
Abstract
Aims/hypothesis
Type 2 diabetes is associated with complications in the central nervous system (CNS), including learning and memory, and an increased risk for neurodegenerative diseases. The mechanism underlying this association is not understood. The aim of this study was to gain greater insight into the possible mechanisms of diabetes-induced cognitive decline.
Methods
We used microarray technology to identify and examine changes in gene expression in the hippocampus of a murine model of type 2 diabetes, the db/db mouse. Bioinformatics approaches were then used to investigate the biological significance of these genes. To validate the biological significance we evaluated mRNA and protein levels.
Results
At 8 and 24 weeks, 256 and 822 genes, respectively, were differentially expressed in the db/db mice. The most significantly enriched biological functions were related to mitochondria, heat shock proteins, or the endoplasmic reticulum (ER), the majority of which were downregulated. The ER-enriched cluster was one of the clusters that contained the highest number of differentially expressed genes. Several of the downregulated genes that were differentially expressed at 24 but not at 8 weeks are directly involved in the unfolded protein response (UPR) pathway and include two heat shock proteins (encoded by Hspa5 and Hsp90b1), a transcriptional factor (x-box binding protein 1, encoded by Xbp1), and an apoptotic mediator (DNA-damage inducible transcript 3, encoded by Ddit3).
Conclusions/interpretation
The changes that we observed in the UPR pathway due to ER stress may play a role in the pathogenesis of CNS complications in diabetes. The results of this study are a foundation for the development of pharmacological targets to reduce ER stress in diabetic hippocampi.
Similar content being viewed by others


Introduction
According to the Centers for Disease Control and Prevention, 25.8 million people in the USA (8.3% of the population) have diabetes mellitus, with 1.9 million new cases diagnosed per year (2010). Diabetes is a metabolic disorder characterised by hyperglycaemia. Type 2 diabetes accounts for approximately 90–95% of all cases and is, in addition, characterised by hyperinsulinaemia and insulin resistance. Type 2 diabetes is associated with obesity, hypertension, hypercholesterolaemia, hyperlipidaemia, and micro- and macrovascular complications including neuropathy, retinopathy, nephropathy and cardiovascular disease [1]. Central nervous system (CNS) complications associated with type 2 diabetes are gaining more attention [2].
Many factors may play a role in diabetes-associated cognitive deficits, such as impaired insulin signalling, glucose metabolism dysregulation, cardiovascular disease and microvascular complications [2]. Cognitive deficits in type 2 diabetes are more pronounced in hippocampal-related tasks and correlate with the degree of glycaemic control [3]. Diabetes is associated with brain atrophy and electrophysiological changes that may result in deficits in learning, memory, attention, executive function and psychomotor efficiency [4]. The diabetes-induced changes in the CNS may, over time, lead to an acceleration in brain ageing and increase the risk of age-related neurodegenerative diseases such as Alzheimer’s disease [5].
Age-related neurodegenerative diseases are characterised by the accumulation of misfolded proteins [6, 7]. The endoplasmic reticulum (ER) plays a vital role in protein synthesis, protein folding, calcium homeostasis and the synthesis of fatty acids, sterols and phospholipids. Physiological or pathological conditions that perturb these processes result in ER stress, which may be either acute (short-term) or chronic (long-term) in nature [8, 9]. The unfolded protein response (UPR) is triggered by an increase in misfolded or unfolded proteins in the ER lumen [8, 10, 11]. The purpose of the UPR is to relieve the burden on the ER by increasing the folding capacity, decreasing protein synthesis (translation) and/or degrading unfolded proteins through the ubiquitin–proteasome system or autophagy [8, 10]. If this does not relieve ER stress, then apoptosis is activated [12]. ER stress is implicated in age-related neurodegenerative diseases and the UPR is compromised with ageing [13–15].
Diabetes and obesity increase ER stress, impair the compensatory role of the UPR and are involved in the dysregulation of glucose metabolism in the liver, pancreatic beta cells and adipocytes of mice [16–18]. ER stress is involved in the development of peripheral insulin resistance and is implicated in diabetes-related complications such as retinopathy and nephropathy [10, 12, 16–18]. The cellular mechanisms of diabetes-associated cognitive impairment are poorly understood. Data suggestive of a connection between ER stress and cognitive impairment have been reported in a diet-induced obese mouse model [19]. While hippocampal-dependent cognitive deficits have been identified in db/db mice [20, 21], evidence for involvement of ER stress in diabetic hippocampi is lacking. To explore the pathophysiology of diabetes-associated cognitive impairment, we employed microarray technology and bioinformatics approaches to identify changes in gene expression in the hippocampus of a murine model of type 2 diabetes and obesity, the db/db mouse [22].
Methods
Mice
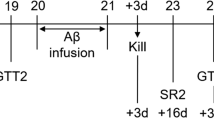
The db + and db/db mice (stock #000642) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and breeding colonies were established at the University of Michigan. Mice were kept in a pathogen-free environment and were cared for according to the University of Michigan Committee on the Care and Use of Animals guidelines. Mice were fed powdered LabDiet 5053 mouse chow (PMI Nutrition International, St Louis, MO, USA). Blood glucose levels were measured every 4 weeks after a 6 h fast using one drop of tail blood and a standard glucometer (OneTouch; LifeScan, Milpitas, CA, USA). The glucometer does not process glucose levels over 33.3 mmol/l; therefore, all measurements were rounded down to 33.3 mmol/l. HbA1c was measured at 8 and 24 weeks using the Helena Laboratories Test Kit, Glycotek Affinity Column Method (Helena Laboratories, Beaumont, TX, USA) by collecting 50 μl of blood prior to the tissue harvest.
Tissue harvest
Twenty-four male db + and db/db mice were killed at 8 and 24 weeks (six mice per group) by sodium pentobarbital overdose and tissues were harvested as previously described [23]. The hippocampus was dissected from each hemisphere with half of the hippocampus stored in 30 μl RNA later (Ambion, Austin, TX, USA) for gene expression analysis. The other half of the hippocampus was flash frozen in liquid nitrogen and stored at −80°C until ready for western immunoblotting.
RNA preparation
Total RNA was isolated from one hemisphere of the hippocampus using the RNeasy Mini Kit (QIAGEN, Valencia, CA, USA), including an on-column deoxyribonuclease digestion, following the manufacturer’s protocol. RNA quality and quantity were assessed by microfluid electrophoresis using an RNA 6000 Pico LabChip on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples with a minimum RNA integrity number of seven were used for microarray hybridisation [24].
Affymetrix microarrays
Six samples from each group that met the RNA quality criteria were analysed by microarray. Total RNA (75 ng) was amplified and biotin-labelled using the Ovation Biotin–RNA Amplification and Labeling System (NuGEN Technologies, San Carlos, CA, USA) according to the manufacturer’s protocol. Amplification and hybridisation were performed by the University of Michigan Comprehensive Cancer Center Affymetrix and Microarray Core Facility (University of Michigan, Ann Arbor, MI, USA) using the Affymetrix GeneChip Mouse Genome 430 2.0 array. The intensities of the target hybridisations to their respective probe features were detected by laser scan of the array. Image files were generated by Affymetrix GeneChip software (MAS5).
The Affymetrix raw data files (CEL files) were analysed using a local copy of GenePattern, a bioinformatics platform from the Broad Institute [25]. The samples were Robust multi-array average (RMA) normalised using the BrainArray custom chip definition file (CDF) version 12 [26]. Microarray quality assessment was performed using probe-level modelling (PLM) and quality metrics provided by the ‘affy’ package of BioConductor [27]. Outliers were excluded using qualitative hierarchical clustering analysis, dChip’s outlier detection function [28] and a principal component analysis (PCA)-based outlier detection script [29]. Arrays were excluded if at least two of the three methods agreed that the array was an outlier.
Microarray data analysis
Identification of differentially expressed genes
Differentially expressed genes (DEGs) between db + and db/db mouse hippocampi at both 8 and 24 weeks were identified using the GenePattern platform and a standard RMA-based probe set approach, which averages normalised expression levels across all probes for a gene. Differential expression of genes was determined using the intensity-based moderated T-statistic (IBMT) test [30] with a false discovery rate (FDR) <0.10.
Functional annotation and enrichment analyses
The Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov) [31] and ConceptGen (http://conceptgen.ncibi.org) [32] were used to identify over-represented biological functions and pathways among the DEGs. From DAVID, the following annotation databases were used: Gene Ontology (GO) Cellular Component, GO Biological Process, GO Molecular Function, and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways. From ConceptGen, annotation from the Medical Subject Headings (MeSH) database was used. The p values for annotation terms in both DAVID and ConceptGen were determined using a modified Fisher’s exact test. DAVID terms with p < 0.05 were included, and ConceptGen terms with both p and Q <0.05 were included.
Since ConceptGen uses human Entrez Gene ID numbers, the mouse gene IDs from our experiment were converted to human gene IDs using a custom script. The script used the Mouse Genome Informatics (MGI) Orthology database [33] to first find orthologous genes, then the National Center for Biotechnology Information (NCBI) HomoloGene database to find homologous genes, and finally the NCBI Entrez Gene database to find genes with the same official symbol (See electronic supplementary material (ESM) Section 1). Only significantly enriched terms annotating four or more genes were included in further analyses.
Literature survey of DEGs for functional relevance
To examine the relevance and importance of our DEGs to the enriched biological functions and pathways, we surveyed the literature using a literature mining tool and the Entrez Gene database. SciMiner (http://jdrf.neurology.med.umich.edu/SciMiner) [34], our in-house web-based literature mining tool, identified genes and the frequency with which they appear in the literature for multiple PubMed queries (ESM Section 2). Search terms with a minimum of 1,000 papers in PubMed were used for further analyses and the top 25 most frequently mentioned genes from each query were compared with the list of DEGs. This gave us an indication of the relative importance of our DEGs related to specific topics, which is not possible with tools such as DAVID and ConceptGen.
A custom script using NCBI Entrez Utils (http://eutils.ncbi.nlm.nih.gov) was developed to search the Entrez Gene entries of a set of genes for certain user-specified search terms. Once terms of interest (biological functions and pathways) were identified using the methods described in the previous sections, the script was run on the list of DEGs to identify a subset of genes that were possibly related to those terms. Individual Entrez Gene entries and the literature were then manually examined to determine the actual relationship between the gene and the search terms. This method is similar to annotation of the genes from a database such as GO, but has the advantage of not only indicating the presence of a relationship between a term and a gene (which an annotation database could provide), but also allowing for the manual curation of the nature of that relationship (ESM Section 3).
Real-time RT-PCR
The expression of eleven 24-week DEGs was confirmed by real-time RT-PCR using the same samples as the microarray. Reverse transcription was performed using the iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA). Real-time PCR amplification and SYBR green fluorescence detection were performed using the iCycler iQ Real-time Detection System (Bio-Rad). The fluorescence threshold value (Ct) was calculated using iCycler iQ system software. The mRNA levels were normalised to an endogenous reference gene (Gapdh, encoding glyceraldehyde-3-phosphate dehydrogenase; ∆Ct) and then relative to a control group (∆∆Ct), and were expressed as \( {2^{{ - \Delta \Delta {{\text{C}}_{\text{t}}}}}} \). The average was calculated from two runs per sample.
Western immunoblotting
Western immunoblotting was performed as previously described [35]. Polyclonal antibodies against heat shock protein 5 (HSPA5, also known as glucose-regulated protein [GRP] 78/BiP; Abcam, Cambridge, MA, USA), x-box binding protein 1 (XBP1; Santa Cruz Biotechnology, Santa Cruz, CA, USA), heat shock protein 90 beta member 1 (HSP90B1, also known as GRP94; Abcam) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore, Billerica, MA, USA) were used.
Statistical analysis
Data analyses were performed using Prism, version 5 (GraphPad Software, La Jolla, CA, USA). Six mice per group at 8 and 24 weeks of age were used for metabolic studies. These six mice were used for microarray, RT-PCR and western immunoblotting. A separate cohort of at least five mice was also evaluated by western blotting. For all experiments, a two-tailed t test was performed to compare the db + mice to db/db mice. All values are reported as the mean ± SEM.
Results
Metabolic variables in db/db mice
The db/db mice gained a significant amount of weight compared with the db + mice at 8 weeks (24.0 ± 0.5 g [db +] and 34.7 ± 0.4 g [db/db]; p < 0.001) and 24 weeks (31.0 ± 0.6 g [db +] and 50.1 ± 2.5 g [db/db]; p < 0.001; Fig. 1a). Fasting blood glucose was markedly elevated in the db/db compared with the db + mice at 8 weeks (9.0 ± 0.4 [db +] and 24.8 ± 1.9 [db/db]; p < 0.001) and 24 weeks (6.4 ± 0.4 [db +] and 32.6 ± 0.5 [db/db]; p < 0.001; Fig. 1b). Furthermore, the HbA1c was significantly increased at 8 weeks in the db/db compared with the db + mice (0.05 ± 0.004% [31.1 mmol/mol; db +] and 0.06 ± 0.005% [42.1 mmol/mol; db/db]; p < 0.05) and at 24 weeks (0.07 ± 0.001% [53.0 mmol/mol; db +] and 0.13 ± 0.007% [119 mmol/mol; db/db]; p < 0.001), indicative of prolonged hyperglycaemia (Fig. 1c).

Metabolic variables were assessed in diabetic mice at 8 and 24 weeks. Measures of (a) weight, (b) glucose and (c) HbA1c in db + (white bars) and db/db (black bars) mice. To convert values for HbA1c in % into mmol/mol, subtract 2.15 and multiply by 10.929. *p < 0.05 and **p < 0.001 based on t test; n = 6
Microarray
Approximately 12,360 and 12,280 genes in the 8 and 24 week arrays, respectively, of the 16,331 genes on the microarray annotated by the custom CDF were expressed above the background in at least one of the samples. Analysis of the changes in gene expression revealed a total of 256 DEGs with 121 genes downregulated and 135 genes upregulated in the db/db mice compared with the db + mice at 8 weeks and a total of 822 DEGs with 469 genes downregulated and 353 genes upregulated in the 24 week db/db mice compared with the db + mice (ESM Section 4).
Functional enrichment
The upregulated and downregulated DEGs were each clustered into functional annotation terms using DAVID and ConceptGen. The 24 week DEGs contained more significantly enriched annotation terms than the 8 week DEGs (Table 1). Many of the significantly enriched terms at 8 weeks have the same or a similar term enriched at 24 weeks in the same direction (ESM Section 5). At 24 weeks, the most significantly enriched terms with p < 0.001 in the downregulated DEGs were related to mitochondria, HSPs, the ER and sterol biosynthesis. The mitochondria- and ER-enriched clusters contained the highest number of associated genes, with 80 and 42 genes, respectively.
Literature survey of DEGs
SciMiner was used to identify genes associated with specific queries in the literature. Of all the search terms (ESM Section 2, 2.1), two queries had the highest number of differentially regulated genes in the top 25: ‘ER stress’ OR ‘endoplasmic reticulum stress’ and chaperones (MeSH). Therefore, further analyses focused on ER stress. Specifically, four of the 24 week DEGs (Hspa5, Hsp90b1, Xbp1 and Ddit3, which encodes DNA-damage inducible transcript 3) including the top two ranked genes (Hspa5 and Ddit3), were among the top 25 genes related to ER stress.
A subset of genes from the 24 week DEGs that were possibly associated with ER stress was selected by searching for genes whose Entrez Gene entries for either the mouse or human gene contained at least one of the following terms: ‘ER stress’, ‘ER-stress’, ‘endoplasmic reticulum stress’ or ‘endoplasmic reticulum-stress’. A subset of these genes was identified as showing strong evidence of a relationship with ER stress by the authors manually reading the Entrez Gene entries and relevant literature. Table 2 contains details of the nine genes that were associated with ER stress, all of which are significantly downregulated at 24 weeks but not significantly regulated at 8 weeks. The four genes from the literature mining (Hspa5, Hsp90b1, Xbp1 and Ddit3) were also present in this list.
RT-PCR
To validate the microarray expression data, RT-PCR was performed on selected DEGs at 24 weeks. Seven genes were chosen because they had a minimum of 1.85-fold change (FC): Sdf2l1, encoding stromal cell-derived factor 2-like 1; Hist2h3c2, encoding histone cluster 2 H3C2; Cth, encoding cystathionase; Zfp354b, encoding zinc finger protein 354B; Klhl5, encoding kelch-like 5; Pla2g4e, encoding phospholipase A2, group IVE; Retnla, encoding resistin-like alpha; Fig. 2a. Four DEGs were selected because they were key ER-stress genes (Hspa5, Hsp90b1, Xbp1 and Ddit3; Fig. 2b). The mRNA expression levels of the two ER chaperones, Hspa5 and Hsp90b1, decreased in the 24 week db/db mice (0.56 and 0.84 FC, respectively). The mRNA expression of a key transcription factor of the UPR, Xbp1, also significantly decreased in the 24 week db/db mice (0.73 FC). Although the mRNA expression levels of Ddit3 did not reach significance, there was a downward trend for the levels in the 24 week db/db mice compared with db +. In total, eight of the 11 tested genes demonstrated significant differential expression, all in parallel with the direction of up-/downregulation in the microarray results (Fig. 2c).

RT-PCR verification of the expression levels of the 24 week DEGs. Relative mRNA expression levels (FC) in db/db (black bars) compared with db + (white bars) mice of (a) seven selected DEGs (>1.85 FC) and (b) four ER stress-related DEGs: Hspa5, Hsp90b1, Xbp1 (spliced [s] and unspliced [u]), and Ddit3. (c) The log2 transformation of the relative mRNA expression levels of all 11 DEGs selected for RT-PCR (black bars); 10 of the 11 parallel the direction of changes in the microarray (white bars). *p < 0.05 based on t test; n = 6
Western immunoblotting
To determine the biological relevance of the ER-stress-related downregulated DEGs, western immunoblotting and densitometry analysis was used to evaluate the protein levels (expressed as the ratio normalised to GAPDH). The relative protein levels of HSPA5 decreased 19% in the 24 week db/db mice compared with the db + mice (0.91 ± 0.04 and 1.1 ± 0.07, respectively; Fig. 3a). Similarly, the relative levels of HSP90B1 decreased 15% in the 24 week db/db mice compared with the db + mice (0.96 ± 0.03 and 1.1 ± 0.05; Fig. 3b). The relative levels of XBP1s (spliced XBP1) in the 24 week db/db mice (0.52 ± 0.16) decreased 67% compared with the db + mice (1.6 ± 0.32) and XBP1u (unspliced XBP1) decreased 48% in the db/db mice compared with the db + mice (0.71 ± 0.09 and 1.4 ± 0.15, respectively; Fig. 3c). The ratio of XBP1s/XBP1u decreased 60% in the 24 week db/db mice compared with the db + mice (0.58 ± 0.22 and 1.5 ± 0.16, respectively; Fig. 3d). Unfortunately, DDIT3 levels were undetectable in the db + and db/db mouse hippocampi at 24 weeks (data not shown).

ER stress response was assessed in diabetic mice at 24 weeks by western immunoblotting. Representative blot and densitometry in the db + (left two lanes) and db/db (right two lanes) mice; (a) HSPA5, (b) HSP90B1 and (c) XBP1s (upper blot) and XBP1s (centre blot). The values represent the ratio normalised to GAPDH (lower blot), the loading control. (d) The ratio of XBP1s to XBP1u in db + (white bars) and db/db (black bars) mice. *p < 0.05 and **p < 0.01 based on t test; n = 10 for db + and n = 11 for db/db mice
Discussion
Type 2 diabetes is associated with complications in the CNS; however, the pathophysiology is not well understood. Here we report the first evaluation of the genome-wide effects of type 2 diabetes in the hippocampus of db + and db/db mice at 8 and 24 weeks, using microarrays to assess the alterations in gene expression in the hippocampus. The most significantly enriched terms in the 24 week downregulated DEGs were related to mitochondria, HSPs, the ER and sterol biosynthesis. The ER-enriched cluster included 42 genes. Literature mining revealed that four of our DEGs were in the top 25 genes related to ER stress and directly involved in the UPR. Thus, we validated the biological significance of these four downregulated genes—Hspa5, Hsp90b1, Xbp1 and Ddit3.
Hspa5 and Hsp90b1 are ER molecular chaperones that interact with misfolded proteins and are associated with cell survival [8, 9]. The downregulation of these chaperones may leave cells particularly vulnerable to further stress [36]. The expression levels of Hspa5 and Hsp90b1 decline in the ageing hippocampus [14, 37]. Thus, one limitation in our study is the inability to determine whether the downregulation is a direct result of the obesity or diabetes, or is associated with brain ageing, which can be accelerated by diabetes [38]. Further studies are required to fully elucidate the mechanisms contributing to ER stress in the hippocampus during diabetes.
Xbp1, a transcription factor and master regulator of ER folding capacity, is transcriptionally upregulated during the typical UPR to acute stress [9]. In response to this stress, an intron is excised from the unspliced variant of Xbp1 mRNA yielding spliced Xbp1 mRNA [11]. Spliced Xbp1 mRNA encodes XBP1s, a transcriptional activator of Hspa5 and Hsp90b1, whereas unspliced Xbp1 mRNA encodes XBP1u, an inhibitor of the UPR and chaperone induction [11]. XBP1u shuttles between the nucleus and cytoplasm and may function as a negative feedback regulator of XBP1s [39].
Chronic stressors, such as diabetes or obesity, lead to years of persistent ER stress and UPR activation and cells must adapt to survive [10, 40]. Adaptation to ER stress is a complex cell-type dependent mechanism that involves maintenance of cellular function and avoidance of apoptosis. This adaptation may involve partial activation or suppression of one of more branches of the UPR [9]. Xbp1s mRNA declines after prolonged ER stress and is not activated in cells that have adapted to this stress [41, 42]. Furthermore, Ddit3, a mediator of apoptosis, is rapidly increased at the initial onset of stress, but is quickly reduced owing to the instability of the mRNA and protein during adaptation [42]. Thus, the downregulation in the gene (Hspa5, Hsp90b1, Xbp1 and Ddit3) and mRNA (Hspa5, Hsp90b1, Xbp1s and Xbp1u) expression levels, and protein levels (HSPA5, HSP90B1, XBP1s and XBP1u) provide evidence that hippocampal cells adapt to ER stress during diabetes.
XBP1s plays a role in inhibiting insulin signalling and contributes to the development of peripheral insulin resistance [18]. Treatment with small molecules that act as chaperones for proper protein folding results in the resolution of ER stress, normalisation of blood glucose and restoration of insulin sensitivity in the liver of ob/ob mice [43]. Similarly, overexpression of an ER chaperone, GRP150, results in improvements in glucose tolerance and insulin sensitivity in db/db mice [16]. Therefore, ER stress undoubtedly plays a role in insulin resistance. Insulin resistance is associated with cognitive decline in rodents and in epidemiological studies [44, 45]. Pharmacological inhibition of ER stress using chemical chaperones not only improves insulin sensitivity but also protects against cognitive deficits in mouse models of diabetes [19]. Thus, ER stress may contribute to cognitive deficits.
Collectively, our data suggest that hippocampal cells adapt to type 2 diabetes-induced prolonged ER stress with partial suppression of Xbp1 (Fig. 4). The decrease in the ratio of XBP1s to XBP1u, which indicates that there are higher levels of XBP1u compared with XBP1s, may be responsible for the suppression of the induction of Hspa5 and Hsp90b1 and contribute to the development of insulin resistance, which we have demonstrated in the hippocampi of db/db mice [46]. Although this study does not reveal the mechanism underlying the connection between ER stress and cognitive decline, it does provide a foundation for further exploration of the role of adaptation to hippocampal ER stress in vivo in neuronal insulin resistance and cognitive deficits. Our results provide the first evidence of ER stress in the hippocampus of a murine model of type 2 diabetes.

Representation of our proposed model of adaptation to ER stress in the diabetic hippocampus. An intron of the unspliced (u) variant of Xbp1 mRNA is excised during the response to ER stress, yielding spliced (s) Xbp1 mRNA. Xbp1s mRNA encodes the protein XBP1s, which activates the synthesis of ER chaperones, whereas Xbp1u mRNA encodes the protein XBP1u, which inhibits the synthesis of ER chaperones. During adaptation to prolonged ER stress in the hippocampus, the ratio of XBP1s to XBP1u is altered to inhibit ER chaperone synthesis
Abbreviations
- CDF:
-
Chip definition file
- CNS:
-
Central nervous system
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- DEG:
-
Differentially expressed gene
- ER:
-
Endoplasmic reticulum
- FC:
-
Fold change
- FDR:
-
False discovery rate
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GO:
-
Gene Ontology
- GRP:
-
Glucose-regulated protein
- HSP90B1:
-
Heat shock protein 90 beta member 1
- HSPA5:
-
Heat shock protein 5
- NCBI:
-
National Center for Biotechnology Information
- RMA:
-
Robust multi-array average
- UPR:
-
Unfolded protein response
- XBP1:
-
X-box binding protein 1
- XBP1s:
-
Spliced XBP1
- XBP1u:
-
Unspliced XBP1
References
Li L, Holscher C (2007) Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 56:384–402
Sims-Robinson C, Kim B, Rosko A, Feldman EL (2010) How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 6:551–559
Gold SM, Dziobek I, Sweat V et al (2007) Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 50:711–719
Wrighten SA, Piroli GG, Grillo CA, Reagan LP (2009) A look inside the diabetic brain: contributors to diabetes-induced brain aging. Biochim Biophys Acta 1792:444–453
Biessels GJ, van der Heide LP, Kamal A, Bleys RL, Gispen WH (2002) Ageing and diabetes: implications for brain function. Eur J Pharmacol 441:1–14
Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22
Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292:1552–1555
Schroder M (2008) Endoplasmic reticulum stress responses. Cell Mol Life Sci 65:862–894
Rutkowski DT, Kaufman RJ (2007) That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci 32:469–476
Lin JH, Walter P, Yen TS (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 3:399–425
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Naidoo N (2009) The endoplasmic reticulum stress response and aging. Rev Neurosci 20:23–37
Naidoo N (2009) ER and aging-protein folding and the ER stress response. Ageing Res Rev 8:150–159
Scheper W, Hoozemans JJ (2009) Endoplasmic reticulum protein quality control in neurodegenerative disease: the good, the bad and the therapy. Curr Med Chem 16:615–626
Nakatani Y, Kaneto H, Kawamori D et al (2005) Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem 280:847–851
Hotamisligil GS (2010) Endoplasmic reticulum stress and atherosclerosis. Nat Med 16:396–399
Ozcan U, Cao Q, Yilmaz E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Lu J, Wu DM, Zheng YL et al (2011) Ursolic acid improves high fat diet-induced cognitive impairments by blocking endoplasmic reticulum stress and IkappaB kinase beta/nuclear factor-kappaB-mediated inflammatory pathways in mice. Brain Behav Immun 25:1658–1667
Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T (2002) Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 113:607–615
Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP (2008) Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 11:309–317
Chua SC Jr, Chung WK, Wu-Peng XS et al (1996) Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 271:994–996
Sullivan KA, Hayes JM, Wiggin TD et al (2007) Mouse models of diabetic neuropathy. Neurobiol Dis 28:276–285
Schroeder A, Mueller O, Stocker S et al (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3
Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP (2006) GenePattern 2.0. Nat Genet 38:500–501
Dai M, Wang P, Boyd AD et al (2005) Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 33:e175
Gautier L, Cope L, Bolstad BM, Irizarry RA (2004) affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20:307–315
Jones L, Goldstein DR, Hughes G et al (2006) Assessment of the relationship between pre-chip and post-chip quality measures for Affymetrix GeneChip expression data. BMC Bioinformatics 7:211
Shieh AD, Hung YS (2009) Detecting outlier samples in microarray data. Stat Appl Genet Mol Biol 8, Article 13
Sartor MA, Tomlinson CR, Wesselkamper SC, Sivaganesan S, Leikauf GD, Medvedovic M (2006) Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinformatics 7:538
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Sartor MA, Mahavisno V, Keshamouni VG et al (2010) ConceptGen: a gene set enrichment and gene set relation mapping tool. Bioinformatics 26:456–463
Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT (2011) The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res 39:D842–848
Hur J, Schuyler AD, States DJ, Feldman EL (2009) SciMiner: web-based literature mining tool for target identification and functional enrichment analysis. Bioinformatics 25:838–840
Kim B, Backus C, Oh S, Hayes JM, Feldman EL (2009) Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 150:5294–5301
Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M (2004) Induction of neuronal death by ER stress in Alzheimer's disease. J Chem Neuroanat 28:67–78
Paz Gavilan M, Vela J, Castano A et al (2006) Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging 27:973–982
Launer LJ (2005) Diabetes and brain aging: epidemiologic evidence. Curr Diab Rep 5:59–63
Yoshida H, Oku M, Suzuki M, Mori K (2006) pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol 172:565–575
Harding HP, Ron D (2002) Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51(Suppl 3):S455–461
Lin JH, Li H, Yasumura D et al (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318:944–949
Rutkowski DT, Arnold SM, Miller CN et al (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 4:e374
Ozcan U, Yilmaz E, Ozcan L et al (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140
Convit A (2005) Links between cognitive impairment in insulin resistance: an explanatory model. Neurobiol Aging 26(Suppl 1):31–35
Greenwood CE, Winocur G (2005) High-fat diets, insulin resistance and declining cognitive function. Neurobiol Aging 26(Suppl 1):42–45
Kim B, Sullivan KA, Backus C, Feldman EL (2011) Cortical neurons develop insulin resistance and blunted Akt signaling: a potential mechanism contributing to enhanced ischemic injury in diabetes. Antioxid Redox Signal 14:1829–1839
Nakanishi K, Kamiguchi K, Torigoe T et al (2004) Localization and function in endoplasmic reticulum stress tolerance of ERdj3, a new member of Hsp40 family protein. Cell Stress Chaperones 9:253–264
Wang X, Venable J, LaPointe P et al (2006) Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127:803–815
Acknowledgements
The authors acknowledge A. Heacock for her expert editorial advice and the expertise of J. Hayes, S.S. Oh and C. Backus with regard to animal care and metabolic profiling. The authors also acknowledge the Michigan Diabetes Research and Training Center for HbA1c measurements.
Funding
This work was supported by the National Institutes of Health (U54-DA021519 (NCIBI) to ELF); the Animal Models of Diabetic Complications Consortium (AMDCC U01-DK076160 to ELF); the Program for Neurology Research and Discovery; a Juvenile Diabetes Research Foundation Postdoctoral Fellowship (to JH) and an Aging Training Grant (NIA T32-AG000114 to CS-R).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
CS-R and SZ contributed to the conception and design of the study and the acquisition, analysis and interpretation of the data, drafted/revised the article and gave final approval of the version to be published. JH contributed to the analysis of data, revised the article and approved the final version. ELF, the guarantor, contributed to the conception of the study and interpretation of the data, revised the article and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Sims-Robinson and S. Zhao contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Entrez Gene ID species conversion tool (PDF 7 kb)
ESM 2
SciMiner tables (PDF 10 kb)
ESM 3
Entrez Gene search tool (PDF 9 kb)
ESM 4
Complete differentially expressed genes (DEG) list (PDF 111 kb)
ESM 5
Complete DAVID and ConceptGen results (PDF 28 kb)
Rights and permissions
About this article
Cite this article
Sims-Robinson, C., Zhao, S., Hur, J. et al. Central nervous system endoplasmic reticulum stress in a murine model of type 2 diabetes. Diabetologia 55, 2276–2284 (2012). https://doi.org/10.1007/s00125-012-2573-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2573-6