells from ADPKD1 patients. Eur. J. Hum. Genet. 8: 487–492.
Pei, Y., Watnick, T., He, N., Wang, K., Liang, Y., Parfrey, P., Germino, G. and St George-Hyslop, P. (1999) Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 10: 1524–1529.
Koptides, M., Mean, R., Demetriou, K., Pierides, A. and Deltas, C. C. (2000) Genetic evidence for a trans-heterozygous model for cystogenesis in autosomal dominant polycystic kidney disease. Hum. Mol. Genet. 9: 447–452.
Wu, G., Tian, X., Nishimura, S., Markowitz, G. S., D'Agati, V., Park, J. H., Yao, L., Li, L., Geng, L., Zhao, H., Edelmann, W. and Somlo, S. (2002) Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease. Hum. Mol. Genet. 11: 1845–1854.
Lantinga-van Leeuwen, I. S., Dauwerse, J. G., Baelde, H. J., Leonhard, W. N., van de Wal, A., Ward, C. J., Verbeek, S., Deruiter, M. C., Breuning, M. H., de Heer, E. and Peters, D. J. (2004) Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum. Mol. Genet. 13: 3069–3077.
Pritchard, L., Sloane-Stanley, J. A., Sharpe, J. A., Aspinwall, R., Lu, W., Buckle, V., Strmecki, L., Walker, D., Ward, C. J., Alpers, C. E., Zhou, J., Wood, W. G. and Harris, P. C. (2000)A human PKD1 transgene generates functional polycystin-1 in mice and is associated with a cystic phenotype. Hum. Mol. Genet. 9: 2617–2627.
Thivierge, C., Kurbegovic, A., Couillard, M., Guillaume, R., Cote, O. and Trudel, M. (2006) Overexpression of PKD1 causes polycystic kidney disease. Mol. Cell. Biol. 26: 1538–1548.
Ward, C. J., Hogan, M. C., Rossetti, S., Walker, D., Sneddon, T., Wang, X., Kubly, V., Cunningham, J. M., Bacallao, R., Ishibashi, M., Milliner, D. S., Torres, V. E. and Harris, P. C. (2002) The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 30: 259–269.
Onuchic, L. F., Furu, L., Nagasawa, Y., Hou, X., Eggermann, T., Ren, Z., Bergmann, C., Senderek, J., Esquivel, E., Zeltner, R., Rudnik-Schoneborn, S., Mrug, M. et al. (2002) PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 70: 1305–1317.
Rossetti, S., Torra, R., Coto, E., Consugar, M., Kubly, V., Malaga, S., Navarro, M., El-Youssef, M., Torres, V. E. and Harris, P. C. (2003) Acomplete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int. 64: 391–403.
Bergmann, C., Senderek, J., Kupper, F., Schneider, F., Dornia, C., Windelen, E., Eggermann, T., Rudnik-Schoneborn, S., Kirfel, J., Furu, L., Onuchic, L. F., Rossetti, S. et al. (2004) PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum. Mutat. 23: 453–463.
Bergmann, C., Senderek, J., Windelen, E., Kupper, F., Middeldorf, I., Schneider, F., Dornia, C., Rudnik-Schoneborn, S., Konrad, M., Schmitt, C. P., Seeman, T., Neuhaus, T. J. et al. (2005) Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 67: 829–848.
Ibraghimov-Beskrovnaya, O., Bukanov, N. O., Donohue, L. C., Dackowski, W. R., Klinger, K.W. and Landes, G. M. (2000) Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum. Mol. Genet. 9: 1641–1649.
Streets, A. J., Newby, L. J., O'Hare, M. J., Bukanov, N. O., Ibraghimov-Beskrovnaya, O. and Ong, A. C. (2003) Functional analysis of PKD1 transgenic lines reveals a direct role for polycystin-1 in mediating cell-cell adhesion. J. Am. Soc. Nephrol. 14: 1804–1815.
Wilson, P. D., Geng, L., Li, X. and Burrow, C. R. (1999) The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab. Invest. 79: 1311–1323.
Qian, F., Boletta, A., Bhunia, A. K., Xu, H., Liu, L., Ahrabi, A. K., Watnick, T. J., Zhou, F. and Germino, G. G. (2002) Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. USA 99: 16981–16986.
Wei, W., Hackmann, K., Xu, H., Germino, G. and Qian, F. (2007) Characterization of cis-autoproteolysis of polycystin-1, the product of human polycystic kidney disease 1 gene. J. Biol. Chem. 282: 21729–21737.
Chauvet, V., Tian, X., Husson, H., Grimm, D. H., Wang, T., Hiesberger, T., Igarashi, P., Bennett, A. M., Ibraghimov-Beskrovnaya, O., Somlo, S. and Caplan, M. J. (2004) Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Invest. 114: 1433–1443.
Gonzalez-Perret, S., Kim, K., Ibarra, C., Damiano, A. E., Zotta, E., Batelli, M., Harris, P. C., Reisin, I. L., Arnaout, M. A. and Cantiello, H. F. (2001) Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc. Natl. Acad. Sci. USA 98: 1182–1187.
Hanaoka, K., Qian, F., Boletta, A., Bhunia, A. K., Piontek, K., Tsiokas, L., Sukhatme, V. P., Guggino, W. B. and Germino, G. G. (2000) Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408: 990–994.
Babich, V., Zeng, W. Z., Yeh, B. I., Ibraghimov-Beskrovnaya, O., Cai, Y., Somlo, S. and Huang, C. L. (2004) The N-terminal extracellular domain is required for polycystin-1-dependent channel activity. J. Biol. Chem. 279: 25582–25589.
Koulen, P., Cai, Y., Geng, L., Maeda, Y., Nishimura, S., Witzgall, R., Ehrlich, B. E. and Somlo, S. (2002) Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 4: 191–197.
Nauli, S. M., Alenghat, F. J., Luo, Y., Williams, E., Vassilev, P., Li, X., Elia, A. E., Lu, W., Brown, E. M., Quinn, S. J., Ingber, D. E. and Zhou, J. (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33: 129–137.
Qian, F., Germino, F. J., Cai, Y., Zhang, X., Somlo, S. and Germino, G. G. (1997) PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet. 16: 179–183.
Tsiokas, L., Kim, E., Arnould, T., Sukhatme, V. P. and Walz, G. (1997) Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. USA 94: 6965–6970.
Ward, C. J., Yuan, D., Masyuk, T. V., Wang, X., Punyashthiti, R., Whelan, S., Bacallao, R., Torra, R., LaRusso, N. F., Torres, V. E. and Harris, P. C. (2003) Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 12: 2703–2710.
Menezes, L. F., Cai, Y., Nagasawa, Y., Silva, A. M., Watkins, M. L., Da Silva, A. M., Somlo, S., Guay-Woodford, L. M., Germino, G. G. and Onuchic, L. F. (2004) Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 66: 1345–1355.
Kaimori, J. Y., Nagasawa, Y., Menezes, L. F., Garcia-Gonzalez, M. A., Deng, J., Imai, E., Onuchic, L. F., Guay-Woodford, L. M. and Germino, G. G. (2007) Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum. Mol. Genet. 16: 942–956.
Hiesberger, T., Gourley, E., Erickson, A., Koulen, P., Ward, C. J., Masyuk, T. V., Larusso, N. F., Harris, P. C. and Igarashi, P. (2006) Proteolytic cleavage and nuclear translocation of fibrocystin is regulated by intracellular Ca2+ and activation of protein kinase C. J. Biol. Chem. 281: 34357–34364.
Wang, S., Zhang, J., Nauli, S. M., Li, X., Starremans, P. G., Luo, Y., Roberts, K. A. and Zhou, J. (2007) Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol. Cell. Biol. 27: 3241–3252.
Wilson, P. D. (2004) Polycystic kidney disease: new understanding in the pathogenesis. Int. J. Biochem. Cell Biol. 36: 1868–1873.
Torres, V. E. and Harris, P. C. (2003) Autosomal dominant polycystic kidney disease. Nefrologia 23Suppl 1: 14–22.
Sullivan, L. P., Wallace, D. P. and Grantham, J. J. (1998) Epithelial transport in polycystic kidney disease. Physiol. Rev. 78: 1165–1191.
Avner, E. D. (1993) Epithelial polarity and differentiation in polycystic kidney disease. J. Cell Sci. Suppl. 17: 217–222.
Lowden, D. A., Lindemann, G.W., Merlino, G., Barash, B. D., Calvet, J. P. and Gattone, V. H. 2nd (1994) Renal cysts in transgenic mice expressing transforming growth factoralpha. J. Lab. Clin. Med. 124: 386–394.
Torres, V. E. (1999) Apoptosis in cystogenesis: hands on or hands off? Kidney Int. 55: 334–335.
Sullivan, L. P., Wallace, D. P. and Grantham, J. J. (1998) Chloride and fluid secretion in polycystic kidney disease. J. Am. Soc. Nephrol. 9: 903–916.
Grantham, J. J. (1997) Mechanisms of progression in autosomal dominant polycystic kidney disease. Kidney Int. Suppl. 63: S93–S97.
Ong, A. C. (2000) Polycystin expression in the kidney and other tissues: complexity, consensus and controversy. Exp. Nephrol. 8: 208–14.
Kim, K., Drummond, I., Ibraghimov-Beskrovnaya, O., Klinger, K. and Arnaout, M. A. (2000) Polycystin 1 is required for the structural integrity of blood vessels. Proc. Natl. Acad. Sci. USA 97: 1731–1736.
Lu, W., Peissel, B., Babakhanlou, H., Pavlova, A., Geng, L., Fan, X., Larson, C., Brent, G. and Zhou, J. (1997) Perinatal lethality with kidney and pancreas defects in mice with a targeted Pkd1 mutation. Nat. Genet. 17: 179–181.
Bukanov, N. O., Husson, H., Dackowski, W. R., Lawrence, B. D., Clow, P.A., Roberts, B.L., Klinger, K.W. and Ibraghimov-Beskrovnaya, O. (2002) Functional polycystin-1 expression is developmentally regulated during epithelial morphogenesis in vitro: downregulation and loss of membrane localization during cystogenesis. Hum. Molec. Genet. 11: 923–936.
Huan, Y. and van Adelsberg, J. (1999) Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J. Clin. Invest. 104: 1459–1468.
Scheffers, M. S., van der Bent, P., Prins, F., Spruit, L., Breuning, M. H., Litvinov, S. V., de Heer, E. and Peters, D. J. (2000) Polycystin-1, the product of the polycystic kidney disease 1 gene, co- localizes with desmosomes in MDCK cells. Hum. Mol. Genet. 9: 2743–2750.
Roitbak, T., Ward, C. J., Harris, P. C., Bacallao, R., Ness, S. A. and Wandinger-Ness, A. (2004) A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol. Biol. Cell 15: 1334–1346.
Russo, R. J., Husson, H., Joly, D., Bukanov, N. O., Patey, N., Knebelmann, B. and Ibraghimov-Beskrovnaya, O. (2005) Impaired formation of desmosomal junctions in ADPKD epithelia. Histochem. Cell Biol. 124: 487–497.
Prozialeck, W. C., Lamar, P. C. and Appelt, D. M. (2004) Differential expression of E-cadherin, N-cadherin and betacatenin in proximal and distal segments of the rat nephron. BMC Physiol. 4: 10.
Joly, D., Morel, V., Hummel, A., Ruello, A., Nusbaum, P., Patey, N., Noel, L. H., Rousselle, P. and Knebelmann, B. (2003) Beta4 integrin and laminin 5 are aberrantly expressed in polycystic kidney disease: role in increased cell adhesion and migration. Am. J. Pathol. 163: 1791–1800.
Lo, S. H., Yu, Q. C., Degenstein, L., Chen, L. B. and Fuchs, E. (1997) Progressive kidney degeneration in mice lacking tensin. J. Cell Biol. 136: 1349–1361.
Shannon, M. B. and Miner, J. H. (2004) Insertional mutation in laminin alpha 5: a new mousemodel for polycystic kidney disease. In: American Society of Nephrology, vol. 15. pp. 652A, Couser, W. G. (ed.), LW & W, St. Louis, MO, USA.
Pan, J., Wang, Q. and Snell, W. J. (2005) Cilium-generated signaling and cilia-related disorders. Lab. Invest. 85: 452–463.
Smith, L. A., Bukanov, N. O., Husson, H., Russo, R. J., Barry, T. C., Taylor, A. L., Beier, D. R. and Ibraghimov-Beskrovnaya, O. (2006) Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. J. Am. Soc. Nephrol. 17: 2821–2831.
Marshall, W. F. and Nonaka, S. (2006) Cilia: tuning in to the cell's antenna. Curr. Biol. 16: R604–R614.
Singla, V. and Reiter, J. F. (2006) The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313: 629–633.
Barr, M. M. and Sternberg, P.W. (1999) A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 401: 386–389.
Lin, F., Hiesberger, T., Cordes, K., Sinclair, A. M., Goldstein, L. S., Somlo, S. and Igarashi, P. (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 100: 5286–5291.
Pazour, G. J., Dickert, B. L., Vucica, Y., Seeley, E. S., Rosenbaum, J. L., Witman, G. B. and Cole, D. G. (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 151: 709–718.
Praetorius, H. A. and Spring, K. R. (2001) Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 184: 71–79.
Bhunia, A. K., Piontek, K., Boletta, A., Liu, L., Qian, F., Xu, P. N., Germino, F. J. and Germino, G. G. (2002) PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 109: 157–168.
Robert, A., Margall-Ducos, G., Guidotti, J. E., Bregerie, O., Celati, C., Brechot, C. and Desdouets, C. (2007) The intra-flagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J. Cell Sci. 120: 628–637.
Li, X., Luo, Y., Starremans, P. G., McNamara, C. A., Pei, Y. and Zhou, J. (2005) Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat. Cell Biol. 7: 1202–1212.
Garcia-Gonzalez, M. A., Menezes, L. F., Piontek, K. B., Kaimori, J., Huso, D. L., Watnick, T., Onuchic, L. F., Guay-Woodford, L. M. and Germino, G. G. (2007) Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum. Mol. Genet. 16: 1940–1950.
Yamaguchi, T., Nagao, S., Wallace, D. P., Belibi, F. A., Cowley, B. D., Pelling, J. C. and Grantham, J. J. (2003) Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 63: 1983–1994.
Yamaguchi, T., Wallace, D. P., Magenheimer, B. S., Hempson, S. J., Grantham, J. J. and Calvet, J. P. (2004) Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 279: 40419–40430.
Torres, V. E. and Harris, P. C. (2007) Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J. Intern. Med. 261: 17–31.
Bukanov, N. O., Smith, L. A., Klinger, K.W., Ledbetter, S. R. and Ibraghimov-Beskrovnaya, O. (2006) Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 444: 949–952.
Kim, E., Arnould, T., Sellin, L. K., Benzing, T., Fan, M. J., Gruning, W., Sokol, S. Y., Drummond, I. and Walz, G. (1999) The polycystic kidney disease 1 gene product modulates Wnt signaling. J. Biol. Chem. 274: 4947–4953.
Saadi-Kheddouci, S., Berrebi, D., Romagnolo, B., Cluzeaud, F., Peuchmaur, M., Kahn, A., Vandewalle, A. and Perret, C. (2001) Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene 20: 5972–5981.
Qian, C. N., Knol, J., Igarashi, P., Lin, F., Zylstra, U., Teh, B. T. and Williams, B. O. (2005) Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J. Biol. Chem. 280: 3938–3945.
Simons, M., Gloy, J., Ganner, A., Bullerkotte, A., Bashkurov, M., Kronig, C., Schermer, B., Benzing, T., Cabello, O. A., Jenny, A., Mlodzik, M., Polok, B. et al. (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 37: 537–543.
Kleymenova, E., Ibraghimov-Beskrovnaya, O., Kugoh, H., Everitt, J., Xu, H., Kiguchi, K., Landes, G., Harris, P. and Walker, C. (2001) Tuberin-dependent membrane localization of polycystin-1: a functional link between polycystic kidney disease and the TSC2 tumor suppressor gene. Mol. Cell 7: 823–832.
Shillingford, J. M., Murcia, N. S., Larson, C. H., Low, S. H., Hedgepeth, R., Brown, N., Flask, C. A., Novick, A. C., Goldfarb, D. A., Kramer-Zucker, A., Walz, G., Piontek, K. B. et al. (2006) the mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 103: 5466–5471.
Guay-Woodford, L. M. (2003) Murine models of polycystic kidney disease: molecular and therapeutic insights. Am. J. Physiol. Renal Physiol. 285: F1034–F1049.
Piontek, K. B., Huso, D. L., Grinberg, A., Liu, L., Bedja, D., Zhao, H., Gabrielson, K., Qian, F., Mei, C., Westphal, H. and Germino, G. G. (2004) A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol. 15: 3035–3043.
Wu, G., D'Agati, V., Cai, Y., Markowitz, G., Park, J. H., Reynolds, D. M., Maeda, Y., Le, T. C., Hou, H. Jr., Kucherlapati, R., Edelmann, W. and Somlo, S. (1998) Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93: 177–188.
Sweeney, W. E., Chen, Y., Nakanishi, K., Frost, P. and Avner, E. D. (2000) Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 57: 33–40.
Torres, V. E., Sweeney, W. E., Wang, X., Qian, Q., Harris, P. C., Frost, P. and Avner, E. D. (2003) EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int. 64: 1573–1579.
Torres, V. E., Sweeney, W. E. Jr., Wang, X., Qian, Q., Harris, P. C., Frost, P. and Avner, E. D. (2004) Epidermal growth factor receptor tyrosine kinase inhibition is not protective in PCK rats. Kidney Int. 66: 1766–1773.
Lin, H. H., Yang, T. P., Jiang, S. T., Yang, H. Y. and Tang, M. J. (1999) Bcl-2 overexpression prevents apoptosis-induced Madin-Darby canine kidney simple epithelial cyst formation. Kidney Int. 55: 168–178.
Fedorov, L. M., Schmittwolf, C., Amann, K., Thomas, W. H., Muller, A. M., Schubert, H., Domen, J. and Kneitz, B. (2006) Renal failure causes early death of bcl-2 deficient mice. Mech. Ageing Dev. 127: 600–609.
Moser, M., Dahmen, S., Kluge, R., Grone, H., Dahmen, J., Kunz, D., Schorle, H. and Buettner, R. (2003) Terminal renal failure in mice lacking transcription factor AP-2 beta. Lab. Invest. 83: 571–578.
Tao, Y., Kim, J., Faubel, S., Wu, J. C., Falk, S. A., Schrier, R.W. and Edelstein, C. L. (2005) Caspase inhibition reduces tubular apoptosis and proliferation and slows disease progression in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 102: 6954–6959.
Omori, S., Hida, M., Fujita, H., Takahashi, H., Tanimura, S., Kohno, M. and Awazu, M. (2006) Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J. Am. Soc. Nephrol. 17: 1604–1614.
Torres, V. E. (2005) Vasopressin antagonists in polycystic kidney disease. Kidney Int. 68: 2405–2418.
Walz, G. (2006) Therapeutic approaches in autosomal dominant polycystic kidney disease (ADPKD): is there light at the end of the tunnel? Nephrol. Dial. Transplant. 21: 1752–1757.
Nagao, S., Nishii, K., Katsuyama, M., Kurahashi, H., Marunouchi, T., Takahashi, H. and Wallace, D. P. (2006) Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J. Am. Soc. Nephrol. 17: 2220–2227.
Ruggenenti, P., Remuzzi, A., Ondei, P., Fasolini, G., Antiga, L., Ene-Iordache, B., Remuzzi, G. and Epstein, F. H. (2005) Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 68: 206–216.
Tao, Y., Kim, J., Schrier, R.W. and Edelstein, C. L. (2005) Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J. Am. Soc. Nephrol. 16: 46–51.
Meijer, L., Borgne, A., Mulner, O., Chong, J. P., Blow, J. J., Inagaki, N., Inagaki, M., Delcros, J. G. and Moulinoux, J. P. (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243: 527–536.
Bach, S., Knockaert, M., Reinhardt, J., Lozach, O., Schmitt, S., Baratte, B., Koken, M., Coburn, S. P., Tang, L., Jiang, T., Liang, D. C., Galons, H., et al. (2005) Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 280: 31208–31219.
Benson, C., White, J., De Bono, J., O'Donnell, A., Raynaud, F., Cruickshank, C., McGrath, H., Walton, M., Workman, P., Kaye, S., Cassidy, J., Gianella-Borradori, A. et al. (2007) A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 96: 29–37.
Leuenroth, S. J., Okuhara, D., Shotwell, J. D., Markowitz, G. S., Yu, Z., Somlo, S. and Crews, C. M. (2007) Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc. Natl. Acad. Sci. USA 104: 4389–4394.
Li, H., Findlay, I. A. and Sheppard, D. N. (2004) The relationship between cell proliferation, Cl-secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int. 66: 1926–1938.
Ibraghimov-Beskrovnaya, O. and Bukanov, N. O. (2006) In vitro cystogenesis: the search for drugs antagonizing cyst development. Nephrol. Ther. 2Suppl 2: S109–S114.
Magenheimer, B. S., St John, P. L., Isom, K. S., Abrahamson, D. R., De Lisle, R. C., Wallace, D. P., Maser, R. L., Grantham, J. J. and Calvet, J. P. (2006) Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(−) Co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 17: 3424–3437.
Chapman, A. B. (2007) Autosomal dominant polycystic kidney disease: time for a change? J. Am. Soc. Nephrol. 18: 1399–1407.
Grantham, J. J., Torres, V. E., Chapman, A. B., Guay-Woodford, L. M., Bae, K. T., King, B. F. Jr., Wetzel, L. H., Baumgarten, D. A., Kenney, P. J., Harris, P. C., Klahr, S., Bennett, W. M. et al. (2006) Volume progression in polycystic kidney disease. N. Engl. J. Med. 354: 2122–2130.
Gumbiner, B. M. (1996) Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84: 345–357.
Acknowledgement
The authors would like to thank Steve Ledbetter, Ryan Russo, Marlon Pragnell and Tom Natoli for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Received 8 August 2007; received after revision 19 September 2007; accepted 2 October 2007
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ibraghimov-Beskrovnaya, O., Bukanov, N. Polycystic kidney diseases: From molecular discoveries to targeted therapeutic strategies. Cell. Mol. Life Sci. 65, 605–619 (2008). https://doi.org/10.1007/s00018-007-7362-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-007-7362-x