
Abstract
This study presents the development and mapping of simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers in chickpea. The mapping population is based on an inter-specific cross between domesticated and non-domesticated genotypes of chickpea (Cicer arietinum ICC 4958 × C. reticulatum PI 489777). This same population has been the focus of previous studies, permitting integration of new and legacy genetic markers into a single genetic map. We report a set of 311 novel SSR markers (designated ICCM—ICRISAT chickpea microsatellite), obtained from an SSR-enriched genomic library of ICC 4958. Screening of these SSR markers on a diverse panel of 48 chickpea accessions provided 147 polymorphic markers with 2–21 alleles and polymorphic information content value 0.04–0.92. Fifty-two of these markers were polymorphic between parental genotypes of the inter-specific population. We also analyzed 233 previously published (H-series) SSR markers that provided another set of 52 polymorphic markers. An additional 71 gene-based SNP markers were developed from transcript sequences that are highly conserved between chickpea and its near relative Medicago truncatula. By using these three approaches, 175 new marker loci along with 407 previously reported marker loci were integrated to yield an improved genetic map of chickpea. The integrated map contains 521 loci organized into eight linkage groups that span 2,602 cM, with an average inter-marker distance of 4.99 cM. Gene-based markers provide anchor points for comparing the genomes of Medicago and chickpea, and reveal extended synteny between these two species. The combined set of genetic markers and their integration into an improved genetic map should facilitate chickpea genetics and breeding, as well as translational studies between chickpea and Medicago.
Similar content being viewed by others

Introduction
Chickpea (Cicer arietinum L.) is an annual, self-pollinated diploid (2n = 2x = 16) species with a relatively small genome of 740 Mbp (Arumuganathan and Earle 1991). Chickpea is also the World’s third most widely grown food legume. Over 95% of chickpea production area and consumption occur in developing countries, with India contributing the largest share (65%), followed by Pakistan (9%), Iran (7%), and Turkey (4%) (FAOSTAT database http://faostat.fao.org/site/567/default.aspx#ancor, 2007). Cytogenetic and seed protein analyses are consistent with C. reticulatum as the wild progenitor of domesticated C. arietinum, with southeastern Turkey as the presumed center of origin (Ladizinsky and Adler 1976). Cultivated chickpea is composed of two genetically distinct sub-types that are readily distinguished based on seed size and color: Desi, composed of small, brown seeded varieties, and Kabuli, composed of large, cream seeded varieties. Due to relatively low rates of polymorphism between cultivated chickpea accessions, inter-specific crosses between C. arietinum and C. reticulatum have been the primary focus for genetic studies of agronomic traits (see Singh et al. 2008).
A diverse array of technologies is available to identify and monitor DNA polymorphism and as a consequence molecular markers are now routinely used in the breeding programs of several crop species (Varshney et al. 2006, 2007). In the case of chickpea, molecular markers reported in the literature are almost entirely simple sequence repeat (SSR) loci (Choudhary et al. 2006; Hüttel et al. 1999; Lichtenzveig et al. 2005; Sethy et al. 2003, 2006a, b; Winter et al. 1999). Despite considerable effort, low rates of both intra- and inter-specific polymorphism have limited the number of these SSR markers that have been integrated into chickpea genetic maps. A primary goal of the current study was to screen additional molecular markers and thereby enhance the marker density of chickpea genetic maps.
Chickpea is a close relative of the model legume system Medicago truncatula (Fig. 1, reproduced according to Choi et al. 2004b), and thus should benefit from the increasingly detailed description of the structure and function of the M. truncatula genome (Cannon et al. 2006). A pressing task for chickpea researchers is to use knowledge gained from the study of reference legumes, such as M. truncatula, Lotus japonicus, and soybean, to advance genetic improvement of chickpea. Comparative genomics based on orthologous genetic markers offers means to bridge model and crop legumes. Alignment of linkage maps and sequenced orthologous regions between several legume species has revealed an extensive network of macro- and micro-synteny between legume species; importantly, genomic and genetic comparisons of orthologous nodulation genes in several legumes suggests that comparison of genome structure and function may have practical applications to cross-species gene prediction and isolation (see Zhu et al. 2005). The lack of infrastructure (knowledge and physical capacity) in chickpea, however, has limited the potential for cross-genome comparisons and has hampered progress in the area of genomics-assisted breeding (Varshney et al. 2009a).

Phylogenetic relationships: Papilionoideae family. This figure (taken from Choi et al. 2004b) illustrates the phylogenetic relation of chickpea with other legumes. Chickpea and Medicago belong to inverted repeat lacking clade (IRLC) of Papilionoideae and constituted the cool season legumes
In view of the above, we sought to enhance marker repertoire and density of genetic maps in chickpea using a combination of several molecular marker sets. We developed two novel sets of molecular markers based on an SSR-enriched genomic DNA library and gene-based single nucleotide polymorphism (SNP) markers derived from comparison of Medicago and chickpea ESTs. These novel genetic markers were analyzed together with published genetic markers to develop a dense genetic map of chickpea. We anticipate that these resources will serve as tools for genomics-assisted breeding in chickpea, and enhance prospects for transfer of knowledge about the structure and function of the Medicago genome to chickpea with a final objective of chickpea improvement.
Materials and methods
Plant material and DNA extraction
The cultivated chickpea germplasm line ICC 4958, belonging to Desi type and a parent line of inter-specific reference mapping population (C. arietinum ICC 4958 × C. reticulatum PI 489777) was used to construct microsatellite-enriched library. While two genotypes of chickpea (ICC 4958 and ICC 1882) were used to optimize the polymerase chain reaction (PCR) conditions for newly developed SSR markers, an array of 48 genotypes which includes 33 genotypes from cultivated chickpea (C. arietinum) and 15 from wild species of chickpea (7 genotypes from C. reticulatum, 2 genotypes from C. echinospermum and one each from C. bijugum, C. cuneatum, C. judaicum, C. microphyllum, C. pinnatifidum, and C. yamashitae) was used to assess the polymorphism potential of new set of SSR markers (Table 1).
For integrating the markers into the genetic map, the inter-specific reference mapping population ICC 4958 × PI 489777 comprising of a total of 131 RILs was used. While all 131 RILs were used to score genotyping data for the SSR markers isolated in this study as well as reported by Lichtenzveig et al. (2005), a subset of 94 RILs was used with gene-based markers.
Total genomic DNA was extracted by employing the standardized high throughput mini DNA extraction protocol (as mentioned in Cuc et al. 2008). The quality and quantification of extracted DNA was checked on 1.2% agarose. The DNA was normalized to 5 ng/μl for further use.
Construction of SSR-enriched library
To construct a size-fractioned chickpea genomic DNA library, purified genomic DNA (100 μg) of ICC 4958 was completely digested with MboI or Sau3AI in combination with TaqI enzyme. The restricted fragments were separated on low-melting agarose gels, and the gel zone containing the fragments of DNA of size 800–1200 bp were excised and ligated into Promega pGEM 3Z(f) vector (Promega, Madison, WI, USA). The vector was transformed into E. coli Sure strain—DH10B (Stratagene, Heidelberg, Germany) by electroporation. Approximately 400,000 clones were plated at a density of 20,000 colonies per plate. The masterplates generated were replica-plated on positively charged PVDF macroarrays. Macroarrays were printed using contact printing technology at RZPD GmbH, Berlin, Germany.
For enriching the genomic DNA library, synthetic oligos (GA)10 and (TAA)10 were enzymatically 3′ end-labeled with digoxigenated oligonucleotides (DIG Oligonucleotide 3′-End Labeling Kit; Roche, Mannheim, Germany). Subsequently, macroarrays/filters were hybridized with above-mentioned oligo-probes in Roti-Hybri-Quick buffer (Carl Roth GmbH, Karlsruhe, Germany) including 10 μg/ml sheared, denatured E. coli DNA to minimize non-specific binding. Filters were hybridized at 55°C overnight and washed three times each for 10 min in 1:2, 1:5, and 1:10 dilutions of the hybridization buffer at 60°C. The digoxigen was detected in a “direct detection assay” performed with the DIG Wash and Block buffer set, and DIG Luminescent detection Kit (Roche, Mannheim, Germany) for chemiluminescent detection with a monospecific antibody coupled to alkaline phosphatase in the presence of CSPD. Filters were exposed to X-ray films (Amersham, Buckinghamshire, UK) with intensifying screens for 4 h or overnight, and the colonies giving strong signals were scraped from the master plates; re-grown; spotted on Hybond N membranes (Amersham, Buckinghamshire, England) to fix the DNA by lysis. Hybridization and chemiluminescent detection was done repeatedly to pick the clones with positive signals. These clones were grown on LB agar plates with ampicillin (100 μg/ml) overnight at 37°C. Aliquots of these colonies were used for colony PCR.
Development of genomic SSR markers
The colonies with high level of signal were used to isolate plasmid DNA using standard alkaline lysis method (Sambrook and Russell 2001). After checking the quality of the plasmid DNA on 0.8% agarose gel, the clones were sequenced using the BigDye Terminator cycle sequencing kit on an ABI3700/ABI3730XL (Applied Biosystems Inc., Foster City, CA, USA). 288 clones were sequenced in both directions using standard T7 promoter and SP6 primers and 19 clones in one direction by using M13-forward sequencing primer at Macrogen (www.macrogen.com) and ICRISAT.
The sequences generated were subjected to CAP3, a contig assembly program (http://pbil.univ-lyon1.fr/cap3.php) in order to define unigenes. These unigenes were subjected to MIcroSAtellite (MISA, Varshney et al. 2002) tool to search microsatellites considering minimum ten repeat units of mono- (N), and four repeats of di- (NN), tri- (NNN), tetra- (NNNN), penta- (NNNNN) and hexa- (NNNNNN) nucleotides and compound microsatellites present within a distance of 100 bp. Primer pairs for SSRs were designed using Primer3 program (http://frodo.wi.mit.edu/) in batch file, and the SSR markers developed were designated as ICCM (ICRISAT Chickpea Microsatellite) markers (Table 2).
Development of gene-based SNP markers
PCR primers derived from Medicago ESTs (expressed sequence tags), Medicago BAC (bacterial artificial chromosome)-end sequences, and M. sativa cDNA sequences have been described in previous studies (Choi et al. 2004a, b, 2006). To design PCR primers based on chickpea ESTs (Buhariwalla et al. 2005), candidate chickpea transcripts were compared to sequenced Medicago BAC clones (http://www.medicago.org/genome), and transcripts with high nucleotide identity and low copy representation to the Medicago genome were selected for primer designing. Primers were designed from highly conserved coding sequences, to amplify across intron regions (Choi et al. 2004a), using the Lasergene PrimerSelect software package (DNAStar Inc., Madison, WI, USA). Details of these primer pairs are given in Table 3.
The polymorphic gene-based markers between the parents of mapping population were identified essentially as mentioned in Choi et al. (2004a). Each pair of corresponding sequences from genotypes ICC 4958 and PI 489777 was aligned using Sequencher software (Gene Codes, Ann Arbor, MI, USA) to detect SNPs. The sequences with SNPs were transferred to DNA Strider 1.2 (Douglas 2008) to identify restriction site that is coincident with SNPs and cleavable amplified polymorphic sequence (CAPS) assay for genotyping the corresponding SNPs were developed. In cases where a suitable restriction enzyme site was not identified, oligonucleotide primers were designed immediately adjacent to the SNP position, which allows for a single base extension of the SNP site using ABI SNaPshot Multiplexing Kits (Applied Biosystems Inc., Foster City, CA, USA).
Genotyping assay
For both ICCM as well as H-series SSR markers, the forward primers were anchored with M13 tail (CACGACGTTGTAAAACGAC). PCR amplicons generated by SSR markers were analyzed on capillary electrophoresis, while for gene-based SNP markers the CAPS or SNaPshot assays were used for genotyping. For SSR genotyping, PCR was carried out in 5 μl reaction volume in GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA, USA). The reaction mixture contained final concentration of 5 ng/μl of template DNA, 0.5 mM dNTPs, 0.5 μM of M13 tailed forward, 1 μM of reverse primer, 1 μM of M13 labeled primer, 0.75 mM of MgCl2, 0.1 U of Taq DNA polymerase (AmpliTaq Gold), and 1× PCR buffer (AmpliTaq Gold). An initial denaturation was given for 15 min at 94°C. Subsequently, ten touch-down PCR cycles comprising of 94°C for 20 s, 61/60/55°C (depending on the marker as given in Table 2, ESM Table 1) for 20 s, and 72°C for 30 s were performed. These cycles were followed by 35 cycles of 94°C for 10 s with constant annealing temperature of 54/56/48°C (depending on marker and touch-down profiles as given in Table 2, ESM Table 1) for 20 s, and 72°C for 30 s, and a final extension was carried out at 72°C for 20 min. The amplified products were separated by capillary electrophoresis using ABI PRISM® 3700 DNA analyzer, and allele calling was carried out as given in Varshney et al. (2009b).
For SNP genotyping, in CAPS assay, 1.5 μl PCR product of 94 RILs was digested with the corresponding restriction enzymes. Each digestion reaction contained 2–5 U of the corresponding restriction enzyme and 1× compatible buffer in a total volume of 10 μl. Enzyme digestions were incubated at the appropriate temperature for at least 4 h. Digestion products were separated and scored as mentioned in Choi et al. (2004a). In case of SNaPshot assay, the ABI SNaPshot Multiplexing Kits was used following the same protocol as suggested by the manufacturer, except that 0.5 μl SNaPshot mix for a single marker was used (see Choi et al. 2004a).
Polymorphism assessment of SSR markers
While ICCM-series markers were screened on the panel of 48 diverse genotypes including the parents of the inter-specific mapping population (Table 1), the H-series markers were screened on only two parental genotypes (ICC 4958 and PI 489777). Allelic data obtained for the SSR markers were subjected to AlleloBin program (http://www.icrisat.org/gt-bt/download_allelobin.htm) for allele calling based on the repeat units of SSR motif for corresponding markers. In case of ICCM markers, the binned allelic data were used to calculate polymorphic information content (PIC) value of the markers by using the PowerMarker V3.25 program (http://statgen.ncsu.edu/powermarker/).
Linkage analysis and map construction
Genotyping data for both ICCM- and H-series polymorphic markers were generated on 131 recombinant inbred lines (RILs) of the mapping population and for 94 RILs in case of gene-based SNP markers. In addition, marker genotyping data for 407 marker loci were compiled (Huettel et al. 2002; Pfaff and Kahl 2003; Tekeoglu et al. 2000; Winter et al. 1999, 2000).
Marker genotyping data were analyzed using the χ2 test to assess the goodness-of-fit to the expected 1:1 segregation ratio for each marker. Subsequently, genotyping data for all the markers, including those with distorted segregation, were used for linkage analysis using MAPMAKER/EXP 3.0 (Lander et al. 1987). Marker loci were first divided into linkage groups at a LOD score of 16 and a recombination fraction of 0.37 by two-point analysis using the ‘group’ command. Marker order in the linkage groups was determined using the multi-point analysis ‘try’ command of the program. Most likely order of the loci within the group was determined using multipoint ‘compare’ command. The ungrouped marker loci were also attempted to integrate into genetic map at a smaller LOD value (up to 6). The map distances were calculated by applying the ‘Kosambi’ mapping function (Kosambi 1944) as per MAPMAKER/EXP 3.0 program. Residual heterozygosity was not considered in linkage mapping.
Results
Isolation and characterization of simple sequence repeats
A genomic DNA library composed of ca. 400,000 clones was constructed from the ICC 4958 genotype. Hybridization of this library with GA and TAA oligo probes yielded 359 clones that were sequenced and assembled into a set of 115 contig and 342 singleton DNA sequences, which we refer to as genome survey sequences (GSS). These sequences were submitted to National Centre for Biotechnology Information (NCBI) and respective GenBank accessions are mentioned in Table 2.
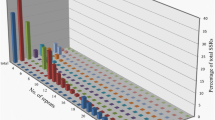
Two hundred and ninety-nine of the 457 GSSs were determined to contain a total of 643 SSRs, with 165 GSSs containing more than one SSR. As depicted in Fig. 2, di- and tri-nucleotide repeats were the most abundant (39 and 40%, respectively), with mono-nucleotide and tetra-nucleotide repeats representing 16 and 3% of cases, respectively. Other types of SSRs had <1% representation. In terms of repeat motifs, the tri-nucleotide repeat motif TAA/ATT was most common, accounting 36.8% of all repeat, followed by the di-nucleotide repeat GA/CT at 19.2%.

Frequency of microsatellites based on type of repeat motifs in microsatellite-enriched library of chickpea. Frequency of tri-nucleotide repeats were higher among the chickpea microsatellite markers followed by di-nucleotide repeats. N, mono-nucleotide repeats; NN, di-nucleotide repeats; NNN, tri-nucleotide repeats; NNNN, tetra-nucleotide repeats; NNNNN, penta-nucleotide repeats, NNNNNN, hexa-nucleotide repeats
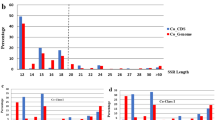
These SSR loci were categorized into two groups based on the length of their SSR tracts: Class I SSRs (>20 nucleotides in length) and Class II containing SSRs (>12 but <20 nucleotides in length) (Fig. 3). Considering only perfect SSRs, which is the set of SSRs that contain a single motif (e.g., TAA), we observed uneven distribution between Classes I and II. In particular, the longer Class I SSRs were substantially enriched for tri-nucleotide repeats, which represented 77% of all Class I repeats. A similar uneven distribution was noted for other repeats, but most notably the penta-nucleotide repeats, which comprised 55% of all Class II repeats and less than 2% of all Class I repeats.

Distribution of Class I and Class II repeats in newly isolated chickpea microsatellites. Class I microsatellites are with >20 nucleotides in length and Class II repeats contain perfect SSRs with >12 but <20 nucleotides in length. Among Class I repeats, tri-nucleotide repeats were abundant followed by di-nucleotide repeats, while in Class II repeats, penta-nucleotide repeats contributed highest, followed by hexa-repeats. N, mono-nucleotide repeats; NN, di-nucleotide repeats; NNN, tri-nucleotide repeats; NNNN, tetra-nucleotide repeats; NNNNN, penta-nucleotide repeats, NNNNNN, hexa-nucleotide repeats
Similarity analysis was performed for all 457 GSSs using BLASTN and BLASTX algorithms, and significant similarity was determined at an Expect value threshold of ≤1E–05 (Table 4). Relatively few of the GSS sequences had E values that surpassed this score, irrespective of the species data set under analysis. This is consistent with the expectation that randomly selected short genomic sequences only occasionally correspond to gene coding regions that will match EST data sets. Nevertheless, in cases where BLAST hits with e-value lower than 1E–05 threshold were recorded, the degree of similarity, expressed as either nucleotide identity of deduced protein similarity, was highest for phylogenetically related species, decreasing in rank order of phylogenetic distance (i.e., Medicago > lotus > soybean = cowpea = common bean > poplar > Arabidopsis > rice). Among these sequences, 40 were identified as related sequences in all three analyzed cool season legumes, i.e., chickpea, Medicago, and Lotus (Hologalegina clade; see Fig. 1), while 29 sequences had similarity with all three analyzed warm season legumes, i.e., soybean, common bean, and cowpea (Phaseoleae clade). Only 21 sequences were identified as similar sequences in both Hologalegina and Phaseoleae species. Two of these GSSs (FI856609 and FI856659) showed significant similarity with sequences of all the plant species analyzed in the present study (see ESM Table 2).
With the objective of annotating these newly isolated GSSs, all 457 GSSs were analyzed for BLASTX analysis using UniProt database. 137 of these GSSs (29.9%) showed homology to the UniProt database at a relatively relaxed cutoff value of ≤ 1E–05. Among these, 84 unique protein sequences were used for deriving respective gene ontology (GO) (see ESM Table 3). The GO studies permitted assignment of 64 sequences to biological process, 64 to cellular component, and 67 to molecular function ontologies. According to the GO schema, single proteins typically have more than one Ontology assignment.
Development of novel SSR genetic markers
All SSR containing GSSs (299) were analyzed by means of Primer3, yielding a list of potential oligonucleotide primers from which 311 primer pairs were selected and synthesized. Where feasible primer pairs were designed for more than one SSR in a single GSS with the goal of increasing the conversion of GSSs into useable genetic markers.
Primer pairs were screened for amplification of DNA from two chickpea genotypes, i.e., ICC 4958 and ICC 1882 (Table 2). This analysis provided a set of 234 markers (75%) with scorable amplicons. Screening of these 234 markers on 48 genotypes of chickpea further defined a subset of 147 polymorphic markers (62.82%), with allele content ranging from 2 to 21 and an average of five alleles per marker. Among these 147 polymorphic sites, 56 were polymorphic exclusively in wild species, 8 were polymorphic exclusively in cultivated and 83 of them were polymorphic across wild and cultivated species of chickpea.
We refer to these new polymorphic SSR markers as ICCM (ICRISAT Chickpea Microsatellite) markers. Allelic data obtained from 48 genotypes were used to calculate the PIC value of each ICCM marker, and thus infer the discriminatory power of these ICCM markers. PIC values ranged from 0.04 to 0.92 with an average of 0.26. Twenty-six markers displayed the minimum PIC value of 0.04 each, while marker ICCM0160 had both the highest PIC value (0.92) and the highest number of alleles (21), followed by marker ICCM0022 with 18 alleles and a PIC value of 0.89 (Table 2). As has been observed in previous studies of SSRs from plant species (Temnykh et al. 2001), Class I SSRs (41 of 57) were on average more polymorphic that Class II SSRs (106 of 177), with mean PIC values of 0.38 and 0.22, respectively. Nevertheless, a higher fraction of the polymorphic SSRs identified in this study were from Class II (106) compared to Class I (41), owing to the increased abundance of Class II SSRs in our data set. Consistent with their overall abundance in Class I SSRs (Fig. 3), tri-nucleotide repeats (20) constituted major part of the Class I polymorphic sites, with compound repeats (18) comprising the next largest fraction of Class I ICCM markers. In contrast, di-nucleotide repeats were relatively rare in the total Class II data set, but comprised the largest fraction of polymorphic Class II ICCM markers (47); similar to Class I markers, compound repeats (30) constituted of the second most common fraction of Class II polymorphic sites.
In addition to the ICCM markers developed in this study, we also analyzed a set of 233 markers developed primarily by Lichtenzveig et al. (2005); these are the so-called “H-series” SSR markers. One-hundred fifty-three H-series markers yielded scorable amplicons in two PCR profiles (ESM Table 1). Both the ICCM and H-series SSR markers were tested for polymorphism between chickpea ICC 4958 and PI 489777, the parents of the inter-specific mapping population. From this analysis we identified 104 SSRs (52 ICCM and 52 H-series) that were suitable as genetic markers in the inter-specific cross, with polymorphism rates of 33.9 and 22.2% for the H-series and ICCM SSR markers, respectively.
Development of gene-based SNP markers
A set of 246 gene-specific primers, developed earlier by Choi et al. (2004a) based on gene sequences of M. truncatula and M. sativa, were used to amplify DNA of the parental genotypes of the inter-specific mapping population of chickpea. One-hundred four (~42%) of these primer pairs showed strong single fragments on 1% agarose gels; these amplicons were re-sequenced in both mapping parents of the inter-specific cross (ICC 4958 and PI 489777), quality-scored, and trimmed to yield 96 pairs of high quality sequences. Additional 25 primer pairs were designed based on chickpea EST sequences that possessed high similarity to previously mapped Medicago genes, yielding 18 additional high-quality sequence pairs. Alignment of the 114 ICC 4958 and PI 489777 sequence pairs revealed SNPs in 80 (~70%) genes. Seventy-one of these genes contained SNPs that could be converted to reliable genotyping assays using either CAPS or SNaPshot protocols (Table 3). Two additional gene-based markers, P40 and chitinase II, were also used for genetic analysis; these genes were previously mapped in chickpea by Pfaff and Kahl (2003), while their putative orthologs have been mapped in M. truncatula by Choi et al. (2004a).
Construction and features of the genetic map
The inter-specific cross between ICC 4958 × PI 489777 is maintained as an advanced recombinant inbred population that has been used in numerous genetic studies (Huettel et al. 2002; Pfaff and Kahl 2003; Winter et al. 2000). Although the number of markers previously analyzed in this population is relatively large (407 loci), a high percentage of the markers are anonymous sequences (e.g., RFLP) and/or exhibit dominant patterns of inheritance (e.g., AFLP). Thus, in many cases, these legacy genetic maps are based on molecular markers that are either difficult to apply or to reproduce. With the intent of extending this genetic map, and enhancing the number of easily scorable markers, we genotyped the 123 new molecular markers (52 ICCM SSR loci and 71 gene-based SNP loci) and 52 previously published H-series SSR loci described above, and combined the genotype data with that of the 407 previously published loci. Linkage relationships were evaluated using MAPMAKER/EXP 3.0.
As shown in Fig. 4, 47 (90.3%) of 52 ICCM marker loci, 46 (88.4%) of 52 H-series SSR loci, all (100%) of 71 gene-based marker loci, and 357 (87.7%) of 407 legacy marker loci coalesced to yield eight linkage groups, in agreement with eight chickpea chromosomes. The linkage groups were numbered according to Winter et al. (2000), using marker loci that were common to both studies. This revised genetic map contains 521 marker loci, with an average inter-marker distance of 4.99 cM and spanning 2,602.1 cM. Considering the 740-Mbp physical size of the chickpea genome (Arumuganathan and Earle 1991), and ignoring the fact that rates can vary widely within the genome, 1 cM distance in the present map equates to roughly 285 kbp. With the exception of linkage group (LG) 8, which has relatively few genetic markers (25 markers), the average number of markers per linkage group was 71 ± 8.9. LG 8 was also the shortest linkage group based on genetic distance, spanning 124.7 cM; however, in general LG size was not well correlated with the number of markers. As described below, comparative mapping with Medicago truncatula revealed that the entirety of chickpea LG8 corresponds to one arm of Medicago Chr5, adding further credibility to its assignment as a physically short linkage group.


An integrated genetic map of chickpea based on recombinant inbred lines of C. arietinum (ICC 4958) × C. reticulatum (PI 489777). Map was constructed using MAPMAKER/EXP 3.0 with Kosambi mapping function. Distances between the loci (in cM) are shown to the left of the linkage group and all the loci are at the right side of the map. Newly developed SSR markers developed from microsatellite-enriched library (ICCM-series) are bold and italicized; SSR markers taken from Lichtenzveig et al. (2005) are bold, italicized, and underlined; SNP markers which were used as the anchor markers in comparative mapping of chickpea and Medicago were depicted as bold and underlined. Linkage groups (LGs) are designated according to the map of Winter et al. (2000)
Comparative linkage analysis between Medicago and chickpea genomes
As shown in Fig. 5, the 71 gene-based SNP markers are distributed among eight major linkage groups of chickpea, facilitating comparison of genome structure between M. truncatula and chickpea. The respective M. truncatula and chickpea LGs are numbered according to Choi et al. (2004a) and Winter et al. (2000). Alignment of conserved genes between the two genetic maps reveals a high level of synteny between the two genomes. In particular, the M. truncatula linkage groups 1, 2, 3, 4, 7, and 8 correspond to chickpea linkage groups 4, 1, 5, 6, 3, and 7, respectively. Despite the overall high level of synteny between these six pairs of linkage groups, intra-chromosomal segment rearrangements reduce co-linearity (but not synteny) between M. truncatula LG1 (MtLG1) and chickpea LG4 (CaLG4). In contrast to the conserved synteny noted for Mt–Ca linkage group pairs 1–4, 2–1, 3–5, 4–6, 7–3, and 8–7, one-to-one relationships do not hold true for M. truncatula linkage groups 5 and 6 and chickpea linkage groups 2 and 8. In particular, M. truncatula LG5 can be aligned with both chickpea LG2 and LG8. We note that CaLG8 appears to be derived entirely from one arm of MtLG5, consistent with its short genetic distance and small number of genetic markers, described above. In several cases, conserved markers mapped to non-syntenic positions between the two genomes (e.g., CDC2 and TC88727 on MtLG1, DNABP on MtLG4, and TCMO on MtLG5), which may reflect translocation or duplication events involving single genes or small chromosomal segments, or the mapped loci may correspond to paralogous genes. Mt-LG6 could not be effectively aligned to any of the chickpea linkage groups (Fig. 5), consistent previous reports describing Mt LG6 as rich in heterochromatin (Kulikova et al. 2001) and having a relatively low content of transcribed genes (Choi et al. 2004a).

Comparative map of Medicago and chickpea. Gene-based SNP markers (marked in red color) were used as the anchor markers in comparative analysis of chickpea and Medicago genome. The resistance gene homologs (RGH) are depicted as oval structures and their homologs in Medicago are shown with connecting dotted lines. Solid lines show the macrosynteny observed across chickpea and Medicago with respect to 71 gene-based markers
Comparison of resistance gene homologs (RGH) between Medicago and chickpea
The majority of functionally characterized disease resistance (R) genes encode a nucleotide-binding site (NBS) and a leucine-rich repeat (LRR) region (Hulbert et al. 2001). NBS-LRR genes have been deeply surveyed and characterized in M. truncatula (Zhu et al. 2002; Ameline-Torregrosa et al. 2008), with >330 NBS-LRR genes having known genetic positions. In contrast, chickpea RGHs are not thoroughly surveyed, and only a limited number of sequences from degenerate PCR are available in the public databases (Meyers et al. 1999; Huettel et al. 2002). Nevertheless, several phylogenetically distinct RGH classes have been placed on the genetic map of chickpea (Huettel et al. 2002), thus facilitating the comparative genome analysis presented here.
Comparative phylogenetic analysis of RGH sequences from M. truncatula with those from chickpea is illustrated in Fig. 6. To highlight the comparison, only those M. truncatula sequences that are relevant to the mapped chickpea sequences are shown. In the TIR-NBS-LRR subfamily, chickpea RGH-G (CAC86496 on CaLG6; Huettel et al. 2002) is highly similar to several M. truncatula TIR-NBS-LRR genes located on MtLG4, in a region syntenic to Cicer LG6 that also contains chickpea RGH-G (Huettel et al. 2002). Similarly, chickpea RGH-B (CAC86491; Huettel et al. 2002) is a CC-NBS-LRR gene that is closely related to several CC-NBS-LRR genes located in a cluster at the top of MtLG3, in a region of the Medicago genome syntenic with the terminus of CaLG5 that contains RGH-B (Huettel et al. 2002). A lack of synteny was observed for chickpea RGH-D (TIR-NBS-LRRs represented by sequences CAC86454, CAC86455, CAC86493, AF186626, and AF186629; Huettel et al. 2002), which is located at the top of CaLG2; the closest homologs of RGH-D in M. truncatula (i.e., BAC AC144658) are localized to the distal region of MtLG4. We note that the bottom of CaLG2 harbors numerous active resistance genes against two of the most important diseases of chickpea (Fusarium wilt and Ascochyta blight). At present, no RGHs have been reported mapped close to these resistance phenotypes (Winter et al. 2000; Pfaff and Kahl 2003; Sharma et al. 2004). Moreover, the low frequency of comparative molecular markers around these R gene regions in both M. truncatula and chickpea complicate precise statements regarding the relationship of these genome regions.

Comparison of RGH sequences in Medicago and chickpea. To highlight the comparison between the chickpea and Medicago RGHs, only those Medicago sequences that are relevant to the mapped chickpea sequences have been shown in this figure. In the TIR-NBS-LRR subfamily, chickpea RGH-G (CAC86496 on Ca-LG6) was found highly similar to several Medicago TIR-NBS-LRR genes (a) located on BAC clones AC144502 and AC135160. AC144502 and AC135160 were closely linked on Mt-LG4, in a region syntenic to Ca-LG6 that also contained chickpea RGH-G. In the CC-NBS-LRR (b) subfamily (Ca-LG5), chickpea RGH-B (CAC86491) was closely related to several CC-NBS-LRR genes located on Medicago BAC clones AC145027, AC142396, AC130810, AC146744, and AC131249
Discussion
SSR markers have become common place for plant genetics and breeding applications. Despite the fact that hundreds of SSR markers have been identified and tested in chickpea (Hüttel et al. 1999; Sethy et al. 2006a, 2006b; Winter et al. 1999; Lichtenzveig et al. 2005), the narrow genetic background of cultivated chickpea germplasm has limited their application, and thus there exists a need to develop a larger set of novel genetic markers. With the objective of enriching the marker repertoire of chickpea, we have contributed novel SSR markers derived from a genomic library enriched for GA and TAA repeat motifs and a set of gene-based SNP markers. The basis of our marker discovery work was C. arietinum genotype ICC 4958, which is being used as a reference genotype for genomic and genetic resource by the chickpea community.
In the present study, 65.4% of hybridizing genomic clones in our SSR-enriched library yielded 643 SSRs. This rate of SSR recovery is comparable with previous studies, for example in peanut where 68% of hybridizing clones yielded SSRs (Cuc et al. 2008). Moreover, the relatively high abundance of tri- and di-nucleotide repeats that we observed is consistent with previous studies in chickpea (Hüttel et al. 1999; Lichtenzveig et al. 2005; Winter et al. 1999). Among the SSRs identified here, the most common SSR motifs were TAA/ATT repeats and GA/CT repeats. This result reflects the fact that our enrichment targeted TAA and GA motifs, and it is consistent with previous studies in chickpea (Hüttel et al. 1999; Lichtenzveig et al. 2005; Winter et al. 1999), other legume species (Akkaya et al. 1992; Cregan et al. 1994; Mun et al. 2006), and even in cereal species (Varshney et al. 2002; Jayashree et al. 2006).
Temnykh et al. (2001) developed a scheme to classify SSRs according to length, in which Class I and Class II SSRs are greater than or less than 20 bp, respectively. This division based on sequence length has practical utility, because Class I SSRs are generally more polymorphic and thus more desirable as genetic markers. The majority of SSRs isolated from our SSR-enriched library belong to Class II, though as expected the Class I SSRs had higher rates of polymorphism. A useful measure of polymorphic potential for any genetic marker is its polymorphism information content value, or PIC value. PIC values provide information on the probability that a given marker will be polymorphic between any two individuals in a population, and thus are a function both of allele frequencies and allele number. Screening of the ICCM-series markers on 48 genotypes revealed that average PIC value of SSR markers having Class I repeats (0.38) was higher than that of Class II repeats (0.22). The majority of the Class I repeats were tri-nucleotide repeats, consistent with the known utility of tri-nucleotide repeats as genetic markers in plants (Varshney et al. 2005).
Polymorphic information content value was also analyzed in relation to repeat unit type and length. Among di-, tri-, and tetra-nucleotide repeats, tri-nucleotide repeats showed higher polymorphism (average PIC = 0.33) with average allele number of 5.7 per marker. Markers with mono-nucleotide repeats showed the least polymorphism (average PIC = 0.197). Relatively longer repeats appear to have contributed to the higher level of polymorphism as compared to di-nucleotide repeats (Gupta and Varshney 2000). It was also observed that among tri-nucleotide SSRs, the SSR markers based on (TAA/TTA) repeat motifs displayed higher polymorphism (average PIC = 0.35) with an average allele number of 6.12 per marker. Similarly, among di-nucleotide repeats SSR markers based on TA/AT repeat motifs had a higher average PIC value (0.27) compared to others with an average of 6.1 alleles. In fact, the earlier studies in chickpea also revealed the abundance of TAA/TTA (tri-nucleotide) and TA/GA (di-nucleotide) SSR motifs and the extensive polymorphism found with markers containing these repeat motifs (Hüttel et al. 1999; Lichtenzveig et al. 2005). PIC values of compound SSRs (average PIC = 0.29) were comparable with tri-nucleotide repeats with 5.68 alleles per marker. This can be attributed to the fact that the markers with compound SSRs have more than one SSR motif, which increases their chances to be polymorphic markers.
We assessed the potential identity of SSR-related sequences by performing BLAST analyses versus plant EST data sets, and based on GeneOntology analysis through UniProt. Less than one-third of the SSR-associated GSS sequences had significant hits in these databases, though were hits were recorded the derived annotations add a potentially useful data type to the marker metadata. Not surprisingly, chickpea GSS sequences (from which the SSRs were derived) had higher similarity to ESTs from other legume species, and overall higher similarity to dicot outgroups (i.e., poplar and Arabidopsis) than to monocot (i.e., rice) data sets.
Comprehensive genetic map of chickpea
An inter-specific mapping population derived from ICC 4958 (C. arietinum) and PI 489777 (C. reticulatum) was used to incorporate novel microsatellite and gene based markers. This mapping population has been widely used in past by chickpea community in order to incorporate several hundred microsatellite markers (Winter et al. 2000) and gene-based markers (Pfaff and Kahl 2003). The diverse genetic background of the parents provides for higher rates of polymorphism not only at the genetic level but also at phenotypic levels such as resistance to Fusarium wilt (Winter et al. 2000) and Ascochyta blight (Rakshit et al. 2003), facilitating trait mapping. Therefore, this population is generally considered as the international reference mapping population.
The present genetic map of chickpea represents 521 marker loci, spanning 2,602 cM with an average inter-marker distance of 4.99 cM. The order of common marker loci defined in present map agrees with earlier reports from Winter et al. (2000). However, the current map differs considerably from that of Winter et al. (2000) in having eight linkage groups, in agreement with eight chromosomes, whereas the Winter et al. (2000) map was composed of 16 linkage groups. There are probably at least two factors that contribute to this condensation of linkage groups: first, the new markers identified in the present study act as bridge points between the Winter et al. linkage groups, and second, essentially all of the markers mapped in the current study behave a co-dominant genetic features, which adds considerable power to the genetic evaluation compared to a high fraction of dominant markers in earlier studies. Importantly, the comparative analyses to Medicago support a simple assignment of eight chickpea linkage groups to eight chromosomes.
Comparative mapping of chickpea and Medicago
Mappig of the gene-based markers from Medicago in the genetic map of chickpea showed not only a high level of macrosynteny but also revealed features of structural divergence between the two genomes. Six of the eight linkage groups display a one-to-one correspondence between the Medicago and chickpea, suggesting that these linkage groups reflect the genome of the common Galegoid clade legume ancestor. Medicago LG5 and LG6, and chickpea LG2 and LG4, appear to have a more complicated ancestry, consisting of a minimum of several chromosomal translocation events. Thus, Mt-LG 5 is essentially a composite of portions of LG2 and LG8 of chickpea. Several research groups have compared genome structure between Medicago and various crop legumes (see Zhu et al. 2005). Our current results extend the comparative network to include chickpea, by demonstrating broad conservation of genome macrostructure between chickpea and Medicago.
One goal of comparative genetic analyses is to transfer information from well-characterized reference species to less well-characterized crops with an eye toward crop improvement. Among the agronomic targets in chickpea is resistance to several economically important pathogens; candidate genes for disease resistance are the conserved family of NBS-LRR resistance gene homologs (RGH). Several phylogenetically distinct RGH classes have been placed on the genetic map of chickpea (Huettel et al. 2002), thus facilitating the comparative genome analysis between chickpea and Medicago. In particular, we have documented two cases of syntenic NBS-LRR clusters that contain co-phyletic genes in each species. Interestingly, Ca-LG2 is known to harbor active resistance genes against Fusarium wilt and Ascochyta blight. At present, no RGHs have been reported mapped close to these resistance phenotypes. Nevertheless, the facts that a single conserved gene (TC87369) maps to the top terminal region of both Mt-LG6 and Ca-LG2, and that both linkage groups are rich in NBS-LRR genes and/or active disease resistance genes (Sharma et al. 2004; Zhu et al. 2002), may suggest shared ancestry of Mt-LG6 and Ca-LG2, though such speculation needs to be verified by more detailed study of the respective genome regions.
Similar observations of NBS-LRR synteny have been made for resistance gene homologs within the Solanaceae (Grube et al. 2000) and between Medicago and pea (Pisum sativum) (Zhu et al. 2002). However, the limited numbers of comparative molecular markers (gene-based SNPs) around these R gene regions in both Medicago and chickpea precludes precise statements regarding the relationship of these genome regions. Although the current analysis is based on a relatively small number of comparative markers, the potential of more detailed analyses to predict gene content and chromosomal structure in chickpea by reference to Medicago seems clear.
Conclusion
A set of 311 novel microsatellite markers were developed from microsatellite-enriched library in order to increase the genomic resources in chickpea. In total 147 potential SSR marker loci were found based on diversity pattern of SSR loci on a panel of 48 diverse chickpea genotypes. These markers should have utility for genetic analysis of a range of chickpea mapping populations and as anchor markers in comparative mapping to other legumes.
References
Akkaya MS, Bhagwat AA, Cregan PB (1992) Length polymorphism of simple sequence repeat DNA in soybean. Genetics 132:1131–1139
Ameline-Torregrosa C, Wang BB, O’Bleness MS, Deshpande S, Zhu H, Roe B, Young ND, Cannon SB (2008) Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol 146:5–21
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Rep 9:208–219
Buhariwalla HK, Jayashree B, Eshwar K, Crouch JH (2005) Development of ESTs from chickpea roots and their use in diversity analysis of the Cicer genus. BMC Plant Biol 5:16
Cannon SB, Sterck L, Rombauts S, Sato S, Wang X, Mudge J, Vasdewani J, Cheung F, Gouzy JP, Schiex T, Spanngl M, Schoof H, Nicholson C, Humphays S, Mayer K, Rogers J, Quetier F, Oldroyd GED, Debelle F, Cook DR, Town CD, Roe BA, Tabata S, Van de Peer Y, Young ND (2006) Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc Natl Acad Sci USA 103:14959–14964
Choi HK, Kim D, Uhm T, Limpens E, Lim H, Mun JH, Kalo P, Penmetsa RV, Seres A, Kulikova O, Roe BA, Bisseling T, Kiss GB, Cook DR (2004a) A sequence-based genetic map of Medicago truncatula and comparison of marker colinearity with M. sativa. Genetics 166:1463–1502
Choi HK, Mun JH, Kim DJ, Zhu H, Baek JM, Mudge J, Roe B, Ellis N, Doyle J, Kiss GB, Young ND, Cook DR (2004b) Estimating genome conservation between crop and model legume species. Proc Natl Acad Sci USA 101:15289–15294
Choi HK, Luckow MA, Doyle J, Cook DR (2006) Development of nuclear gene-derived molecular markers linked to legume genetic maps. Mol Genet Genomics 276:56–70
Choudhary S, Sethy NK, Shokeen B, Bhatia S (2006) Development of sequence-tagged microsatellite site markers for chickpea (Cicer arietinum L.). Mol Ecol Notes 6:3–95
Cregan PB, Bhagwat AA, Akkaya MS, Rongwen J (1994) Microsatellite fingerprinting and mapping of soybean. Meth Mol Cell Biol 5:49–61
Cuc LM, Mace E, Crouch J, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8:55
Douglas SE (2008) DNA strider: an inexpensive sequence analysis package for the Macintosh. Mol Biotechnol 3:37–45
Grube RC, Radwanski ER, Jahn M (2000) Comparative genetics of disease resistance within the Solanaceae. Genetics 155:873–887
Gupta PK, Varshney RK (2000) The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
Huettel B, Santra D, Muehlbauer J, Kahl G (2002) Resistance gene analogues of chickpea (Cicer arietinum L.): isolation, genetic mapping and association with a Fusarium resistance gene cluster. Theor Appl Genet 105:479–490
Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39:285–312
Hüttel B, Winter P, Weising K, Choumane W, Weigand F (1999) Seguence-tagged microsatellite site markers for chickpea (Cicer arietinum L.). Genome 42:210–217
Jayashree B, Punna R, Prasad P, Bantte K, Hash CT, Chandra S, Hoisington DA, Varshney RK (2006) A database of simple sequence repeats from cereal and legume expressed sequence tags mined in silico: survey and evaluation. In Silico Biol 6:0054
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen 12:172–175
Kulikova O, Gualtieri G, Geurts R, Kim DJ, Cook D, Huguet T, de Jong JH, Fransz PF, Bisseling T (2001) Integration of the FISH pachytene and genetic maps of Medicago truncatula. Plant J 27:49–58
Ladizinsky G, Adler A (1976) The origin of chickpea (Cicer arietinum L.). Euphytica 25:211–217
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Lichtenzveig J, Scheuring C, Dodge J, Abbo S, Zhang HB (2005) Construction of BAC and BIBAC libraries and their applications for generation of SSR markers for genome analysis of chickpea, Cicer arietinum L. Theor Appl Genet 110:492–510
Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Sobral BW, Young ND (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. Plant J 20:317–332
Mun JH, Kim DJ, Choi HK, Gish J, Debellé F, Mudge J, Denny R, Endré G, Dénarié J, Kiss GB, Roe B, Young ND, Cook DR (2006) Distribution of microsatellites in the genome of Medicago truncatula: a resource of genetic markers that integrate genetic and physical maps. Genetics 172:2541–2555
Pfaff T, Kahl G (2003) Mapping of gene-specific markers on the genetic map of chickpea (Cicer arietinum L.). Mol Genet Genomics 269:243–251
Rakshit S, Winter P, Tekeoglu M, Juarez Muñoz J, Pfaff T, Benko-Iseppon AM, Muehlbauer FJ, Kahl G (2003) DAF marker tightly linked to a major locus for Ascochyta blight resistance in chickpea (Cicer arietinum L.). Euphytica 132:23–30
Sambrook J, Russell DR (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sethy NK, Shokeen B, Bhatia S (2003) Isolation and characterization of sequence-tagged microsatellite sites markers in chickpea (Cicer arietinum L.). Mol Ecol Notes 3:428–430
Sethy NK, Choudhary S, Shokeen B, Bhatia S (2006a) Identification of microsatellite markers from Cicer reticulatum: molecular variation and phylogenetic analysis. Theor Appl Genet 112:347–357
Sethy NK, Shokeen B, Edwards KJ, Bhatia S (2006b) Development of microsatellite markers and analysis of intraspecific genetic variability in chickpea (Cicer arietinum L.). Theor Appl Genet 112:1416–1428
Sharma KD, Winter P, Kahl G, Muehlbauer FJ (2004) Molecular mapping of Fusarium oxysporum f. sp. ciceris race 3 resistance gene in chickpea. Theor Appl Genet 108:1243–1248
Singh R, Sharma P, Varshney RK, Sharma SK, Singh NK (2008) Chickpea improvement: role of wild species and genetic markers. Biotechnol Genet Eng Rev 25:267–314
Tekeoglu M, Tullu A, Kaiser WJ, Muehlbauer FJ (2000) Inheritance and linkage of two genes that confer resistance to fusarium wilt in chickpea. Crop Sci 40:1247–1251
Temnykh S, Declerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Varshney RK, Thiel T, Stein N, Langridge P, Graner A (2002) In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell Mol Biol Lett 7:537–546
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Varshney RK, Hoisington DA, Tyagi AK (2006) Advances in cereal genomics and applications in crop breeding. Trends Biotechnol 24:490–499
Varshney RK, Mahender T, Aggrawal RK, Börner A (2007) Genic molecular markers in plants: development and applications. In: Varshney RK, Tuberosa R (eds) Genomic assisted crop improvement: genomics approaches and platforms. Springer, The Netherlands, pp 13–30
Varshney RK, Close TJ, Singh NK, Hoisington DA, Cook DR (2009a) Orphan legume crops enter the genomics era!. Curr Opin Plant Biol 12:202–210
Varshney RK, Hoisington DA, Nayak SN, Graner A (2009b) Molecular plant breeding: methodology and achievements. In: Somers D, Langridge P, Gustafson PJ (eds) Plant genomics: methods and protocols. The Humana Press, Totowa, pp 283–304
Winter P, Pfaff T, Udupa SM, Hüttel B, Sharma PC, Sahi S, Arrequin-Espinoza R, Weigand F, Muehlbauer FJ, Kahl G (1999) Characterization and mapping of sequence-tagged microsatellite sites in the chickpea (C. arietinum L.). Mol Gen Genet 262:90–101
Winter P, Benko-Iseppon AM, Hüttel B, Ratnaparkhe M, Tullu A, Sonnante G, Pfaff T, Tekeoglu M, Santra D, Sant VJ, Rajesh PN, Kahl G, Muehlbauer FJ (2000) A linkage map of the chickpea (Cicer arietinum L.) genome based on the recombinant inbred lines from a C. arietinum × C. reticulatum cross: localization of resistance genes for Fusarium races 4 and 5. Theor Appl Genet 101:1155–1163
Zhu H, Cannon SB, Young ND, Cook DR (2002) Phylogeny and genomic organization of the TIR and non-TIR NBS-LRR resistance gene family in Medicago truncatula. Mol Plant Microbe Interact 15:529–539
Zhu H, Choi HK, Cook DR, Shoemaker RC (2005) Bridging model and crop legumes through comparative genomics. Plant Physiol 137:1189–1196
Acknowledgments
Thanks are due to Prathima Juvvadi, Gudipati Srivani and Abdul Gafoor for their technical assistance. Financial support from Tropical Legume I of Generation Challenge Program (GCP, http://www.generationcp.org) of CGIAR and National Fund of Indian Council of Agricultural Research of Government of India are gratefully acknowledged. The financial support by National Science Foundation Grant 0110206 to DRC is acknowledged. Council of Scientific and Industrial Research (CSIR), Government of India is acknowledged for providing fellowship to SNN and the Indian Council of Research NATP-HRD program for providing fellowship to SD. We thank Dr. Fred Muehlbauer of the USDA-ARS for providing the chickpea recombinant inbred population and genetic marker data.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. T. Nguyen.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nayak, S.N., Zhu, H., Varghese, N. et al. Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120, 1415–1441 (2010). https://doi.org/10.1007/s00122-010-1265-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1265-1