
Abstract
Objective
Sepsis-associated delirium is a common and poorly understood neurological complication of sepsis. This review provides an update of the diagnostic criteria and treatment strategies and the current knowledge about the mechanisms involved in sepsis associated brain dysfunction.
Data sources
Articles published between 1981 and 2006 were identified through a Medline search for “encephalopathy” and “sepsis” and by hand searching of articles cited in the identified publications. The immune response to sepsis results in multiorgan failure including brain dysfunction.
Discussion
The potential mechanisms for sepsis-associated delirium include vascular damage, endothelial activation, breakdown of the blood-brain barrier, metabolic disorders, brain inflammation and apoptosis. On the other hand, there is evidence for distinct neuroprotective factors, such as anti-inflammatory mediators and glial cell activity.
Conclusions
The diagnosis of sepsis-associated delirium relies mainly on clinical and electrophysiological criteria, and its treatment is entirely based on general management of sepsis.
Similar content being viewed by others

Introduction
Brain dysfunction is a serious complication of sepsis. In current diagnostic manuals (Diagnostic and Statistical Manual, DSM IV; International Classification of Diseases, ICD 10) the term “encephalopathy” has been replaced by “delirium”, which indicates an acute and potentially reversible organic brain dysfunction. In this contribution we therefore use the term sepsis-associated delirium (SAD) or brain dysfunction, and we propose abandoning the term “sepsis-induced encephalopathy”. SAD often occurs early in the course of sepsis and may be transient or may reflect irreversible brain damage. Its prevalence varies from 9% to 71% of patients with severe sepsis, depending on how it is defined [1–5]. No large, multicenter, prospective cohort studies has investigated the clinical signs and laboratory tests of SAD to allow an accurate estimate of its prevalence. One multicenter study found that 307 of 1,333 patients (23%) with severe sepsis had an acute alteration in mental status [2]. However, this investigation did not systematically use a clinical scoring system or objective testing. When sensitive diagnostic tools of brain dysfunction such as electrophysiological testing are used, SAD may be detected in almost all patients [1, 4–6]. The severity of SAD, whether assessed clinically [2, 3] or using electrophysiological testing [1, 5] is correlated with the global severity of illness. Nevertheless, one multicenter sepsis trial reported SAD an independent predictor of death [2]. In addition, a prospective study of 50 patients with sepsis showed that mortality increased (from 16% to 63%) with decreasing Glasgow Coma Score (GCS; from 15 to 3 to 8), confirming the independent prognostic value of SAD [3]. In patients with positive blood cultures a normal electroencephalography (EEG) was associated with full recovery, and mortality increased with severity of EEG abnormalities (19% with theta, 36% with delta, 50% with triphasic waves, and 67% with burst suppression) [1]. While SAD was first identified in a case of typhus, brain dysfunction may complicate sepsis due to any pathogen, whether Gram-positive or Gram-negative bacteria, virus, fungi, or parasites [2].
Diagnosis
The diagnostic approach of SAD requires recognition of brain dysfunction and relating it to infection or systemic inflammation. In theory, the diagnosis of SAD includes clinical, electrophysiological or biochemical criteria [1, 3, 5, 6] (Table 1). In practice, the diagnostic approach varies according to sedation (Fig. 1).

Proposed decision tree for the diagnosis of sepsis-associated delirium
Definition and diagnosis of sepsis-associated delirium
SAD is characterized by the acute onset of impaired cognitive function ranging from slowing of mentation, inattention, disorientation, and agitation to stupor or coma. SAD is a diagnosis of exclusion and requires the demonstration of absence of direct infection of the central nervous system, of head trauma, fat embolism, and of drugs side effects.
In nonsedated patients mental status is straightforwardly assessed by the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU), and delirium is likely when there is an acute onset of changes or fluctuations in the course of mental status, and inattention, and either disorganized thinking or an altered level of consciousness [7]. The Assessment to Intensive Care Environment (ATICE) score, which enables assessment of awakening, comprehension, and calmness, may indicate delirium when the score is less than 10 (maximum value being 20) [8], and the GCS allows determination of the depth of coma. Evaluation of timing of occurrence of symptoms is part of the CAM-ICU scale and not of ATICE and GCS. The ATICE score relies heavily on assessment of eye-opening to a variety of stimuli whereas the CAM-ICU and the GCS use a broader range of responses in determining the presence of delirium or coma. The assessment of comprehension by ATICE or GCS relies on responses to simple verbal orders whereas the CAM-ICU score explores in more detail attention and organization in thinking. The ATICE score may provide a better evaluation of patient's tolerance to his environment, particularly synchrony with the ventilator.
In sedated patients daily interruption of sedation may facilitate assessment of mental status [9]. However, discontinuation of sedative agents does not always result in successful awakening of the patient, and in many cases awakening is accompanied by agitation. In these situations the persistence of coma or occurrence of agitation cannot be unequivocally ascribed to sepsis and may be due to the accumulation of or withdrawal from sedatives. Moreover, clinicians may not discontinue sedation in severely ill patients to prevent self-extubation or poor patient-ventilator synchronization [10]. In these patients a clinical approach may still be useful, particularly in the evaluation of brainstem responses. If the clinical examination is unrevealing, the diagnosis of SAD should rely on laboratory investigations, which may include EEG and sensory evoked potentials (SEP). However, sedatives may interfere with the interpretation of signal abnormalities on the EEG. Although SEP are not affected by continuous sedation, evaluation of SEP may be too cumbersome to be used routinely in the ICU [5]. Serum levels of neuron-specific enolase (NSE) and S-100 β-protein, markers of brain injury, have been shown to be correlated with poor outcome in septic shock [11]. In the future our comprehension and the diagnosis of SAD may improve with advances in neuroimaging. Spectroscopic magnetic resonance imaging (MRI), positron emission tomography, and functional MRI may permit quantification of alterations in brain metabolism and function, but these modalities are not widely available and remain to be rigorously studied in the ICU setting.
Approach to the diagnosis of sepsis-associated delirium
Neurological examination
Neurological examination should systematically assess neck stiffness, motor responses, motor movements (asterixis, tremor, or myoclonus), muscular tonus (paratonic rigidity), deep tendon reflexes, and plantar reflex, and cranial nerves, including eye position and movement, pupillary size, blinking to strong light, light response, corneal reflex, grimacing to painful stimulation, oculocephalic response, and cough reflex. This examination allows the detection of focal lesions, as illustrated for oculomotor function in Figs. 2 and 3.

Common abnormal eye positions in unconscious patients. a Normal eye position. Pupillary size and papillary light response must be assessed. b Horizontal conjugate deviation indicates hemispheric lesions. The gaze is directed toward the lesion. Lesions in the pons below the oculomotor nuclei and thalamus damage may produce tonic deviation away from the lesion. Differential diagnosis includes seizures. c Upward deviation indicates bilateral hemispheric damage, such as that seen after extensive hypoxic-ischemic insult, after cardiac resuscitation or asphyxia. d Downward eye deviation indicates lesions to the thalamus or to the dorsal midbrain, often caused by a massive thalamic hemorrhage extending in the mesencephalon. e Skew deviation in the resting position is indicative of primary brainstem lesion, possibly in the region of the interstitial nucleus of Cajal. The higher eye often corresponds to site of damaged midbrain or pons. f Caloric stimulation with ice water, while the head is 30° upright, stimulates horizontal canals and produces a tonic deviation toward the ear, but it may also reveal adduction paralysis (internuclear ophtalmoplegia). (Adapted from [22])

Common eye movement abnormalities. Spontaneous eye movements rarely have localizing value and are most commonly seen in hypoxic-ischemic coma. a Roving, without particular significance. b Periodic alternating gaze: predominantly in hepatic encephalopathy, bilateral midbrain or vermis lesions. c Ping-pong: bihemispheric alterations. d Convergence nystagmus: lesions in the mesencephalon. e Bobbing: rapid downward conjugate movement with slower return to baseline position is found mostly in pontine lesions (primary due to compressions from lesions in the cerebellum). f Dipping: corresponds to bihemispheric damage, often of hypoxic-ischemic nature. (Adapted from [22])
Drug toxicity
Various drugs commonly used in critically ill patients may induce brain dysfunction and should be discontinued as soon as possible, and their doses should be adjusted in the presence of impaired hepatic or renal function.
Biochemical variables
Standard biochemical testing is necessary for ruling out metabolic abnormalities. As mentioned above, plasma NSE and S-100 β-protein levels can be measured in a delirious patients. Neuroimaging is suggested to rule out structural lesions. Cerebrospinal fluid analysis is usually normal. Lumbar puncture should be performed only when meningitis is suspected.
Electrophysiology testing
The EEG may be normal or may show excessive theta, predominantly delta, triphasic waves or burst suppression, according to the severity of SAD [1]. Based on our practice, we recommend that in sedated patients EEG be performed systematically whenever abnormal movements occur, or if delirium is suspected to rule out subtle epilepsy. SEP may show a prolongation of subcortical and cortical pathways but cannot be considered as a routine investigation in ICU [4, 5].
Brain imaging
Although often more difficult to obtain than computed tomography, brain MRI provides better and often earlier information on brain damage, especially of the white matter and blood-brain barrier. Common findings include ischemic or hemorrhagic lesions in about 10% of patients, and lesions of leukoencephalopathy surrounding Virshow–Robin spaces. These lesions, ranging from small multiple areas to diffuse lesions and characterized by hyperintensity on fluid-attenuated inversion recovery images, indicate blood-brain barrier breakdown [12] (Fig. 4). Damage to the gray matter may include bilateral lesions of basal ganglia and thalami [13]. Neuroimaging may still underestimate brain injuries as suggested by postmortem studies showing ischemic lesions in all cases, hemorrhage in 26%, microthrombi in 9%, microabscesses in 9%, and leukoencephalopathy in 9% [14] (Fig. 5). Optimal timing of brain MRI in septic shock remains unknown. We suggest that brain MRI be performed whenever focal neurological signs or alteration in mental status occur, particularly if plasma NSE and S-100 β-protein levels are increased. Knowing the nature and extent of brain lesions may result in therapeutic interventions, for example, anticoagulant therapy, or may contribute to decisions to withhold or withdraw life-support treatments.

Day 3 (left) and day 30 (right) magnetic resonance imaging of the brain in a 79-year-old woman with Streptococcus pneumoniae lung infection complicated by septic shock and acute respiratory distress syndrome. Multiple bright signal areas visible bilaterally at the level of the centrum semiovale indicate leukoencephalopathy

Neuropathological changes in patients who died from septic shock. a Large leptomeningeal hemorrhage adjacent to the right scissure of Sylvius. b Recent petechial hemorrhage in the right nucleus paraventricularis; H&E, × 100. c Fibrinous microthrombi from disseminated intravascular coagulation; H&E, × 200. d Nonbacterial thrombotic endocarditis, gross appearance of the heart. e Distal fibrinocruoric emboli in small leptomeningeal arteries; recent ischemia in the underlying cortex; H&E, × 40. f Septic emboli within necrotic area; H&E, × 40. g Multifocal necrotizing leukoencephalopathy; horizontal section of the upper pons; Luxol fast blue/cresyl violet. h Multifocal necrotizing leukoencephalopathy, recent necrotic changes in the transverse pontine fibers; H&E, × 60. (Adapted from [15] with permission)
Mechanisms of sepsis-associated delirium
Most studies investigating potential mechanisms of SAD have used animals or cell cultures. Although these studies may have expanded our understanding of central nervous system cells' response to endotoxin or cytokines, the way in which these mechanisms relate to clinical brain injury remains obscure.
It is important to note that homeostasis requires a balanced interaction between the central nervous system and the immune system. The brain mediates via the autonomic nervous system and neurohormones the growth and proliferation of most if not all tissues involved in immunity, and all immune cells have membrane or cytosolic receptors for a number of neuromediators [15]. The systemic inflammatory response to infection results in brain activation, which subsequently generates appropriate anti-inflammatory response. However, excess in proinflammatory mediators entering the brain can cause cerebral damage [14]. In turn, dysfunction of the autonomic nervous and neuroendocrine systems may alter immunity in a vicious circle resulting in metabolic derangements and organ failure [15, 16].
Brain signaling during sepsis
Two major pathways allow neuroimmune communications (a) circumventricular organs (CVOs) located in the midline ventricular system, and (2) the vagus nerve. CVOs lack a blood-brain barrier. Thus, they permit a direct communication between brain and blood stream. Some CVOs are located in the vicinity of neuroendocrine structures (e.g., organum subfornicale, organum subcommisurale, corpus pineale, neurohypohysis and organum vasculosum laminae terminalis), and others are located close to brainstem autonomic centers (i.e., area postrema) [17]. Thus blood-borne cytokines enter the brain through these areas, which express receptors for cytokines and bacterial fragments during systemic inflammation. In addition, specific carriers for cytokines allow blood-borne cytokines to reach hypothalamic nuclei inside the blood-brain barrier [17]. Circulating interleukin-1β activates afferent vagal fibers terminating in the nucleus tractus solitarius, with subsequent stimulation of the hypothalamic-pituitary-adrenal axis [18]. Efferent activity in the vagus nerve, termed “cholinergic anti-inflammatory pathway”, releases acetylcholine in the vicinity of macrophages in the reticuloendothelial system and leads to cellular deactivation and inhibition of cytokine release [19, 20]. The sympathetic and parasympathetic systems are thought to work hand-in-hand to modulate inflammatory responses.
Inflammation of the brain
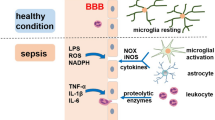
Endothelial activation and blood-brain barrier breakdown
Diffuse endothelial activation induced by sepsis may result in blood-brain barrier breakdown [21], allowing circulating mediators to enter into the brain. Microbial products and pro-inflammatory cytokines induce expression of CD40 and adhesion molecules on endothelial cells from the human brain [22–24]. Through nuclear factor κB activation overexpression of inducible nitric oxide synthase (iNOS) further increases blood-brain barrier permeability [25].
Brain expression of inflammatory mediators
Components of innate and adaptive immune systems are expressed in the brain during lipopolysaccharide (LPS) challenge [26]. They are first expressed within the CVOs and then spread to deeper areas of the brain controlling neuroendocrine and autonomic functions. Toll-like receptors (TLR) 4, 2, and 9 are expressed by glial cells, such as microglia, astrocytes and oligodendrocytes at rest and after exposure to LPS [27]. Whether TLRs are expressed in neurons remains controversial. TLR4 function in resident cells may be important for sustained brain-specific inflammation during endotoxemia [26].
Endotoxin also upregulates the iNOS in the brain, causing accumulation of nitric oxide which behaves as a neurotoxic effector [28]. Prostaglandins and purines, such as ATP and adenosine, are further key mediators in brain response to sepsis, particularly inducing fever. Following LPS stimulation astrocytes and microglia release a significant amount of prostaglandin E [29], and microglia expresses prostaglandin receptors and cyclo-oxygenase 2 [30]. A number of other mediators may be involved in the cerebral immune response, such as macrophage migrating inhibitory factor, macrophage inflammatory protein, endothelin 1, angiotensin II, platelet-activating factor, superoxide radicals, and carbon monoxide.
Ischemic lesions and cerebral flux alterations
Inadequate perfusion pressure and disturbances of the cerebral microcirculation may play a role in the development of SAD. Studies of cerebral blood flow, endothelial reactivity, and oxygen consumption show impaired [31] or preserved cerebral perfusion in sepsis [32]. Ischemic events likely contribute to the development of SAD. Endotoxin aggravated hypoxic-ischemic insult in a dose-dependent manner in neonatal rats [33]. In septic shock postmortem studies show that ischemic lesions in parts of the brain susceptible to ischemia are always present [14]. In our unpublished work in 330 patients with septic shock systematic screening for cerebrovascular events included computed tomography or MRI guided by careful neurological examination prior to ICU discharge and showed ischemic and hemorrhagic lesions in 7% and 10% of cases, respectively.
Dysfunction of intracellular metabolism and apoptosis
Exposure to LPS may cause apoptosis of brain cells via dysfunction of mitochondrial respiration [34], activation of mitogen-activated protein kinase and nuclear factor κB pathways [35], and release or accumulation of calcium and reactive oxygen species [36]. Nevertheless, during exposure to LPS the 70-kDa heat shock proteins may prevent iNOS triggered cells premature death [37]. In septic shock postmortem studies show iNOS induced neuron and glial apoptosis, mainly in cardiovascular autonomic centers and the hippocampus [38]. Cell culture experiments indicate an important role for microglial cells. These cells play important role in immune surveillance, host defense, and tissue repair by clearing apoptotic cells and downregulating cytokines and encephalitogenicT-lymphocytes [39]. In coculture the presence of microglial cells attenuated LPS-induced neuronal apoptosis.
Neurohormones and neurotransmitters deregulation
During systemic inflammation NO, cytokines, and prostaglandins modulate brain neurotransmission, especially β-adrenergic system, GABAergic synapses, central muscarinic cholinergic regulation, corticotrophin releasing factor, adrenocorticotrophin hormone, vasopressin synthesis, and medullary autonomic center output [40–43]. Neurotoxic substances such as ammonium, or amino acids may also be involved [3]. In sepsis, plasma and CSF levels of tyrosine, tryptophan, and phenylalanine which impair neurotransmitters synthesis are increased following muscle proteolysis and reduced hepatic clearance. Prolonged LPS exposure impaired synaptic transmission and neuronal excitability of pyramid neurons in the hippocampus, involved in behavioral and emotional systems [44]. In rats the brain may become tolerant to endotoxin in a brain region dependent manner, suggesting plastic modulation of brain functions [45, 46]. Finally, neurotransmitters and neurohormones modulate cerebral expression of inflammatory mediators [15]. The final behavioral, neuroendocrine, and autonomic responses are therefore variable because they depend on a complex and spatiotemporally organized processes that involve both stimulatory and inhibitory factors, which themselves depend on activation of brain cells. Various neuroendocrine dysfunctions have been observed in septic shock, for example, impairment of hypothalamic-pituitary-adrenal axis, vasopressin deficiency [15, 38], and autonomic dysfunction [16, 38].
Outcome
Mortality related to SAD varies from 16% to 63% according to the intensity of brain dysfunction [3]. Although it has been shown that SAD is an independent prognostic factor [3], it remains unclear whether it causes death or reflects severity of illness. Persistent or relapsing delirium often indicates uncontrolled sepsis. SAD may be reversible with recovery from sepsis or may result in long-term cognitive disturbances [47].
Treatment of sepsis-associated delirium
Currently there is no specific treatment for SAD. Thus, treatment should focus on the underlying systemic illness and supportive measures. Nevertheless, when ischemic lesions are diagnosed, anticoagulant should be considered. Alternately, evidence of brain hemorrhage should prompt interruption of any molecule with anticoagulant activity. The demonstration of severe brain damage and the likelihood that they are irreversible (e.g., leukoencephalopathy or extensive brain ischemia) may inform decisions to withhold or withdraw further intensive care.
There is some evidence of an important role of iNOS in SAD. Preclinical findings suggest that inhibition of NOS may prevent LPS-induced neuronal apoptosis [48], but it had no effect on consciousness in rats with cecal ligature and puncture induced peritonitis [49]. A clinical trial found nonselective NOS inhibition to be associated with an increase in cardiovascular deaths [50]. Given the potential role of neuronal apoptosis in the development of SAD, strategies to prevent premature cell death may be relevant. However, no clinical or experimental data are available. Various pathways are known to lead to glial cell activation and subsequent upregulation of proinflammatory mediators and cell death. Several drugs may interact with these pathways (Table 2).
Manipulating the endothelium and blood-brain barrier may be a therapeutic option. In LPS-treated mice serum amyloid P component administration lowered blood-brain barrier permeability [51]. In the CLP model, treatment with magnesium reduced blood-brain barrier permeability and brain edema [52]. In addition, with a pretreatment with nonlethal dose of LPS reduced leukocytes recruitment and attenuated brain inflammation in response to further injuries [53]. Finally, numerous antioxidants have shown neuroprotective effects in endotoxin models [54, 55].
Conclusion
Brain dysfunction is a frequent complication of sepsis and contributes to morbidity and mortality. The mechanisms of delirium are complex and may involve reciprocal immune-brain signaling resulting in prolonged inflammation, brain cells activation, overexpression of NO, dysfunction of intracellular metabolism and cell death. The diagnostic approach relies on neurological examination, which then guides laboratory investigations. At this time there is no specific treatment for SAD. The better understanding of bidirectional pathways of immune-brain signaling may help developing future specific treatments. It is paramount that future researches determine (a) the exact prevalence and timing of occurrence of sepsis induced brain damage, (b) the diagnostic accuracy of biochemical markers, and (c) the impact of recognizing sepsis-associated delirium on therapeutic decisions and prognosis.
References
Young GB, Bolton CF, Archibald YM, Austin TW, Wells GA (1992) The electroencephalogram in sepsis-associated encephalopathy. J Clin Neurophysiol 9:145–152
Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB (1990) Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med 18:801–806
Eidelman LA, Putterman D, Putterman C, Sprung CL (1996) The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA 275:470–473
Zauner C, Gendo A, Kramer L, Kranz A, Grimm G, Madl C (2000) Metabolic encephalopathy in critically ill patients suffering from septic or nonseptic multiple organ failure. Crit Care Med 28:1310–1315
Zauner C, Gendo A, Kramer L, Funk GC, Bauer E, Schenk P, Ratheiser K, Madl C (2002) Impaired subcortical and cortical sensory evoked potential pathways in septic patients. Crit Care Med 30:1136–1139
Straver JS, Keunen RW, Stam CJ, Tavy DL, de Ruiter GR, Smith SJ, Thijs LG, Schellens RG, Gielen G (1998) Nonlinear analysis of EEG in septic encephalopathy. Neurol Res 20:100–106
Ely EW, Truman B, Shintani A, Thomason JW, Wheeler AP, Gordon S, Francis J, Speroff T, Gautam S, Margolin R, Sessler CN, Dittus RS, Bernard GR (2003) Monitoring sedation status over time in ICU patients: reliability and validity of the Richmond Agitation-Sedation Scale (RASS). JAMA 289:2983–2991
De Jonghe B, Cook D, Griffith L, Appere-de-Vecchi C, Guyatt G, Theron V, Vagnerre A, Outin H (2003) Adaptation to the Intensive Care Environment (ATICE): development and validation of a new sedation assessment instrument. Crit Care Med 31:2344–2354
Kress JP, Pohlman AS, O'Connor MF, Hall JB (2000) Daily interruption of sedative infusions in critically ill patients undergoing mechanical ventilation. N Engl J Med 342:1471–1477
Schweickert WD, Gehlbach BK, Pohlman AS, Hall JB, Kress JP (2004) Daily interruption of sedative infusions and complications of critical illness in mechanically ventilated patients. Crit Care Med 32:1272–1276
Nguyen DN, Spapen H, Su F, Schiettecatte J, Shi L, Hachimi-Idrissi S, Huyghens L (2006) Elevated serum levels of S-100beta protein and neuron-specific enolase are associated with brain injury in patients with severe sepsis and septic shock. Crit Care Med 34:1967–1974
Sharshar T, Carlier RY, Bernard F, Guidoux C, Brouland JP, Nardi O, Lorin de la Grandmaison G, Aboab J, Gray F, Menon DK, Annane D (2007) Brain lesions in septic shock—an MRI study. Intensive Care Med (submitted)
Finelli PF, Uphoff DF (2004) Magnetic resonance imaging abnormalities with septic encephalopathy. J Neurol Neurosurg Psychiatry 75:1189–1191
Sharshar T, Annane D, Lorin de la Grandmaison G, Brouland JP, Hopkinson NS, Francoise G (2004) The neuropathology of septic shock. Brain Pathol 14:21–33
Chrousos GP (1995) The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 332:1351–1362
Annane D, Trabold F, Sharshar T, Jarrin I, Blanc AS, Raphael JC, Gajdos P (1999) Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am J Respir Crit Care Med 160:458–465
Roth J, Harre EM, Rummel C, Gerstberger R, Hubschle T (2004) Signaling the brain in systemic inflammation: role of sensory circumventricular organs. Front Biosci 9:290–300
Maier SF, Goehler LE, Fleshner M, Watkins LR (1998) The role of the vagus nerve in cytokine-to-brain communication. Ann N Y Acad Sci 840:289–300
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin, Nature 405:458–462
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–388
Papadopoulos MC, Lamb FJ, Moss RF, Davies DC, Tighe D, Bennett ED (1999) Faecal peritonitis causes oedema and neuronal injury in pig cerebral cortex. Clin Sci (Lond) 96:461–466
Omari KM, Dorovini-Zis K (2003) CD40 expressed by human brain endothelial cells regulates CD4+ T cell adhesion to endothelium. J Neuroimmunol 134:166–178
Hess DC, Bhutwala T, Sheppard JC, Zhao W, Smith J (1994) ICAM-1 expression on human brain microvascular endothelial cells. Neurosci Lett 168:201–204
Hess DC, Thompson Y, Sprinkle A, Carroll J, Smith J (1996) E-selectin expression on human brain microvascular endothelial cells. Neurosci Lett 213:37–40
Mayhan WG (1998) Effect of lipopolysaccharide on the permeability and reactivity of the cerebral microcirculation: role of inducible nitric oxide synthase. Brain Res 792:353–357
Chakravarty S, Herkenham M (2005) Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci 25:1788–1796
Laflamme N, Rivest S (2001) Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J 15:155–163
Duport S, Garthwaite J (2005) Pathological consequences of inducible nitric oxide synthase expression in hippocampal slice cultures. Neuroscience 135:1155–1166
Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y (2005) Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem 94:1546–1558
Caggiano AO, Kraig RP (1999) Prostaglandin E receptor subtypes in cultured rat microglia and their role in reducing lipopolysaccharide-induced interleukin-1beta production. J Neurochem 72:565–575
Terborg C, Schummer W, Albrecht M, Reinhart K, Weiller C, Rother J (2001) Dysfunction of vasomotor reactivity in severe sepsis and septic shock. Intensive Care Med 27:1231–1234
Pollard V, Prough DS, Deyo DJ, Conroy B, Uchida T, Daye A, Traber LD, Traber DL (1997) Cerebral blood flow during experimental endotoxemia in volunteers. Crit Care Med 25:1700–1706
Yang L, Sameshima H, Ikeda T, Ikenoue T (2004) Lipopolysaccharide administration enhances hypoxic-ischemic brain damage in newborn rats. J Obstet Gynaecol Res 30:142–147
Chuang YC, Tsai JL, Chang AY, Chan JY, Liou CW, Chan SH (2002) Dysfunction of the mitochondrial respiratory chain in the rostral ventrolateral medulla during experimental endotoxemia in the rat. J Biomed Sci 9:542–548
Lohrer P, Gloddek J, Nagashima AC, Korali Z, Hopfner U, Pereda MP, Arzt E, Stalla GK, Renner U (2000) Lipopolysaccharide directly stimulates the intrapituitary interleukin-6 production by folliculostellate cells via specific receptors and the p38alpha mitogen-activated protein kinase/nuclear factor-kappaB pathway. Endocrinology 141:4457–4465
Wang T, Qin L, Liu B, Liu Y, Wilson B, Eling TE, Langenbach R, Taniura S, Hong JS (2004) Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J Neurochem 88:939–947
Li FC, Chan JY, Chan SH, Chang AY (2005) In the rostral ventrolateral medulla, the 70-kDa heat shock protein (HSP70), but not HSP90, confers neuroprotection against fatal endotoxemia via augmentation of nitric-oxide synthase I (NOS I)/protein kinase G signaling pathway and inhibition of NOS II/peroxynitrite cascade. Mol Pharmacol 68:179–192
Sharshar T, Gray F, Lorin de la Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, Orlikowski D, Raphael JC, Gajdos P, Annane D (2003) Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 362:1799–1805
Magnus T, Chan A, Grauer O, Toyka KV, Gold R (2001) Microglial phagocytosis of apoptotic inflammatory T cells leads to down-regulation of microglial immune activation. J Immunol 167:5004–5010
Kadoi Y, Saito S, Kunimoto F, Imai T, Fujita T (1996) Impairment of the brain beta-adrenergic system during experimental endotoxemia. J Surg Res 61:496–502
Kadoi Y, Saito S (1996) An alteration in the gamma-aminobutyric acid receptor system in experimentally induced septic shock in rats. Crit Care Med 24:298–305
Pavlov VA, Ochani M, Gallowitsch-Puerta M, Ochani K, Huston JM, Czura CJ, Al Abed Y, Tracey KJ (2006) Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci USA 103:5219–5223
Vallieres L, Rivest S (1999) Interleukin-6 is a needed proinflammatory cytokine in the prolonged neural activity and transcriptional activation of corticotropin-releasing factor during endotoxemia. Endocrinology 140:3890–3903
Hellstrom IC, Danik M, Luheshi GN, Williams S (2005) Chronic LPS exposure produces changes in intrinsic membrane properties and a sustained IL-beta-dependent increase in GABAergic inhibition in hippocampal CA1 pyramidal neurons. Hippocampus 15:656–664
Valles A, Marti O, Harbuz MS, Armario A (2002) A single lipopolysaccharide administration is sufficient to induce a long-term desensitization of the hypothalamic-pituitary-adrenal axis. Neuroscience 112:383–389
Valles A, Marti O, Armario A (2005) Mapping the areas sensitive to long-term endotoxin tolerance in the rat brain: a c-fos mRNA study. J Neurochem 93:1177–1188
Hopkins RO, Jackson JC (2006) Long-term neurocognitive function after critical illness. Chest 130:869–878
Wang H, Wu YB, Du XH (2005) Effect of dexamethasone on nitric oxide synthase and Caspase-3 gene expressions in endotoxemia in neonate rat brain. Biomed Environ Sci 18:181–186
Kadoi Y, Goto F (2004) Selective inducible nitric oxide inhibition can restore hemodynamics, but does not improve neurological dysfunction in experimentally-induced septic shock in rats. Anesth Analg 99:212–220
Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, Silverman MS, Takala J, Donaldson J, Arneson C, Grove G, Grossman S, Grover R (2004) Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med 32:21–30
Veszelka S, Urbanyi Z, Pazmany T, Nemeth L, Obal I, Dung NT, Abraham CS, Szabo G, Deli MA (2003) Human serum amyloid P component attenuates the bacterial lipopolysaccharide-induced increase in blood-brain barrier permeability in mice. Neurosci Lett 352:57–60
Esen F, Erdem T, Aktan D, Orhan M, Kaya M, Eraksoy H, Cakar N, Telci L (2005) Effect of magnesium sulfate administration on blood-brain barrier in a rat model of intraperitoneal sepsis: a randomized controlled experimental study. Crit Care 9:R18–R23
Davis AE, Campbell SJ, Wilainam P, Anthony DC (2005) Post-conditioning with lipopolysaccharide reduces the inflammatory infiltrate to the injured brain and spinal cord: a potential neuroprotective treatment. Eur J Neurosci 22:2441–2450
Abd El-Gawad HM, Khalifa AE (2001) Quercetin, coenzyme Q10, and L-canavanine as protective agents against lipid peroxidation and nitric oxide generation in endotoxin-induced shock in rat brain. Pharmacol Res 43:257–263
Bi XL, Yang JY, Dong YX, Wang JM, Cui YH, Ikeshima T, Zhao YQ, Wu CF (2005) Resveratrol inhibits nitric oxide and TNF-alpha production by lipopolysaccharide-activated microglia. Int Immunopharmacol 5:185–193
Young GB, Bolton CF, Austin TW, Archibald YM, Gonder J, Wells GA (1990) The encephalopathy associated with septic illness. Clin Invest Med 13:297–304
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ebersoldt, M., Sharshar, T. & Annane, D. Sepsis-associated delirium. Intensive Care Med 33, 941–950 (2007). https://doi.org/10.1007/s00134-007-0622-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-007-0622-2