
Abstract
Aims/hypothesis
The aim of this study was to quantify protein damage by glycation, oxidation and nitration in a rat model of diabetes at the sites of development of microvascular complications, including the effects of thiamine and benfotiamine therapy.
Methods
Diabetes was induced in male Sprague–Dawley rats by 55 mg/kg streptozotocin and moderated by insulin (2 U twice daily). Diabetic and control rats were given thiamine or benfotiamine (7 or 70 mg kg−1 day−1) over 24 weeks. Plasma, urine and tissues were collected and analysed for protein damage by stable isotopic dilution analysis MS.
Results
There were two- to fourfold increases in fructosyl-lysine and AGE content of glomerular, retinal, sciatic nerve and plasma protein in diabetes. Increases in AGEs were reversed by thiamine and benfotiamine therapy but increases in fructosyl-lysine were not. Methionine sulfoxide content of plasma protein and 3-nitrotyrosine content of sciatic nerve protein were increased in diabetes. Plasma glycation free adducts were increased up to twofold in diabetes; the increases were reversed by thiamine. Urinary excretion of glycation, oxidation and nitration free adducts was increased by seven- to 27-fold in diabetes. These increases were reversed by thiamine and benfotiamine therapy.
Conclusions/interpretation
AGEs, particularly arginine-derived hydroimidazolones, accumulate at sites of microvascular complication development and have markedly increased urinary excretion rates in experimental diabetes. Thiamine and benfotiamine supplementation prevented tissue accumulation and increased urinary excretion of protein glycation, oxidation and nitration adducts. Similar effects may contribute to the reversal of early-stage clinical diabetic nephropathy by thiamine.
Similar content being viewed by others

Introduction
The current global epidemic of diabetes is expected to lead to a later increased global burden of vascular complications of diabetes [1]. Vascular complications develop progressively after 5–40 years of diabetes, although they may appear earlier in type 2 diabetes associated with a preceding period of undiagnosed diabetes and impaired glucose tolerance. A therapeutic intervention to resist and reverse early-stage vascular complications beyond that achieved with current therapy is high-dose supplementation of thiamine. This counters multiple pathways of biochemical dysfunction linked to the development of vascular complications with doses of the vitamin markedly higher than the dietary reference intake [2]. High-dose therapy with thiamine and the thiamine monophosphate (TMP) derivative benfotiamine prevented the development of microvascular complications in experimental diabetes [3–5]. A pilot-scale trial of type 2 diabetic patients with incipient nephropathy (microalbuminuria) showed that high-dose thiamine therapy improved renal function and induced regression of microalbuminuria [6, 7]. Further understanding of how thiamine supplementation prevents incipient nephropathy and other microvascular complications is now required. Prevention of protein damage in renal glomeruli at the sites of vascular complication development may contribute to the therapeutic effect.
Proteins are damaged in physiological systems by glycation, oxidation and nitration. Early stage reactions form \( {N^\varepsilon } \)-fructosyl-lysine (FL) and other fructosamine derivatives. FL residues degrade slowly to form AGEs. Glyoxal, methylglyoxal and 3-deoxyglucosone (3-DG) are potent glycating agents formed by the degradation of glycolytic intermediates, glycated proteins and lipid peroxidation. They react rapidly with proteins to form AGEs, mainly on arginine residues. The most important AGEs quantitatively are arginine-derived hydroimidazolones formed by glyoxal, methylglyoxal and 3-DG: glyoxal-derived hydroimidazolone \( {N^\delta } \)-(5,5-dihydro-4-imidazolon-2-yl)-ornithine (G-H1), methylglyoxal-derived hydroimidazolone \( {N^\delta } \)-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine (MG-H1) and 3-DG-derived hydro-imidazolone \( {N^\delta } \)-(5-hydro-5-(2,3,4-trihydroxybutyl)-4-imidazolon-2-yl)-ornithine and related structural isomers (3DG-H), respectively. Other widely studied AGEs are \( {N^\varepsilon } \)-carboxymethyl-lysine (CML), \( {N^\varepsilon } \)-(1-carboxyethyl)lysine (CEL) and pentosidine. Oxidative damage to protein forms methionine sulfoxide (MetSO) by the oxidation of methionine and dityrosine by oxidative cross-linking of tyrosine. Nitration damage of proteins forms 3-nitrotyrosine (3-NT) (reviewed by Thornalley [8]; Fig. 1).

Protein glycation, oxidation and nitration adduct residues in physiological systems and biodistribution scheme illustrating flows of formation and removal of protein glycation, oxidation and nitration free adducts. a Glycation adduct residues. b Oxidation adduct residues. c Nitration adduct. Protein glycation, oxidation and nitration adduct residues are shown. For the corresponding free adducts at physiological pH, the N-terminal amino group is protonated –NH +3 and the C-terminal carbonyl is a carboxylate –CO −2 moiety. d Biodistribution scheme illustrating flows of formation and removal of protein glycation, oxidation and nitration free adducts
In this study, we quantified protein damage by glycation, oxidation and nitration adducts in tissues, plasma and urine in a streptozotocin (STZ)-induced model of diabetes in rats and healthy controls to establish the major adducts formed and if high-dose therapy with thiamine or benfotiamine decreases protein damage at sites of development of microvascular complications with similar or different effectiveness.
Methods
Animals
Male Sprague–Dawley rats, 250 g, were purchased from Charles River UK (Ramsgate, UK). They were kept two per cage at 21°C, 50–80% humidity and with a daily 14 h light cycle, and had free access to food and water. Diabetes was induced by injection i.v. with 55 mg/kg STZ. Body weight and moderate hyperglycaemia were stabilised by s.c. injection of 2 U Ultralente insulin (long-acting insulin zinc suspension) every 2 days. Thiamine and benfotiamine were given orally after induction of diabetes, mixed with the chow, at high dose (7 and 70 mg kg−1 day−1) over 24 weeks to STZ diabetic and healthy control rats. Metabolic control (fasting glucose and HbA1), decline in renal function (urinary albumin excretion) and GFR (creatinine clearance), body weight and food consumption were characterised [3, 9]. After 24 weeks, a 24 h urine collection was made and the rats were killed, blood samples collected and plasma prepared. Tissues were immediately excised and renal glomeruli isolated. All procedures were approved by the UK Home Office for work under the Animals (Scientific Procedures) Act 1986; project licence 80/1481.
Preparation of tissue extracts
Renal glomeruli were prepared by differential sieving of fresh renal cortex tissue (100–200 mesh; 0.075–0.150 mm pore fraction size sieve) in saline at 4°C [3]. Excised sciatic nerves were rinsed in PBS, cleaned of connective tissue and small vessels, cut into small pieces, homogenised in 2.0 ml 0.25 mol/l sucrose and centrifuged (900 g, 20 min, 4°C). The supernatant fraction was treated with an equal volume of 20% (wt/vol.) trichloroacetic acid (TCA) at 4°C, centrifuged (5 min, 4°C, 900 g), and the protein pellet washed twice with 2.0 ml 10% TCA at 4°C. The protein pellet was stirred with 1.0 ml acetone at −20°C for 20 min, centrifuged (900 g, 4°C, 5 min) and the lipid-containing supernatant fraction discarded. The protein precipitate was washed further with 10% TCA at 4°C and centrifuged. The pellet was dried under a stream of nitrogen gas and stored at −80°C. Retinas were extracted by the Winkler technique [10]. This and all other tissues were washed free of any blood, cut into small pieces (20–100 mg wet weight), homogenised in 10 mmol/l sodium phosphate buffer, pH 7.4 at 4°C, and membranes sedimented by centrifugation (20,000 g, 30 min, 4°C). All samples were stored at −80°C prior to analysis (<6 months).
Determination of protein glycation, oxidation and nitration adducts
These were determined by liquid chromatography with tandem MS detection (LC–MS/MS). Analytes determined were: FL, CML, CEL, G-H1, MG-H1, 3DG-H, pentosidine, MetSO, 3-NT and related amino acids. Glycation, oxidation and nitration adducts of tissue and plasma proteins were determined after exhaustive enzymatic hydrolysis by consecutive incubation with pepsin, pronase E and finally aminopeptidase with prolidase under nitrogen. Antioxidant and pH conditions in pre-analytical processing prevented artefactual formation or loss of analytes. Tissue and plasma protein extracts (0.5–5 mg protein) were washed by four cycles of dilution and concentration in water over a 10 kDa cut-off microspin filter and 100 µg digested for analysis. Glycation, oxidation and nitration free adducts were determined by direct analysis of ultrafiltrates (10 kDa cut-off) of plasma and urine [11]. Analytes were detected by electrospray positive ionisation multiple reaction monitoring LC–MS/MS and quantified by stable isotopic dilution analysis [12]. Data of MG-H1 and CEL content in renal glomeruli have been published previously [3].
Statistics
Data are means ± SD for parametric data and median (minimum–maximum) for non-parametric data, as judged by the Kolmogorov–Smirnov test. Significance of difference of means in parametric data was assessed by Student’s t test and significance of difference of medians in non-parametric data by the Mann–Whitney U test.
Results
Protein glycation, oxidation and nitration adduct residues in renal glomeruli, retina, sciatic nerve and plasma protein
Glycation adducts
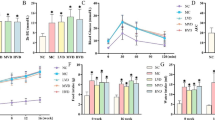
Early-glycation FL adduct content of protein was relatively low in renal glomeruli and retina (0.24 and 0.18 mmol/mol lysine), intermediate in sciatic nerve (0.43 mmol/mol lysine) and highest in plasma protein (1.61 mmol/mol lysine). In diabetic rats, FL content was increased two- to fourfold. Treatment with thiamine or benfotiamine did not prevent these increases. HbA1 was increased twofold in diabetic rats; there was a small, significant decrease of this in diabetic rats receiving 70 mg kg−1 day−1 thiamine but not receiving benfotiamine. CML content of renal glomeruli, retina, sciatic nerve and plasma protein of healthy control rats was relatively low, in the range 0.04–0.27 mmol/mol lysine. This was increased by up to twofold in diabetic rats. These increases were prevented in the retina by thiamine and benfotiamine, by 70 mg/kg thiamine and benfotiamine in sciatic nerve, by 7 mg/kg benfotiamine in renal glomeruli and 70 mg/kg thiamine only in plasma protein. CEL content of renal glomeruli, retina, sciatic nerve and plasma protein of healthy control rats was relatively low at 0.04–0.35 mmol/mol lysine. This was increased by up to twofold in diabetic rats except in the retina. Increases were reversed by thiamine and benfotiamine in renal glomeruli and sciatic nerve and by thiamine only in plasma protein. G-H1 content of healthy control rats was relatively low in renal glomeruli and plasma protein (0.09–0.25 mmol/mol arginine) and higher in retina and sciatic nerve (0.41–0.49 mmol/mol arginine). This was increased two- to threefold in the retina, sciatic nerve and plasma protein of diabetic rats, where increases were reversed by thiamine and benfotiamine. Unexpectedly, G-H1 content was increased by benfotiamine in renal glomeruli of diabetic rats. MG-H1 content of renal glomeruli, retina, sciatic nerve and plasma protein of healthy control rats was 1.29–2.99 mmol/mol arginine. This was increased two- to threefold in tissues and 53% in plasma protein of diabetic rats, where increases were reversed by thiamine and benfotiamine. 3DG-H content of healthy control rats was low in the retina (0.34 mmol/mol arginine), intermediate in plasma protein (0.90 mmol/mol arginine) and high in sciatic nerve and renal glomeruli (2.70–3.18 mmol/mol arginine). In diabetic rats, 3DG-H content was increased 49% in renal glomeruli, two- to threefold in retina and sciatic nerve and unchanged in plasma protein. Increases in renal glomeruli and sciatic nerve were reversed by thiamine and benfotiamine whereas the increase in the retina was reversed by thiamine treatment only (Tables 1, 2, 3 and 4 and Fig. 2). In healthy control rats, pentosidine content was extremely low in renal glomeruli, retina and plasma protein (0.001–0.002 mmol/mol lysine) and higher in sciatic nerve (0.026 mmol/mol lysine). In diabetic rats, pentosidine content was increased fourfold in renal glomeruli and 50% in sciatic nerve and not increased in the retina or plasma protein. Treatment with thiamine reversed the increase in renal glomeruli but not in sciatic nerve (Electronic supplementary material [ESM] Table 1).

Plasma glycation adducts residues in STZ-induced diabetic rats and normal control and effect of thiamine and benfotiamine therapy. a FL. b CML. c CEL. d G-H1. e MG-H1. f 3DG-H. Data are means ± SD. Rat study groups: C, healthy controls (n = 13); CT70, controls +70 mg kg−1 day−1 thiamine (n = 6); CB70, controls +70 mg kg−1 day−1 benfotiamine (n = 8); D, diabetic controls (n = 11); DT7, diabetic +7 mg kg−1 day−1 thiamine (n = 7); DT70, diabetic +70 mg kg−1 day−1 thiamine (n = 7); DB7, diabetic +7 mg kg−1 day−1 benfotiamine (n = 9); and DB70, diabetic +70 mg kg−1 day−1 benfotiamine (n = 8). *p < 0.05, **p < 0.01 and ***p < 0.001 with respect to normal control. † p < 0.05, †† p < 0.01 ††† p < 0.001 with respect to diabetic controls
Oxidation adducts
For oxidative markers, MetSO content of healthy control rats was relatively low in renal glomeruli protein (0.43 mmol/mol methionine), intermediate in plasma protein and retina (1.71–3.35 mmol/mol methionine) and high in sciatic nerve (30.7 mmol/mol methionine). In diabetic rats, this was increased in plasma protein only (about 40%); the increase was reversed by thiamine and benfotiamine (ESM Table 2).
Nitration adduct
3-NT content of healthy control rats was very low (0.0007–0.0021 mmol/mol tyrosine). In diabetic rats, it was only increased threefold in sciatic nerve; the increase was partially reversed by 70 mg/kg benfotiamine (ESM Table 3).
Plasma and urinary glycation, oxidation and nitration free adducts
The plasma concentrations of glycation and oxidation free adducts of healthy control rats were (nmol/l): FL 303, CML 69 ± 16, G-H1 82 ± 39, MG-H1 117 ± 38, 3DG-H 74 ± 22 and MetSO 69 ± 35. In diabetic rats, glycation free adducts were increased about twofold in diabetic rats, whereas MetSO was not increased significantly. Thiamine reversed the diabetes-induced increased in plasma AGE free adducts whereas benfotiamine increased AGE and MetSO free adducts (Fig. 3).

Plasma glycation (a–e) and oxidation (f) free adducts in STZ-induced diabetic rats and normal control and effect of thiamine and benfotiamine therapy. a FL. b CML. c G-H1. d MG-H1. e 3DG-H. f MetSO. Data are means ± SD. Rat study group key as in Fig. 2. *p < 0.05, **p < 0.01 and ***p < 0.001 with respect to normal control. † p < 0.05, †† p < 0.01 ††† p < 0.001 with respect to diabetic controls
Urinary excretion of free adducts (nmol/24 h) in healthy control rats was: FL 300 ± 261, CML 45 ± 25, MG-H1 94 ± 55, pentosidine 0.39 ± 0.15, MetSO 5.21 ± 4.10, dityrosine 0.013 ± 0.007 and 3-NT 0.012 ± 0.008. Urinary free adduct excretion rates were increased markedly in diabetes: FL sevenfold, CML tenfold, MG-H1 27-fold, pentosidine fourfold, MetSO threefold, dityrosine 12-fold and 3-NT sixfold. These diabetes-induced increases were reversed by thiamine and benfotiamine with similar potency for FL, CML, pentosidine and 3-NT; thiamine was more potent than benfotiamine in reversal of urinary MG-H1 increase and benfotiamine more potent than thiamine in reversal of urinary MetSO increase (Fig. 4 and ESM Table 4).

Urinary fluxes of glycation (a–d), oxidation (e) and nitration (f) free adducts in STZ-induced diabetic rats and normal control and effect of thiamine and benfotiamine therapy. a FL. b CML. c MG-H1. d Pentosidine. e MetSO. f 3-NT. Data are means ± SD. Rat study group key as in Fig. 2. *p < 0.05, **p < 0.01 and ***p < 0.001 with respect to normal control. † p < 0.05, †† p < 0.01 ††† p < 0.001 with respect to diabetic controls
GFR and renal clearances of glycation and oxidation free adducts were deduced from plasma and urinary free adduct concentrations. There was a high clearance of 3-DG-H (fractional clearance 115%), intermediate clearances of FL, CML, CEL and MG-H1 (fractional clearances 20–27%), and low clearances of G-H1 and MetSO (fractional clearances 0.3% and 2%). In diabetic rats, clearances of glycation and oxidation free adducts were markedly increased: FL twofold, CML twofold, CEL 11-fold, G-H1 eightfold, MG-H1 15-fold and MetSO fourfold. These increases were partially or completely reversed by treatment with thiamine and benfotiamine (Table 5).
Discussion
In this study we found increased protein damage at sites of development of vascular complications in an established model of experimental incipient diabetic nephropathy, where both thiamine and benfotiamine prevented development of nephropathy [3, 13]. Other studies have found degeneration of sciatic nerve and retina characteristic of early-stage diabetic neuropathy and retinopathy in this model [14, 15]. Increased tissue content of AGEs in diabetes leads to impairment of protein function, linked to vascular cell detachment and anoikis [16, 17], oxidative stress [18], low-grade inflammation [18, 19] and other cell dysfunctions implicated in the development of microvascular complications [20]. Thiamine and benfotiamine probably decrease AGE and oxidation and nitration adduct formation by increasing transketolase expression and activity in tissues [3, 4, 21]; this counters metabolic dysfunction and oxidative stress in hyperglycaemia, maintains antioxidant and dicarbonyl-metabolising enzyme activities and thereby prevents protein damage [2].
This is the first application of quantitative ‘gold standard’ stable isotopic dilution analysis LC–MS/MS to quantify a comprehensive range of glycation, oxidation and nitration adducts in tissues, plasma and urine in experimental diabetes. Independent MS estimates of CML residues of renal glomeruli [22] and immunoassay of CML content of plasma protein of healthy control rats [23] were in agreement with estimates herein. Immunoassay of AGEs in tissue, however, has overestimated increases in experimental diabetes [4, 5, 24].
Increased FL residue content of sciatic nerve and brain of STZ diabetic rats was reported previously using 3H-labelled borohydride reduction [25, 26]. FL residue content of proteins is influenced by glucose concentration, protein reactivity, repair of FL residues by fructosamine 3-phosphokinase (not available in plasma) [27] and protein half-life. The high FL residue content of plasma protein of rats is probably because of lack of de-glycation activity and the relatively long half-life of plasma proteins. The half-life of serum albumin in rats is 42 h and was not affected by glycation [28]. The marked increase in FL content of plasma and glomerular protein of diabetic rats is expected, given the fivefold increased fasting plasma glucose concentration. A high increase was also found in the brain (ESM Table 5). Lower increases of FL content in retinal and sciatic nerve protein and HbA1 are consistent with the de-glycating activity of these tissues and erythrocytes [27]. Although thiamine and benfotiamine therapy did not prevent increases in FL content of tissue proteins in diabetic rats, 70 mg/kg thiamine produced a small decrease in HbA1. The function of residual beta cells of STZ diabetic rats [29] may be improved by thiamine.
The detection of protein glycation, oxidation and nitration free adducts in plasma and urine is consistent with their release from damaged proteins in tissues by proteolysis. Other sources of free adducts are absorption after digestion of damaged proteins in food and glycation, oxidation and nitration of amino acids. In situ rates of proteolysis are expected to increase challenged with the two- to fourfold increase in substrate concentration at sites of increased protein damage. The contribution of glycation free adducts from digestion of food is controversial. Increased AGE and MetSO residue content of peptides in portal venous plasma was found in previous studies [30], consistent with absorption of glycated and oxidised peptides from digested food. The four- to 27-fold increase in urinary excretion of pentosidine, CML and MG-H1 free adducts in diabetic rats without similar proportional increase in food consumption found herein, however, suggests the contribution of AGEs in food to AGEs exposure is low in the diabetic state [9].
The link of urinary excretion of protein glycation, oxidation and nitration free adducts to flux of endogenous adduct formation is compromised where there is metabolism and repair of the free adducts. This applies to the repair of FL and MetSO free adducts by fructosamine 3-phosphokinase [27] and MetSO reductase [31], respectively, and metabolism of 3-NT to 3-nitro-4-hydroxyphenylacetic acid and 3-nitro-4-hydroxyphenyllactic acid [32]. Decreased renal tubular re-uptake and metabolism of glycation and oxidation free adducts in the diabetic state may give rise to increases in urinary excretion of free adducts disproportionate to the increase in de novo formation in tissue proteins. The decrease of urinary excretion of FL free adduct by thiamine and benfotiamine therapy was unexpected as neither agent decreased FL residue content of tissue or plasma protein. This may be because of increased metabolism by fructosamine 3-phosphokinase following increased renal re-uptake of FL associated with improved renal function.
Benfotiamine was more effective in reversal of urinary excretion of MetSO free adduct than thiamine, as indeed it was in improvement of plasma thiols [3]. This is probably related to the thiol intermediate formed by thioester hydrolysis of benfotiamine [2]. This may participate in thiol/disulfide exchange reactions, preserve thiol status and thereby prevent formation of MetSO and maintain levels of thioredoxin cofactor for MetSO reductase [31]. Benfotiamine was also more effective than thiamine in reversal of increased urinary excretion of dityrosine but had similar potency to thiamine in reversal of increased urinary excretion of 3-NT free adduct in diabetic rats. Benfotiamine uses the reduced folate carrier-1 (RFC-1) and phosphatase activity for thiamine delivery [2]. Decreased production of RFC-1 in diabetes and loading of tissues with TMP by benfotiamine approaching levels that inhibit thiamine pyrophosphokinase may impair thiamine delivery by benfotiamine in diabetes [9, 33]. Unexpectedly, benfotiamine therapy increased concentrations of plasma glycation and oxidation free adducts in diabetic rats, relating to decreased clearance and increased tubular re-uptake of AGE and MetSO free adducts. Benfotiamine may activate organic anion and cation transporters in the tubular epithelium and thereby increase retention of AGE and MetSO free adducts in plasma. AGE free adducts are unlikely to be functionally active in vivo, however, as the receptor for AGEs is activated by CML but only at concentrations about 1,000-fold higher than found herein [34].
Abbreviations
- CEL:
-
\( {N^\varepsilon } \)-(1-Carboxyethyl)lysine
- CML:
-
\( {N^\varepsilon } \)-Carboxymethyl-lysine
- 3-DG:
-
3-Deoxyglucosone
- 3DG-H:
-
3-DG-derived hydro-imidazolone \( {N^\delta } \)-(5-hydro-5-(2,3,4-trihydroxybutyl)-4-imidazolon-2-yl)-ornithine and related structural isomers
- FL:
-
\( {N^\varepsilon } \)-Fructosyl-lysine
- G-H1:
-
Glyoxal-derived hydroimidazolone \( {N^\delta } \)-(5,5-dihydro-4-imidazolon-2-yl)-ornithine
- LC–MS/MS:
-
Liquid chromatography–tandem mass spectrometry
- MetSO:
-
Methionine sulfoxide
- MG-H1:
-
Methylglyoxal-derived hydroimidazolone \( {N^\delta } \)-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine
- 3-NT:
-
3-Nitrotyrosine
- STZ:
-
Streptozotocin
- TMP:
-
Thiamine monophosphate
References
Parving HH, Lewis JB, Ravid M, Remuzzi G, Hunsicker LG (2006) Prevalence and risk factors for microalbuminuria in a referred cohort of type II diabetic patients: a global perspective. Kidney Int 69:2057–2063
Thornalley PJ (2005) The potential role of thiamine (vitamin B1) in diabetic complications. Curr Diabetes Res 1:287–298
Babaei-Jadidi R, Karachalias N, Ahmed N, Battah S, Thornalley PJ (2003) Prevention of incipient diabetic nephropathy by high dose thiamine and benfotiamine. Diabetes 52:2110–2120
Hammes H-P, Du X, Edelstein D et al (2003) Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 9:294–299
Stracke H, Hammes HP, Werkmann D et al (2001) Efficacy of benfotiamine vs thiamine on function and glycation products of peripheral nerves in diabetic rats. Exp Clin Endocrinol Diabetes 109:330–336
Rabbani N, Shahzad Alam S, Riaz S et al (2008) High dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: a pilot randomised, double-blind, placebo-controlled study. Diabetologia 52:208–212
Rabbani N, Alam S, Riaz S et al (2009) Thiamine in diabetic nephropathy: a novel treatment modality? Reply to Alkhalaf A, Kleefstra N, Groenier KH et al [letter]. Diabetologia 52:1214–1216
Thornalley PJ (2006) Quantitative screening of protein glycation, oxidation, and nitration adducts by LC–MS/MS: protein damage in diabetes, uremia, cirrhosis, and Alzheimer's disease. In: Dalle-Donne I, Scaloni A, Butterfield DA (eds) Redox proteomics. Wiley, Hoboken, pp 681–728
Babaei-Jadidi R, Karachalias N, Kupich C, Ahmed N, Thornalley PJ (2004) High dose thiamine therapy counters dyslipidaemia in streptozotocin-induced diabetic rats. Diabetologia 47:2235–2246
Winkler BS (1972) The electroretinogram of the isolated rat retina. Vision Res 12:1183–1198
Ahmed N, Argirov OK, Minhas HS, Cordeiro CA, Thornalley PJ (2002) Assay of advanced glycation endproducts (AGEs): surveying AGEs by chromatographic assay with derivatisation by aminoquinolyl-N-hydroxysuccimidyl-carbamate and application to Ne-carboxymethyl-lysine- and Ne-(1-carboxyethyl)lysine-modified albumin. Biochem J 364:1–14
Thornalley PJ, Battah S, Ahmed N et al (2003) Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J 375:581–592
Soulis-Liparota T, Cooper M, Papazoglou DX, Clarke B, Jerums G (1991) Retardation by aminoguanidine of development of albuminuria, mesangial cell expansion, and tissue fluorescence in streptozotocin-induced diabetic rat. Diabetes 40:1328–1334
Coppey LJ, Gellett JS, Davidson EP, Dunlop JA, Yorek MA (2002) Effect of treating streptozotocin-induced diabetic rats with sorbinil, myo-inositol or aminoguanidine on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Int J Diabetes Res 3:21–26
Aizu Y, Oyanagi K, Hu JG, Nakagawa H (2002) Degeneration of retinal neuronal processes and pigment epithelium in the early stage of the streptozotocin-diabetic rats. Neuropathology 22:161–170
Dobler D, Ahmed N, Song LJ, Eboigbodin KE, Thornalley PJ (2006) Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes 55:1961–1969
Pedchenko VK, Chetyrkin SV, Chuang P et al (2005) Mechanism of perturbation of integrin–mediated cell-matrix interactions by reactive carbonyl compounds and its implication for pathogenesis of diabetic nephropathy. Diabetes 54:2952–2960
Rosca MG, Mustata TG, Kinter MT et al (2005) Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol 289:F420–F430
Yao D, Brownlee M (2009) Hyperglycemia-induced reactive oxygen species increase expression of RAGE and RAGE ligands. Diabetes 59:249–255
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414:813–820
Pomero F, Min AM, La Selva M, Allione A, Molinatti GM, Porta M (2001) Benfotiamine is similar to thiamine in correcting endothelial cell defects induced by high glucose. Acta Diabetol 38:135–138
Alderson NL, Chachich ME, Frizzell N et al (2004) Effect of antioxidants and ACE inhibition on chemical modification of proteins and progression of nephropathy in the streptozotocin diabetic rat. Diabetologia 47:1385–1395
Kamata K, Ozawa Y, Kobayashi T, Matsumoto T (2009) Effect of N-epsilon-(carboxymethyl)lysine on coronary vasoconstriction in isolated perfused hearts from control and streptozotocin-induced diabetic rats. J Smooth Muscle Res 45:125–137
Forbes JM, Thomas MC, Thorpe SR, Alderson NL, Cooper ME (2004) The effects of valsartan on the accumulation of circulating and renal advanced glycation end products in experimental diabetes. Kidney Int 66:S105–S107
Williams SK, Howarth NL, Devenny JJ, Bitensky MW (2001) Structural and functional consequences of increased tubulin glycosylation in diabetes mellitus. Proc Natl Acad Sci U S A 79:6546–6550
Cullum NA, Mahon J, Stringer K, Mclean WG (1991) Glycation of rat sciatic nerve tubulin in experimental diabetes mellitus. Diabetologia 34:387–398
Delplanque J, Delpierre G, Opperdoes FR, van Schaftingen E (2004) Tissue distribution and evolution of fructosamine 3-kinase and fructosamine 3-kinase-related protein. J Biol Chem 279:46606–46613
Johnson RN, Easdale RW, Tatnell M, Baker JR (1991) Significance of variation in turnover of glycated albumin on indexes of diabetic control. Clin Chim Acta 198:229–238
Van Gompel J, Mahler T, Paepe M, Kloppel G (1993) Comparison of in situ hybridization and immunocytochemistry for the detection of residual beta cells in the pancreas of streptozotocin-treated diabetic rats. Acta Diabetol 30:118–122
Ahmed N, Thornalley PJ, Luthen R et al (2004) Processing of protein glycation, oxidation and nitrosation adducts in the liver and the effect of cirrhosis. J Hepatol 41:913–919
Weissbach H, Resnick L, Brot N (2005) Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochim Biophys Acta 1703:203–212
Ohshima H, Friesen M, Brouet I, Bartsch H (1990) Nitrotyrosine as a new marker for endogenous nitrosation and nitration of proteins. Food Chem Toxicol 28:647–652
Naggar H, Ola MS, Moore P et al (2002) Downregulation of reduced-folate transporter by glucose in cultured RPE cells and in RPE of diabetic mice. Invest Ophthalmol Vis Sci 43:556–563
Zill H, Bek S, Hofmann T et al (2003) RAGE-mediated MAPK activation by food-derived AGE and non-AGE products. Biochem Biophys Res Commun 300:311–313
Acknowledgements
We thank the Juvenile Diabetes Research Fund and the Wellcome Trust (UK) for support for our diabetes and protein damage research.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Pentosidine content of tissue and plasma protein of STZ-induced diabetic rats and healthy controls with and without thiamine and benfotiamine therapy (PDF 31 kb)
ESM Table 2
MetSO residues of tissue and plasma protein of STZ-induced diabetic rats and healthy controls with and without thiamine and benfotiamine therapy (PDF 31 kb)
ESM Table 3
3-NT residues of tissue and plasma protein of STZ-induced diabetic rats and healthy controls with and without thiamine and benfotiamine therapy (PDF 31 kb)
ESM Table 4
Urinary excretion of dityrosine-free adduct of STZ-induced diabetic rats and controls with and without high dose therapy with thiamine and benfotiamine (PDF 28 kb)
ESM Table 5
Glycation adduct content of brain of STZ-induced diabetic rats and healthy controls with and without thiamine and benfotiamine therapy (PDF 34 kb)
Rights and permissions
About this article
Cite this article
Karachalias, N., Babaei-Jadidi, R., Rabbani, N. et al. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia 53, 1506–1516 (2010). https://doi.org/10.1007/s00125-010-1722-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1722-z