
Abstract
Aims/hypothesis
The list of validated type 2 diabetes susceptibility variants has recently been expanded from three to 19. The variants identified are common and have low penetrance in the general population. The aim of the study is to investigate the combined effect of the 19 variants by applying receiver operating characteristics (ROC) to demonstrate the discriminatory value between glucose-tolerant individuals and type 2 diabetes patients in a cross-sectional population of Danes.
Methods
The 19 variants were genotyped in three study populations: the population-based Inter99 study; the ADDITION study; and additional type 2 diabetic patients and glucose-tolerant individuals. The case–control studies involved 4,093 type 2 diabetic patients and 5,302 glucose-tolerant individuals.
Results
Single-variant analyses demonstrated allelic odds ratios ranging from 1.04 (95% CI 0.98–1.11) to 1.33 (95% CI 1.22–1.45). When combining the 19 variants, subgroups with extreme risk profiles showed a threefold difference in the risk of type 2 diabetes (lower 10% carriers with ≤15 risk alleles vs upper 10% carriers with ≥22 risk alleles, OR 2.93 (95% CI 2.38–3.62, p = 1.6 × 10−25). We calculated the area under a ROC curve to estimate the discrimination rate between glucose-tolerant individuals and type 2 diabetes patients based on the 19 variants. We found an area under the ROC curve of 0.60. Two-way gene–gene interaction showed few nominal interaction effects.
Conclusions/interpretation
Combined analysis of the 19 validated variants enables detection of subgroups at substantially increased risk of type 2 diabetes; however, the discrimination between glucose-tolerant and type 2 diabetes individuals is still too inaccurate to achieve clinical value.
Similar content being viewed by others

Introduction
Type 2 diabetes is a rapidly growing public health problem and although environmental factors are of major importance, genetic risk factors also predispose to the disease. Until recently, only three gene loci, PPARG [1], KCNJ11 [2, 3] and TCF7L2 [4], had convincingly shown replicated association with type 2 diabetes. However, within the last 2 years results of genome-wide association studies (GWA) have revolutionised the field of research and identified 14 new susceptibility type 2 diabetes variants [5–15]. Together with WFS1 [16] and TCF2 (also known as HNF1B) [17, 18], identified by a candidate gene approach, the total number of validated type 2 diabetes susceptibility loci now reaches 19. All identified alleles associated with type 2 diabetes risk are common (minor allele frequency [MAF] > 5%) and have a low penetrance (OR < 1.5) in the general population. Little is known about the molecular mechanisms by which these variants increase diabetes risk; however, physiological studies have demonstrated that the majority may mediate their pathogenic effect through an abnormal beta cell function, which seems to be the case for CDKAL1, SLC30A8, HHEX, CDKN2A, IGF2BP2, TCF7L2, KCNJ11, WFS1, CDC123, JAZF1, MTNR1B and TSPAN8 [3, 8, 14, 19–23]. As for the remaining susceptibility variants, a predisposing effect through obesity affecting an increase in fat accumulation has been demonstrated for variation in FTO [24, 25] and variation in PPARG has shown a potentially pathogenic effect on type 2 diabetes through an impairment of insulin sensitivity [26].
An important question is, to what extent do the combined effect of these variants predict which individuals are at risk of developing type 2 diabetes. Indeed, if the discrimination is successful, the prospect of prediction and application of genotype-based early and individualised prevention and treatment strategies for type 2 diabetes would be of major clinical importance.
The issue of combining the known available type 2 diabetes susceptibility variants has been addressed before in case–control settings. Before the release of GWA studies, Weedon et al. [27] demonstrated that the combined effect of three common genetic variants only moderately enabled discrimination between type 2 diabetes patients and glucose-tolerant individuals (AUC of the receiver operating characteristics [ROC] 0.58). With the recent release of GWA studies and thus the expansion of the number of validated type 2 diabetes susceptibility variants, three studies have investigated the combined effect of 17 independent loci on type 2 diabetes risk and although subgroups of carriers of several risk alleles were considered to be at high risk of developing type 2 diabetes, the overall conclusion pointed towards a low discriminative ability between cases and controls assessed by the AUC of an ROC curve [28–30]. Also, two prospective studies estimated the predictive value of 16 and 18 type 2 diabetes susceptibility variants, respectively. The studies demonstrated that even though the discriminatory power of genetic testing is limited, it increases with duration of follow up [31, 32], suggesting that even genetic risk factors with moderate effects may, on a life-long basis, contribute considerably to diabetes risk.
Recently, two new type 2 diabetes candidate genes were discovered. A Japanese study reported the result of a genome-wide scan of 268,068 single-nucleotide polymorphisms (SNPs) and identified KCNQ1 as a type 2 diabetes susceptibility gene [11, 12]. The rs2237895 variant—located in the intronic region of KCNQ1—showed an OR 1.23 (95% CI 1.18–1.29, p < 1 × 10−16). Additionally, three recent papers reported that variants in the MTNR1B locus strongly associate with type 2 diabetes risk and a meta-analysis of MTNR1B rs10830963 demonstrated OR 1.09 (95% CI 1.05–1.12, p = 3.3×10−7) [13–15].
Here we present an updated study evaluating the combined effect of 19 validated type 2 diabetes susceptibility variants; it includes the newly discovered KCNQ1 and MTNR1B variants that in the Danish population showed to be the fifth and second strongest type 2 diabetes-associated variants, respectively. By applying ROC curves in a large sample of Danes we demonstrate how well the 19 variants discriminate between glucose-tolerant individuals and type 2 diabetes patients alone and in combination with known type 2 diabetes risk factors such as BMI, age and sex. In addition, we investigate whether the combined effect from the variants is explained additively or whether a synergistic effect on diabetes risk (two-way gene–gene interaction) exists between the variants.
Methods
Study population
The 19 type 2 diabetes susceptibility variants were genotyped in 9,395 Danes involving: (1) the population-based Inter99 sample (ClinicalTrials.gov NCT00289237) of middle-aged individuals sampled at the Research Centre for Prevention and Health (n = 4,928) [33]; (2) type 2 diabetes patients and glucose-tolerant individuals sampled through the outpatient clinic at Steno Diabetes Center (n = 2,107 and n = 734, respectively); and (3) screen-detected type 2 diabetes patients from the Danish ADDITION screening cohort (ClinicalTrials.gov NCT00237549) sampled through Department of General Practice at University of Aarhus (n = 1,626) [34]. Study group 1 and glucose-tolerant individuals from study group 2 underwent a standard 75 g oral glucose tolerance test. Informed written consent was obtained from all participants before participation. The study was approved by the Ethical Committee of Copenhagen and Aarhus Counties and was in accordance with the principles of the Helsinki Declaration. Glucose-tolerant individuals and type 2 diabetes patients were defined according to World Health Organization 1999 criteria [35]. Individuals with type 2 diabetes had increased BMI and age (mean BMI 30.6 ± 5.5 kg/m2, mean age 60 ± 10 years) compared with glucose-tolerant individuals (mean BMI 25.6 ± 4.0 kg/m2, mean age 47 ± 9 years).
Statistical analysis
For each variant we estimated the multiplicative effect on type 2 diabetes risk as well as the two-way gene–gene interaction by applying logistic regression with adjustment for sex and age. The two-way interaction was performed by comparing one model including only the main effect (SNP) with an alternative model including a SNP–SNP interaction variable in addition to the main effect. The covariate for each SNP was denoted as the number of disease alleles (i.e. coded as 0, 1 or 2 according to the number of risk alleles) and the pair-wise interaction as the product of the pairs of SNPs (i.e. multiplicative interaction).
When estimating the combined effect of the 19 susceptibility SNPs, each risk allele was defined as the allele associated with increased risk of type 2 diabetes in previous studies [5–18], hence each individual is assigned a risk score ranging from zero to 38. For each risk score the numbers of glucose-tolerant individuals and type 2 diabetic patients were calculated. Fisher’s exact test was applied to test whether the distribution of glucose-tolerant individuals and type 2 diabetes patients was different for subgroups with multiple risk scores and few risk scores.
Receiver operating characteristics
We estimated the discriminatory power between glucose-tolerant individuals and type 2 diabetes patients of the 19 susceptibility variants by applying ROC. We used logistic regression including all variants coded as 0, 1 or 2 according to the number of risk alleles. In order to cross-validate the results, bootstrapping (n = 50) was applied. Cross-validation works by fitting the same model in different bootstraps (subsets of the original data). The remaining subsets (out-of-bag data) are used for the selectivity of the fitted model. For each ROC, an area under the curve is used as a measure of the predictive power of the method. Each ROC curve in the present paper consists of the result of all bootstraps, the mean ROC estimated from the bootstrap samples by taking the mean of the bootstrap sample at each 1–selectivity point and the apparent ROC curve which are estimated from the entire dataset. All analyses were performed using RGui version 2.7.0, applying the per package (http://www.r-project.org, accessed 1 May 2008).
Results
The clinical characteristics of all participants in the three study samples are reported in Electronic supplementary material (ESM) Table 1. We estimated the association of each of the 19 validated variants on type 2 diabetes risk by applying a multiplicative genetic model. Results of the risk of susceptibility variants adjusted for sex, age and BMI can be found in ESM Fig. 1. However, here we focus on results adjusted for sex and age as BMI is an effect modifier compared with sex and age, which are considered confounders (Fig. 1).

ORs and 95% CIs of the 19 validated variants for type 2 diabetes using a genetic multiplicative model adjusted for sex and age. The risk alleles of the 19 variants are defined as the alleles associated with type 2 diabetes in accordance with the literature [5–18]. The 19 variants may exert their diabetogenic effects through: beta cell dysfunction (solid line); altered BMI (dotted line); changes in insulin sensitivity (dashed/dotted line); and unknown mechanisms (dashed line)
The minor alleles of CDKN2A, THADA, JAZF1, HHEX and SLC30A8 variants were associated with a decreased risk of developing type 2 diabetes, whereas the minor alleles of TCF7L2, CDKAL1, KCNQ1, FTO, KCNJ11, TSPAN8, CDC123, MTNR1B, and IGF2BP2 variants were associated with an increased risk of type 2 diabetes. As for the remaining loci, no association was observed although the directions were consistent with previous reports. The OR for each individual variant ranged from 1.04 (95% CI 0.98–1.11; NOTCH2; i.e. no association) to 1.33 (95% CI 1.22–1.45) for TCF7L2, so far the largest risk effect of all common type 2 diabetes loci. As for the result of the single-variant analysis all data have been published previously [8, 12, 13, 21–23, 36–38].
The combined effect of the 19 variants was estimated by calculating the percentage of glucose-tolerant individuals and type 2 diabetic patients stratified according to the number of risk alleles (Fig. 2). We demonstrate that, on average, type 2 diabetic patients carry more risk alleles and thus the curve is shifted right compared with the curve of glucose-tolerant individuals (Fig. 2). By stratifying the number of risk alleles into quartiles we estimated OR 2.13 (95 % CI 1.85–2.47, p = 4.91 × 10−26) between the lowest quartile (number of risk alleles ≤16, n = 1,404) and the uppermost quartile (number of risk alleles ≥20, n = 1,927). In addition, if we stratify the individuals into subgroups of extreme risk allele profiles by comparing the lower 10% (carriers of eight to 15 risk alleles) with the upper 10% (carriers of 22 to 29 risk alleles) we estimate OR 2.93 (95% CI 2.38–3.62, \( p = 1.6 \times 10^{{ - 25}} \)).

The percentage of glucose-tolerant individuals (white bars) and type 2 diabetes patients (grey bars) stratified according to number of risk alleles of the 19 validated type 2 diabetes variants. The OR is calculated using Fisher’s exact test as number of glucose-tolerant individuals and type 2 diabetes patients carrying ≤16 risk alleles (the lowest quartile) vs the number of glucose-tolerant individuals and type 2 diabetes patients carrying ≥20 risk alleles (uppermost quartile). †OR 2.13, p = 4.0 × 10−26
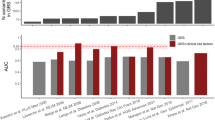
The discriminatory value of a genetic test based on the 19 type 2 diabetes susceptibility variants was calculated as the area under the ROC, which is a graphical plot of the sensitivity vs (1 − specificity). The area under the ROC was estimated to 0.60 (Fig. 3a). We also tested whether the 19 susceptibility variants added to the discriminatory power when accounting for known type 2 diabetes risk factors such as BMI, age and sex (Fig. 3b). A model that includes only BMI, age and sex has an AUC of 0.92 and after applying the 19 susceptibility variants an AUC of 0.93 was achieved (Fig. 3c).

ROC curves for the discrimination between glucose-tolerant individuals and type 2 diabetes patients. The grey shadow represents ROC curves obtained in the process of bootstrap cross-validation (n = 50). The mean of the bootstrapping ROC curves is presented as a thin solid line. The dotted line represents the apparent ROC curve estimated from the original data. a The mean AUC of the bootstrap samples (thick solid line) based on the 19 type 2 diabetes susceptibility variants (AUC 0.60). b The mean AUC of the bootstrap samples when including conventional type 2 diabetes risk factors (BMI, age and sex) (AUC 0.92). c The mean AUC of the bootstrap when including the 19 susceptibility variants and clinical type 2 diabetes risk factors (BMI, age and sex) (AUC 0.93)
Finally we estimated the two-way interaction between each combination of the 19 variants (171 combinations) (ESM Table 1) and the result demonstrated few, probably spurious, associations (p < 0.05). As none of the associations was significant after Bonferroni correction we believe that an additive model between each variant is acceptable. Additionally, we calculated the AUC under an ROC curve in which a model including all variants (additive) is compared with a model including a two-way interaction term in addition to the variants (interaction). The results showed that if interaction is included an AUC of 0.56 is reached, which indicates reduced discriminatory value (ESM Fig. 2).
Discussion
In our analyses, in which we applied ROC to demonstrate the discriminatory value between 5,302 glucose-tolerant individuals and 4,093 type 2 diabetes patients of the combined effect of 19 validated type 2 diabetes susceptibility variants, we were able to identify subgroups with substantial increases in risk of type 2 diabetes. For instance, in a subgroup of individuals carrying more than 22 risk alleles we estimated an odds ratio of 2.93 (95% CI 2.38–3.62, p = 1.6 × 10−25) compared with individuals carrying fewer than 15 risk alleles. However, when evaluating the general ability to discriminate between glucose-tolerant individuals and type 2 diabetes patients, assessed by the area under an ROC curve, we estimated an AUC of 0.60. The ROC analyses in the same study samples of the corresponding discriminatory value of conventional risk factors such as BMI, age and sex resulted in an AUC of 0.92 and when the 19 susceptibility variants were included an AUC of 0.93. Thus, although tremendous progress in finding type 2 diabetes susceptibility genes has recently taken place, the discriminatory value of the common genetic variations is still too limited to be of clinical importance. For illustration, if we screen a sample of individuals for the 19 type 2 diabetes susceptibility genes and we want to detect 80% of the type 2 diabetic patients, we have to account for the fact that 70% of the healthy individuals are misclassified as type 2 diabetic patients (Fig. 3a).
Our results are in line with the conclusions from recent cross-sectional and longitudinal studies assessing the combined impact of several risk alleles on type 2 diabetes risk [27–32]. In the recent prospective study by Lyssenko et al., it was demonstrated that the addition of 16 type 2 diabetes susceptibility variants to clinical risk factors improved the prediction of future type 2 diabetes assessed by the area under an ROC curve from 0.74 to 0.75 [31].
A major limitation in the current and previous comparable cross-sectional studies is the fact that case–control designs are used that comprise approximately equal numbers of glucose-tolerant individuals and type 2 diabetic patients. As a result, the discriminatory value is overestimated as the prevalence rate at the population level constitutes approximately 6% compared with a case–control design which often includes equal numbers of cases and controls. Another limitation when calculating the OR between carriers of multiple risk alleles and few risk alleles is the fact that each risk allele is assigned the same effect. This limitation, however, is overcome when calculating ROC curves where each SNP is assigned an individual effect size. Also, when we run the ROC analyses with age included we introduce an overestimation of the discriminatory value as the majority of the type 2 diabetic patients in most studies are older than the glucose-tolerant individuals.
Gene–gene interaction, the fact that one gene variant masks or enhances the effect of another variant significantly affecting a disease association, has been discussed as one of the promising tools to explain the variation in type 2 diabetes risk. A recent study suggested a significant interaction between variants in IGF2BP2 and LOC38776, SLC30A8 and HHEX on risk of type 2 diabetes [28]; these variants are all known to mediate the pathogenic effect through an abnormal beta cell function. Indeed we also attempted to replicate such findings by estimating the two-way interaction between the 19 variants. The result demonstrated sporadic nominally significant interactions which are most likely to be due to type 1 errors and are not consistent with any of the previous findings [28]. A possible explanation may be a lack of statistical power, as the 19 susceptibility variants investigated here confer a modestly increased risk of type 2 diabetes. Based on 95% confidence interval estimates of the effect size, we can in the current study exclude a gene–gene interaction OR above 2.6 between two variants on type 2 diabetes risk (data not shown). The suggestion that only additivity between the examined type 2 diabetes variants appears to exist is also emphasised in ESM Fig. 2 where the discrimination value is reduced when including possible two-way genetic interaction terms. The results seem to be in line with previous studies [29, 30].
In the present paper we have investigated common variants with a low penetrance in the general population and demonstrated limited success in the discrimination of glucose-tolerant individuals and type 2 diabetes patients based on the genetic profile. Janssens et al. [39] investigated the usefulness of genomic profiling in the general population by simulating a population of 1 million individuals carrying 40 genotypes under different scenarios. The study demonstrated that common variants with low penetrance have little predictive power as we also show in the present paper. In contrast, it has been proposed that accumulation of rare variants with a mildly deleterious effect may substantially increase the relative risk at the individual level [40]. Indeed, with the next generation of sequencing technologies enabling gene-specific re-sequencing of the entire human genome, rare variants may be identified that in combination may contribute substantially to the risk of type 2 diabetes. Such results together with the known common susceptibility variants may increase the discriminative value of genetic risk factors and push the limit towards a threshold acceptable for clinical utility.
Abbreviations
- GWA:
-
Genome-wide association
- MAF:
-
Minor allele frequency
- ROC:
-
Receiver operating characteristic
- SNP:
-
Single-nucleotide polymorphism
References
Altshuler D, Hirschhorn J, Klannemark M et al (2000) The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26:76–80
Gloyn A, Weedon M, Owen K et al (2003) Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52:568–572
Nielsen E, Hansen L, Carstensen B et al (2003) The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes 52:573–577
Grant S, Thorleifsson G, Reynisdottir I et al (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 38:320–323
Saxena R, Voight B, Lyssenko V et al (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Scott L, Mohlke K, Bonnycastle L et al (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345
Sladek R, Rocheleau G, Rung J et al (2007) A genomewide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885
Steinthorsdottir V, Thorleifsson G, Reynisdottir I et al (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39:770–775
Zeggini E, Scott L, Saxena R et al (2008) Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40:638–645
Zeggini E, Weedon M, Lindgren C et al (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316:1336–1341
Yasuda K, Miyake K, Horikawa Y et al (2008) Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 40:1092–1097
Unoki H, Takahashi A, Kawaguchi T et al (2008) SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 40:1098–1102
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proença C et al (2008) A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 41:89–94
Lyssenko V, Nagorny CL, Erdos MR et al (2008) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet 41:82–88
Prokopenko I, Langenberg C, Florez JC et al (2008) Variants in MTNR1B influence fasting glucose levels. Nat Genet 41:77–81
Sandhu M, Weedon M, Fawcett K et al (2007) Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 39:951–953
Gudmundsson J, Sulem P, Steinthorsdottir V et al (2007) Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet 39:977–983
Winckler W, Weedon M, Graham R (2007) Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes 56:685–693
Florez J, Jablonski K, Bayley N et al (2006) TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med 355:241–250
Florez J, Jablonski K, McAteer J et al (2008) Testing of diabetes-associated WFS1 polymorphisms in the Diabetes Prevention Program. Diabetologia 51:451–457
Grarup N, Rose C, Andersson E et al (2007) Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10, 705 Danish subjects: validation and extension of genome-wide association studies. Diabetes 56:3105–3111
Sparsø T, Andersen G, Albrechtsen A et al (2008) Impact of polymorphisms in WFS1 on prediabetic phenotypes in a population-based sample of middleaged people with normal and abnormal glucose regulation. Diabetologia 51:1646–1652
Grarup N, Andersen G, Krarup NT et al (2008) Association testing of novel type 2 diabetes risk alleles in the JAZF1, CDC123/CAMK1D, TSPAN8, THADA, ADAMTS9, and NOTCH2 loci with insulin release, insulin sensitivity, and obesity in a population-based sample of 4, 516 glucose-tolerant middle-aged Danes. Diabetes 57:2534–2540
Frayling T, Timpson N, Weedon M et al (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894
Dina C, Meyre D, Gallina S et al (2007) Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 39:724–726
Ek J, Andersen G, Urhammer SA et al (2001) Studies of the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-gamma2 (PPAR-gamma2) gene in relation to insulin sensitivity among glucose tolerant caucasians. Diabetologia 44:1170–1176
Weedon M, McCarthy M, Hitman G (2006) Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLoS Med 3:e374
Cauchi S, Meyre D, Durand E et al (2008) Post genome-wide association studies of novel genes associated with type 2 diabetes show gene-gene interaction and high predictive value. PLoS ONE 5:e2031
Lango H, Palmer C, Morris A et al (2008) Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes 57:3129–3135
van Hoek M, Dehgan A, Witteman JC et al (2008) Predicting type 2 diabetes based on polymorphisms from genome wide association studies: a population-based study. Diabetes 57:3122–3128
Lyssenko V, Jonsson A, Almgren P et al (2008) Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 20:2220–2232
Meigs JB, Shrader P, Sullivan LM et al (2008) Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med 20:2208–2219
Jørgensen T, Borch-Johnsen K, Thomsen T et al (2003) A randomized non-pharmacological intervention study for prevention of ischaemic heart disease: baseline results Inter99. Eur J Cardiovasc Prev Rehabil 10:377–386
Lauritzen T, Grin S, Borch-Johnsen K et al (2000) The ADDITION study: proposed trial of the cost-effectiveness of an intensive multifactorial intervention on morbidity and mortality among people with Type 2 diabetes detected by screening. Int J Obes Relat Metab Disord 24:6–11
World Health Organization Study Group (1999) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Technical Report Series WHO/NCD/NCS/99.2. WHO, Geneva
Andreasen CH, Stender-Petersen KL, Mogensen MS et al (2008) Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes 57:95–101
Helgason A, Pálsson S, Thorleifsson G et al (2007) Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet 39:218–225
Sparsø T, Bonnefond A, Andersson E et al (2009) The G-allele of intronic rs10830963 in MTNR1B confers increased risk of impaired fasting glycemia and type 2 diabetes through an impaired glucose-stimulated insulin release: studies involving 19,605 Europeans. Diabetes (in press)
Janssens AC, Moonesinghe R, Yang Q et al (2007) The impact of genotype frequencies on the clinical validity of genomic profiling for predicting common chronic diseases. Genet Med 9:528–535
Kryukov GV, Pennacchio LA, Sunyaev SR (2007) Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet 80:727–739
Acknowledgements
This study was supported by the Danish Medical Research Council, the Danish Diabetes Association, the Gerda and Aage Haensch Foundation, the A. P. Møller Foundation for the Advancement of Medical Science, University of Copenhagen and Novo Nordisk. This study also received support from The FOOD Study Group/the Danish Ministry of Food. The authors wish to thank A. Forman, I.-L. Wantzin, T. Lorentzen and M. Stendal for technical assistance, G. Lademann for secretarial support, A. L. Nielsen for database management and M. M. H. Kristensen for grant management.
Duality of interest
K. Borch-Johnsen, T. Hansen and O. Pedersen hold employee shares in Novo Nordisk and have received lecture fees from pharmaceutical companies. All other authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
ORs and 95% CIs of the 19 validated variants on type 2 diabetes using a genetic multiplicative model adjusted for sex, age and BMI. The information given in the box indicates how the 19 variants may exert their diabetogenic effects (PDF 92 kb)
ESM Fig. 2
a p values for the pairwise interaction of the 19 susceptibility variants on type 2 diabetes risk. Below the white diagonal a full model for each SNP has been applied (4 df). Above the white diagonal each variant is coded additively (1 df). b ROC curves for the discrimination of glucose-tolerant individuals and type 2 diabetes patients when two-way interaction is included in the model. The grey shadow represents ROC curves obtained in the process of bootstrap cross-validation (n = 50). The mean of the bootstrapping ROC curves is presented as a solid line (AUC 0.56). For comparison, the additive model is presented as a dotted line (AUC 0.60) (PDF 73 kb)
ESM Table 1
Clinical characteristics of study populations (PDF 51 kb)
Rights and permissions
About this article
Cite this article
Sparsø, T., Grarup, N., Andreasen, C. et al. Combined analysis of 19 common validated type 2 diabetes susceptibility gene variants shows moderate discriminative value and no evidence of gene–gene interaction. Diabetologia 52, 1308–1314 (2009). https://doi.org/10.1007/s00125-009-1362-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1362-3