
Abstract
Cancer survivors often relapse due to evolving drug-resistant clones and repopulating tumor stem cells. Our preclinical study demonstrated that terminal cancer patient’s lymphocytes can be converted from tolerant bystanders in vivo into effective cytotoxic T-lymphocytes in vitro killing patient’s own tumor cells containing drug-resistant clones and tumor stem cells. We designed a clinical trial combining peginterferon α-2b with imatinib for treatment of stage III/IV gastrointestinal stromal tumor (GIST) with the rational that peginterferon α-2b serves as danger signals to promote antitumor immunity while imatinib’s effective tumor killing undermines tumor-induced tolerance and supply tumor-specific antigens in vivo without leukopenia, thus allowing for proper dendritic cell and cytotoxic T-lymphocyte differentiation toward Th1 response. Interim analysis of eight patients demonstrated significant induction of IFN-γ-producing-CD8+, -CD4+, -NK cell, and IFN-γ-producing-tumor-infiltrating-lymphocytes, signifying significant Th1 response and NK cell activation. After a median follow-up of 3.6 years, complete response (CR) + partial response (PR) = 100%, overall survival = 100%, one patient died of unrelated illness while in remission, six of seven evaluable patients are either in continuing PR/CR (5 patients) or have progression-free survival (PFS, 1 patient) exceeding the upper limit of the 95% confidence level of the genotype-specific-PFS of the phase III imatinib-monotherapy (CALGB150105/SWOGS0033), demonstrating highly promising clinical outcomes. The current trial is closed in preparation for a larger future trial. We conclude that combination of targeted therapy and immunotherapy is safe and induced significant Th1 response and NK cell activation and demonstrated highly promising clinical efficacy in GIST, thus warranting development in other tumor types.
Similar content being viewed by others


Introduction
Despite effective treatments achieving remission, cancer survivors often relapse, after which interventions become largely unsuccessful. One culprit is drug-resistant clones—pre-existing and evolving continuously—and another is tumor stem cells—repopulating, resilient, and poorly understood. With their unique features, once outgrowth has occurred, both culprits can evade standard therapies and prevail. The innate and adaptive immunity have been shown to play important roles in protecting the host through tumor immunosurveillance [1–6]. Unfortunately, mechanisms of tumor-induced tolerance enable tumors to escape immunosurveillance [7]. However, the delicate balance can be restored if we can design novel treatment that can break tolerance while promote innate and adaptive antitumor immunity.
Dendritic cells (DCs) capture, process, and cross-present antigens in the context of MHC class-I and costimulatory molecules to subsets of T-lymphocytes, and play critical roles in the regulation and development of distinct immune responses [8–10] including (1) Th1 adaptive cell-mediated immunity (Th1 response) signified by interferon-γ (IFN-γ) secretion and play major roles in protection against pathogens and tumors [1–6], (2) Th2, (3) Th17, and (4) T-regulatory responses (tolerance).
IFN-α is a type-1 IFN and a physiological danger signal [11, 12] can upregulate expression of MHC class-I molecules and costimulatory molecules on DCs, activate innate immunity, modulate DCs, promote Th1 response, help clonal expansion/survival and memory differentiation of T-lymphocytes [9–13], and has been shown to be an effective vaccine adjuvant in animal models [14] and clinical trials [15, 16]. The immunological consequences of tumor cell death induced by individual chemotherapy agents [17] and the subsequent differentiation of DCs and T-lymphocytes in the ensuing 2-week span are of pivotal importance in influencing the development toward the distinct immune responses (Th1, Th2, Th17, or tolerance). Cytotoxic chemotherapy often results in prolonged severe leukopenia depriving DCs of proper maturation (differentiation), thus often resulting in a tolerant/dysfunctional immune state.
With recognition of the IFN-α qualities [9–14], the pivotal role of DCs in the development of distinct immune responses [8–10] and support from our preclinical data, we hypothesize that (1) combining IFN-α with effective non-marrow-suppressive antitumor agent(s) could induce innate immunity and Th1 response; (2) the antitumor immunity can help eradicating tumor cells including the drug-resistant clones and tumor stem cells upfront thus improve response rate; (3) most importantly, antitumor immunity can monitor continuously and eradicate the various continuing-evolving drug-resistant clones and the resilient tumor stem cells when they first emerge at the cellular/subclinical level prior to outgrowth, and this would delay/prevent relapse, ultimately leading to the improvements in progression-free survival (PFS) and overall survival (OS).
To test the hypothesis, we designed a new strategy aiming at developing innate immunity and Th1 response concomitant with partial response (PR) or complete response (CR) achieved by effective non-marrow-suppressive drug therapy. Gastrointestinal stromal tumor (GIST), a sarcoma with incidence of 5000/year in US, represents an excellent model to test our hypothesis for the following reasons. First, imatinib mesylate (IM, Gleevec®, Glivec®) [18], a selective inhibitor of ABL, KIT, PDGFRA/B, is highly effective, induces swift apoptosis/necrosis of GIST within 3–7 days [19], and is non-marrow-suppressive, allowing proper DC and cytotoxic T-lymphocyte differentiation toward Th1 response. Second, GIST cell alterations include cancer/testis antigens [20], tumor-antigens created by activating mutations in KIT (c-kit) or PDGFRA [21–24] and new mutation(s) responsible for IM resistance [25–27]. Third, IM-monotherapy trials in GIST patients have reported response rates (PR + CR) of 54% [28], 52% [29, 30], and 48% [22, 30]. The median PFS remains ≤ 2 years [22, 29, 30] mainly due to the development of IM resistance [25–27]. Discontinuing IM resulted in high rate of relapse due to repopulating stem cells [31]. Thus, better therapies for GIST are needed.
IM was reported to induce DC-mediated natural killer (NK) cell IFN-γ production [32, 33] and potentiate adaptive immunity through IM-off-target inhibition of KIT on DCs [34] and inhibition of Ido [35]; both IM-off-target immunological anti-GIST effects plus IM-inhibition of KIT/PDGFRA signaling contribute to the IM-monotherapy efficacy [22, 28–30] as described above and is less than satisfactory. We intend to bring out the full potential of anti-GIST immunity by a new strategy of combining peginterferon α-2b (PegIFNa2b, Peg-Intron®) [36] with IM and have demonstrated significant Th1 response, innate immunity, and highly promising clinical outcome comparing to IM-monotherapy [22], strongly support all three parts of our hypothesis.
Materials and methods
Preclinical study
Specimens were collected under MD Anderson Institutional Review Board (IRB) protocols LAB_00143. Primary tumor cells were isolated after digesting fresh tumor with collagenase. The chimeric SYN-SSX was sequenced [37]. Peripheral blood mononuclear cell (PBMC)-derived DCs were isolated by plastic adherence and culture supplemented with GM-CSF and IL-4. Cytokine cocktail consisted of TNF-α (R&D), IL-1β (R&D), IL-6 (R&D), and PGE-2 (Sigma) [38]. IL-12-p70 was analyzed using ELISA (Biosources, Camarillo, CA.) and read with UV-900 microplate reader (Bio-Tek Instruments, Winooski, VT). The plastic non-adherent cells were used to positively select CD8+ T-lymphocytes using anti-CD8 monoclonal antibody (mAb) coupled to magnetic microbeads (Miltenyi Biotec, Auburn, CA).
IFN-γ-enzyme-linked immunosorbent spot (IFN-γ-ELISPOT) assay
CD8+ T-lymphocytes were cultured in AIM-V medium supplemented with IL-2 and IL-7 and stimulated with various antigen preparations twice, total 14 days, to generate CTLs. The 96-well ELISPOT plate (Millipore, Billerica, MA) was precoated with anti-IFN-γ antibody, incubated at 4°C overnight, plated with CD8+ T-lymphocytes at 2 × 105 cells/well, and stimulated with 4 × 104 irradiated primary tumor cells for 40 h at 37°C. Biotinylated IFN-γ antibody was added, followed by streptavidin peroxidase. IFN-γ spots were counted using an ELISPOT reader.
51Cr-release assay
Cryopreserved primary tumor cells were used as targets and K562 cells as control. We labeled 2 × 106 target cells with 100 μCi of Na 512 CrO4 (ICN Biomedicals, Irvine, CA) and distributed 3,000 target cells in each well. Blocking experiments were performed using anti-HLA-A.B.C antibody and isotype control (Dako, Carpinteria, CA).
Clinical trial
Refer to "Results".
Genotyping
As described previously [23].
IFN-γ-flow cytometry
PBMCs were cultured with phorbol ester PMA (5 ng/ml) plus ionomycin (745 ng/ml) for 1 h, add brefeldin A (5 mcg/ml) and cultured for additional 4 h. After surface staining with CD4-PerCP, CD8-APC, or CD3-FITC (BD Biosciences), cells were fixed and stained with anti-human IFN-γ-PE (BioLegend, San Diego, CA) [39]. Data were acquired on a FACSCalibur flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star Inc., Ashland).
Immunohistochemical analysis and confocal microscopy
Antigen retrieval with preheated EDTA/Tris buffer pH 9.0 for CD8, CD56, and CD4; and citrate Buffer pH 6.0 for IFN-γ, FasL, and granzyme B. Antibodies included CD4, CD8, CD56 (Dako, Carpinteria, CA); IFN-γ (Abcam, Cambridge, MA); and FasL and granzyme B (Novus Biologicals, Littleton, CO), goat anti-rabbit IgG antibody conjugated with Texas Red, and goat anti-mouse IgG conjugated with Alexa Fluor 488 (Novus Biologicals). Images were acquired using Fluo View software on an Olympus FV1000 confocal laser scanning microscope.
Results
Pre-clinical study
Cancer patient’s anergic T-lymphocytes in vivo can be converted into effective cytotoxic T-lymphocytes capable of killing patient’s own primary tumor cells in vitro
A stage III synovial sarcoma patient (HLA-A24/A29) received standard neoadjuvant chemotherapy (doxorubicin and ifosfamide), achieved PR, and underwent surgery to achieve disease-free status. The patient later relapsed with wild spread metastasis and rapid progression failing all systematic treatment; leukapheresis was performed to collect PBMC. At the time of surgery, the post-neoadjuvant chemotherapy residual tumor consisting of resilient tumor stem cells and chemotherapy-resistant cells—as evidenced by clinical relapse and metastasis shortly after surgery along with drug resistance—were cryopreserved to serve as targets for in vitro cytotoxic T-lymphocytes (CTL) analysis as opposed to using cell lines as targets [37]. Two tumor-specific-9-mer-peptides encompassing the joining of SYT-SSX1 (Peptides #1 and 2) (Fig. 1a) exhibit high predicted binding affinity [40] with HLA-A24 class 1 molecule (Fig. 1b). DCs that were matured with a cytokine cocktail (TNF-α, IL-1β, IL-6, and PGE-2) [38] showed significant secretion of IL-12–p70 (P < 0.01, Fig. 1c). Autologous PBMC-derived CD8+ T-lymphocytes were stimulated with four antigen preparations (Fig. 1d, I–IV) twice in vitro to generate CTLs. When antigen-specific peptides #1 & 2 were presented by mature DCs, we demonstrated significant cytotoxicity against primary tumor cells by both IFN-γ–ELISPOT (P < 0.01, Fig. 1d, III & IV) and 51Cr release assays (P < 0.05, Fig. 1e, III & IV). The percent tumor-specific lysis exhibited a dose-dependent relationship with effector-to-target ratio. 51Cr release was completely blocked by neutralizing anti-HLA-A.B.C antibody and cold temperature at 4°C (Fig. 1e, right panel) indicating the essential role of the HLA class-I molecules and specificity of tumor lysis.

CTLs targeted at patient’s own tumor cells. a Chimeric SYT-SSX1 in a HLA-A24/A29 synovial sarcoma. b Predicted binding of two tumor-specific-9-mer-peptides with HLA class-I molecules. c IL-12-p70 secretion by mature autologous DCs. d IFN-γ–ELISPOT assay. e 51Cr-release assay. CTLs that were generated by stimulation with antigen preparations III1, III2, III1+2, IV1, IV2, IV1+2 can induce significant primary tumor cell lysis using IFN-γ-ELISPOT (P < 0.01) and 51Cr-release assay (P < 0.05). The specific lysis of primary tumor cells can be abrogated by neutralizing antibody against HLA-A.B.C loci and at 4°C (e, right panel)
To the best of our knowledge, this is the first report using a patient’s own post-chemotherapy residual drug-resistant primary tumor cells—a source of micrometastasis and recurrence—as targets for CTL assays and provided direct and convincing evidence that a terminal cancer patient’s T-lymphocytes can be converted from tolerant bystanders ignoring the tumor growth in vivo into tumor-specific effective CTLs in vitro (Fig. 1d, e). Our preclinical studies served two purposes. First, these encouraging results help to justify immunotherapy approach to overcome relapse especially when no other treatment option is in sight due to current poor understanding of the culprits causing relapse. Second, our preclinical study methods are readily applicable for testing the development of tumor-specific immunity during clinical trial by collecting PBMC and applying 51Cr release or IFN-γ–ELOSPOT assays using primary tumor cells (if available) as CTL targets. Without further dwelling on in vitro studies, we move toward our ultimate goal of restoring/stimulating antitumor immunity in vivo in cancer patients.
Aiming at developing antitumor immunity in parallel with achieving PR/CR by drug therapy, we initiated a new strategy of combining PegIFNa2b (3rd signal/danger signal) with IM (killing tumor cells to undermine tumor-induced tolerance and provide 1st and 2nd signals—antigen and co-stimulation) in GIST to test our hypothesis.
Clinical study
GIST clinical trial design is summarized in Fig. 2
Combination treatment of PegIFNa2b plus IM-induced significant IFN-γ-producing-lymphocytes
IFN-γ serves critical function in innate, and adaptive cell-mediate immunity [2–6] is tightly regulated and produced predominantly by four cell types only, activated NK, NKT cells, Th1 CD4+, and Th1 CD8+ CTLs [6] and is the signature of Th1 response. Five of the eight patients enrolled consented to donate PBMC. Total IFN-γ-producing-lymphocytes and subgroups of IFN-γ-producing -CD8+, -CD4+, and -CD4−CD8−-lymphocyte (most likely NK cells) were barely detectable before treatment (Fig. 3a, black columns), and they increased significantly to 18 ± 1.3% (P < 0.0004), 32 ± 4.8% (P < 0.003), 15 ± 2.5% (P < 0.006), and 7.3 ± 0.3% (P < 0.0001), respectively (Fig. 3a, red columns) after four HD-PegIFNa2b (3 mcg/kg/week) with continuing IM. IFN-γ–production of individual patients at single cell level by flow cytometry is shown in Fig. 3b. Subgroup analyses for Pt#7 showed that the percentage induction of IFN-γ-producing -CD8+, -CD4+, and -NK cells was 31.57% (Fig. 3b, u), 11.28% (v), and 6.8% (w), respectively―signifies significant induction of Th1 response and NK cell activation; similar pattern was observed in Pt#3 (Fig. 3b, c, d, e). In case of Pt#7, comparing with pretreatment level (Fig. 3b, k), the kinetics of induction of IFN-γ–producing-lymphocytes demonstrated 102-fold induction after 2 HD-PegIFNa2b (l), reached a peak (861-fold) after 4 HD-PegIFNa2b (m), notably, 4 weeks after stopping PegIFNa2b (n) while continuing IM showed rapid decline (22-fold). After four weekly HD-PegIFNa2b, the treatment was switched to 18 maintenance weekly LD-PegIFNa2b (1.5 mcg/kg/week), and we observed that four of five patients’ IFN-γ-producing-lymphocytes gradually fell to non-detectable level suggesting a PegIFNa2b dose-dependent effect. Our results indicate that combining IM plus 4 weekly HD-PegIFNa2b is both necessary and sufficient to induce significant generation of IFN-γ–producing-lymphocytes consisting of CD8+, CD4+ T-lymphocytes, and NK cells in all patients (Fig. 3a, b, b, g, i, m, o).

Clinical trial design

Flow cytometry analysis of IFN-γ-producing-lymphocytes. a Bar graph demonstrating induction of IFN-γ-producing-lymphocytes before (black bars) and after (red bars) combination treatment with IM plus PegIFNa2b. b Flow cytometry at single cell level. IFN-γ-producing-lymphocytes were barely detectable before treatment (a, f, h, k) and were induced significantly (P < 0.003) in total lymphocytes (b, g, i, m), subtype of CD8+ lymphocytes (c, j, u), CD4+ lymphocytes (d, v), and CD8−CD4− cells (most likely NK) (e, w) after IM plus 4HD-PegIFNa2b
Post-combination-treatment residual tumor showed complete remission and nearly all tumor-infiltrating-lymphocytes produce IFN-γ
All eight GIST patients enrolled in the clinical trial were diagnosed with fine needle aspiration (FNA) or biopsy, so no primary GIST cells were available to serve as targets for IFN-γ-ELISPOT or 51Cr release assay to confirm “GIST-specific” cytotoxicity. However, we studied the post-combination-treatment residual tumor from Pt#4 to compared it with the same patient’s pretreatment biopsy sample and three post-IM-monotherapy residual tumors as controls (Fig. 4a, controls #1–3). The post-combination-treatment residual tumor showed pathologic CR, hyaline degeneration, necrosis, and abundant tumor-infiltrating-lymphocytes (TILs) (Fig. 4b, m) consisting of CD8+ (n) and CD4+ T-lymphocytes (p) and CD56+ NK cells (o). Most TILs expressed CD45RO—a memory T-lymphocyte marker (q) with negative isotype control (r). A small fraction of TILs were positive for granzyme B and FasL (v, w); on confocal microscopy, such TILs mostly co-localized with CD56 (Fig. 4c, cc, gg).

Immunohistochemical studies. a TILs of pretreatment biopsy of Pt#4 and three post-IM-monotherapy GISTs (Controls #1–3) showed no IFN-γ production. b Pt#4 post-combination-treatment residual tumor. It showed pathologic CR with extensive hyaline degeneration, necrosis, and numerous TILs (m), consisting of CD8+ (n), CD56+ (o), and CD4+ (p) lymphocytes. Strikingly, nearly all TILs produced IFN-γ in situ (s), and most TILs expressed the memory T-cell marker CD45RO (q). c Confocal microscopy on Pt#4 post-combination-treatment residual tumor
Strikingly, almost all of the TILs actively produce IFN-γ (Fig. 4b, s), in sharp contrast to the totally negative IFN-γ-staining in the pretreatment GIST biopsy and all three post-IM-monotherapy controls (Fig. 4a, c–f). Uninvolved adjacent lymph nodes showed rare IFN-γ-positive cells (u) suggesting that combination treatment may have induced primarily GIST-reactive TILs.
Our translational research results showed significant induction of IFN-γ-producing -CD8+, -CD4+, -NK cell, and IFN-γ-producing TILs—signifies induction of Th1 response and NK cell activation, thus strongly support part one of our hypothesis.
Patients
Eight patients were enrolled (Table 1), four had stage III and four had stage IV GIST with metastasis to liver, lungs, and or peritoneum. Primary tumors (6–16 cm) originated from stomach, small intestine or rectum. The GIST natural history and response to IM treatment have been shown to correlate with histologic features, stage, anatomical site [24], and genotype [21, 22]. Three GISTs harbored KIT exon 11 mutations, one had KIT exon 9 mutation, two had wild-type (WT) KIT and PDGFRA, and two had insufficient material from FNA for genotyping.
Side effects
Pts#1–3 initially received HD-PegIFNa2b at 4 mcg/kg/week, developed grade 3 neutropenia, and requiring dose reduction to 3 mcg/kg/week. Later the protocol was amended and the HD-PegIFNa2b was reduced to 3 mcg/kg/week (Pts#4–8), and we observed occasional grade 1 or grade 2 neutropenia with quick recovery. Two patients (Pt#1 and 8) developed grade 3 skin rash requiring short-term steroid. All patients experienced transient low-grade fever and mild flu-like symptoms as expected.
Response rate
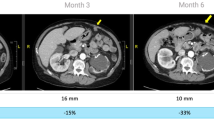
Despite multiple poor prognostic factors, combination treatment of IM plus PegIFNa2b achieved a response rate (PR + CR) of 100% by all three evaluation criteria, RECIST, PET-CT scan criteria [41], and Choi criteria [42, 43], contrasting to the reported GIST IM-monotherapy response rates of 54% [28], 52% [29, 30], and 48% [22, 30]. Pt#2 achieved PR by Choi criteria at week 8 and was not assessable (NA) by PET-CT criteria because the tumor was not FDG–avid or by RECIST, because the tumor was surgically removed 6 weeks after completion of HD-PegIFNa2b when tumor size had reduced by 28% and became amenable to surgery, resulting in insufficient time for RECIST evaluation [44]. In comparison, 13% (49/382) of genotyped GIST patients were categorized as NA by RECIST in the S0033 trial [22]. Pt#4 had a large rectal primary (9.3 cm; Fig. 5, top row), homozygous mutation in KIT exon 11 [23], and high mitotic count (20/50 high power field), nonetheless achieved pathologic CR with abundant IFN-γ-producing TILs (Fig. 4b, s). Pt#5 (9.8 cm primary gastric GIST and extensive liver lesions) and Pt#7 (11.4 cm primary gastric GIST) achieved radiographic near-CR of primary GIST (Fig. 5, 2nd and 3rd row), such that surgeries were no longer indicated—great clinical benefit. Pt#6 harboring WT GIST with extensive liver metastasis, bilateral lung metastasis, and peritoneal implants achieved PR by all three criteria at week 8 (Table 1; Fig. 5, 4th row); however, on day 369 of PR—7 months after completion of PegIFNa2b—while continuing IM, four existing lesions showed increased SUV. HD-PegIFNa2b was then re-initiated with continuation of IM 400 mg/day and resulted in a second PR (Fig. 5, last 2 rows), which lasted 430 days while off both IM and PegIFNa2b. Induction of a second PR after tumor progression (due to IM resistance) is unprecedented.

PET-CT and CT scans. Top three rows show that the combination treatment resulted in swift radiographic CR or near-CR of large primary GIST of Pts#4, 5, and 7 (9.3, 9.8, and 11.4 cm). Pt#6 showed mixed PR and CR (fourth row). Last two rows illustrate the second PR (after emergence of IM resistance) induced by re-initiation of PegIFNa2b
Progression-free survival and overall survival
After a median follow-up of 3.6 years (3.2–4.3 years), the OS is 100% with PS = ECOG 0 for all seven evaluable patients. Pt#1 had unsuccessful genotyping and was grouped with KIT exon 11—the most favorable group—for the evaluation. Pt#7 died at age 85 of unrelated illness while in remission with a radiographic near-CR (Table 1; Fig. 5, 3rd row). The PFS of Pt#6 and the continuing PR/CR of Pts#1, 2, 4, 5, and 8—a total of 6 out of 7 evaluable patients—exceed not only the genotype-specific median PFS [22] but also the upper limit of the 95% confidence level of the genotype-specific PFS of S0033 IM-monotherapy trial (Table 1, last column; S0033 Study data are in brackets) [Personal communication with Dr. Michael C. Heinrich, PI of IM-monotherapy phase III S0033 Study]. Pt#3 harbored aggressive GIST with extensive liver metastasis and achieved PR at week 8 by all three criteria, but three out of the numerous liver lesions showed evidence of increased SUV on PET-CT scan without new lesions on day 765, and the genotype-specific PFS was slightly longer than that of S003 IM-monotherapy trial [22].
Taken together, combination treatment with IM plus PegIFNa2b was well tolerated, safe, demonstrated a 100% response rate (PR + CR), 100% OS rate, and substantially prolonged continuing PR/CR (5 patients) and PFS (2 patients) after a median follow-up of 3.6 years (3.2–4.3 years), and strongly supports part two and three of our hypothesis.
Discussion
In CML, combining two active agents, peginterferon-alfa-2a (Pegasys®) and IM, demonstrated tolerability and improved efficacy over IM-monotherapy [45], but the immunological implications of IFN-α was not addressed. Our GIST study of adding PegIFNa2b (immunotherapy) to the current standard IM (targeted therapy) demonstrated significant induction of innate immunity and Th1 response and highly promising clinical outcome comparing to IM-monotherapy albeit small group study, and strongly supported our hypothesis. Although IM has off-target effect of activating innate [32, 33] and potentiating adaptive immunity [34, 35], the significant induction of IFN-γ-producing-lymphocytes (Fig. 3a) in this study is mainly attributed to PegIFNa2b rather than IM alone because stopping HD-PegIFNa2b (Fig. 3b, n) or switching to maintenance LD-PegIFNa2b while continuing IM resulted in a sharp decline of IFN-γ-producing-lymphocytes to barely detectable level. This new concept/strategy of combining immunotherapy with effective non-marrow-suppressive treatments might be beneficial to other cancer types as well. Combining IFN-α or peginterferon α with radiation, hormone/hormone antagonist, small molecule targeted therapies, or monoclonal antibodies in radiosensitive tumors, prostate, breast, pancreatic, melanoma, hepatocellular, colorectal, and sarcoma may help delay/prevent relapse and warrant further investigations.
References
Fridman WH, Mlecnik B, Bindea G, Pagès F, Galon J (2011) Immunosurveillance in human non-viral cancers. Curr Opin Immunol 23:272–278
Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ et al (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410:1107–1111
Dunn GP, Koebel CM, Schreiber RD (2006) Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6:836–848
Dighe AS, Richards E, Old LJ, Schreiber RD (1994) Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1:447–456
Becker C, Pohla H, Frankenberger B, Schüler T, Assenmacher M, Schendel DJ et al (2001) Adoptive tumor therapy with T lymphocytes enriched through an IFN-gamma capture assay. Nat Med 7:1159–1162
Schoenborn JR, Wilson CB (2007) Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 96:41–101
Rabinovich GA, Gabrilovich D, Sotomayor EM (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25:267–296
Trombetta ES, Mellman I (2005) Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol 23:975–1028
Kadowaki N, Antonenko S, Lau JY, Liu YJ (2000) Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med 192:219–226
Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V (2011) Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol 29:163–183
Rizza P, Capone I, Moretti F, Proietti E, Belardelli F (2011) IFN-α as a vaccine adjuvant: recent insights into the mechanisms and perspectives for its clinical use. Expert Rev Vaccines 10:487–498
Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL et al (2009) Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci USA 106:9010–9015
Narumi K, Udagawa T, Kondoh A, Kobayashi A, Hara H, Ikarashi Y, et al. (2011) In vivo delivery of interferon-α gene enhances tumor immunity and suppresses immunotolerance in reconstituted lymphopenic hosts. Gene Ther, doi:10.1038/gt.2011.73
Sikora AG, Jaffarzad N, Hailemichael Y, Gelbard A, Stonier SW, Schluns KS et al (2009) IFN-alpha enhances peptide vaccine-induced CD8+ T cell numbers, effector function, and antitumor activity. J Immunol 182:7398–7407
Miquilena-Colina ME, Lozano-Rodríguez T, García-Pozo L, Sáez A, Rizza P, Capone I et al (2009) Recombinant interferon-α2b improves immune response to hepatitis B vaccination in haemodialysis patients: results of a randomised clinical trial. Vaccine 27:5654–5660
Hance KW, Rogers CJ, Zaharoff DA, Canter D, Schlom J, Greiner JW (2009) The antitumor and immunoadjuvant effects of IFN-α in combination with recombinant poxvirus vaccines. Clin Cancer Res 15:2387–2396
Ma Y, Conforti R, Aymeric L, Locher C, Kepp O, Kroemer G et al (2011) How to improve the immunogenicity of chemotherapy and radiotherapy. Cancer Metastasis Rev 30:71–82
Manley PW, Cowan-Jacob SW, Buchdunger E, Fabbro D, Fendrich G, Furet P, et al. (2002) Imatinib: a selective tyrosine kinase inhibitor. Eur J Cancer 38(Suppl 5):S19–S27
McAuliffe JC, Hunt KK, Lazar AJ, Choi H, Qiao W, Thall P et al (2009) A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol 16:910–919
Ayyoub M, Taub RN, Keohan ML, Hesdorffer M, Metthez G, Memeo L et al (2004) The frequent expression of cancer/testis antigens provides opportunities for immunotherapeutic targeting of sarcoma. Cancer Immunol 4:7–20
Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT et al (2006) KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 42:1093–1103
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD et al (2008) Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 26:5360–5367
Chen LL, Holden JA, Choi H, Zhu J, Wu EF, Jones KA et al (2008) Evolution from heterozygous to homozygous KIT mutation in gastrointestinal stromal tumor correlates with the mechanism of mitotic nondisjunction and significant tumor progression. Mod Pathol 21:826–836
Miettinen M, Lasota J (2006) Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 23:70–83
Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W et al (2004) A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 64:5913–5919
McLean SR, Gana-Weisz M, Hartzoulakis B, Frow R, Whelan J, Selwood D et al (2005) Imatinib binding and cKIT inhibition is abrogated by the cKIT kinase domain I missense mutation Val654Ala. Mol Cancer Ther 4:2008–2037
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ et al (2006) Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 24:4764–4774
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ et al (2002) Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 347:472–480
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY et al (2004) Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 364:1127–1134
Patel S, Zalcberg JR (2008) Optimizing the dose of imatinib for treatment of gastrointestinal stromal tumours: lessons from the phase 3 trials. Eur J Cancer 44:501–509
Blay JY, Le Cesne A, Ray-Coquard I, Bui B, Duffaud F, Delbaldo C et al (2007) Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol 25:1107–1113
Borg C, Terme M, Taieb J, Ménard C, Flament C, Robert C et al (2004) Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest 114:379–388
Ménard C, Blay JY, Borg C, Michiels S, Ghiringhelli F, Robert C et al (2009) Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res 69:3563–3569
Ray P, Krishnamoorthy N, Oriss TB, Ray A (2010) Signaling of c-kit in dendritic cells influences adaptive immunity. Ann N Y Acad Sci 1183:104–122
Balachandran1 VP, Cavnar1 MJ, Zeng1 S, Bamboat1 ZM, Ocuin1 LM, Obaid1 H, et al. (2011) Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 17:1094–1100
Bukowski RM, Tendler C, Cutler D, Rose E, Laughlin MM, Statkevich P (2002) Treating cancer with PEG Intron: pharmacokinetic profile and dosing guidelines for an improved interferon-alpha-2b formulation. Cancer 95:389–396
Sato Y, Nabeta Y, Tsukahara T, Hirohashi Y, Syunsui R, Maeda A et al (2002) Detection and induction of CTLs specific for SYT-SSX-derived peptides in HLA-A24+ patients with synovial sarcoma. J Immunol 169:1611–1618
Berger TG, Feuerstein B, Strasser E, Hirsch U, Schreiner D, Schuler G et al (2002) Large-scale generation of mature monocyte-derived dendritic cells for clinical application in cell factories. J Immunol Methods 268:131–140
Chen LC, Delgado JC, Jensen PE, Chen X (2009) Direct expansion of human allospecific FoxP3+CD4+ regulatory T cells with allogeneic B cells for therapeutic application. J Immunol 183:4094–4102
Cano P, Fan B (2001) A geometric and algebraic view of MHC-peptide complexes and their binding properties. BMC Struct Biol 1:2–15
Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA et al (1999) Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer 35:1773–1782
Benjamin RS, Choi H, Macapinlac HA, Burgess MA, Patel SR, Chen LL et al (2007) We should desist using RECIST, at least in GIST. J Clin Oncol 25:1760–1764
Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR et al (2007) Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 25:1753–1759
Gleevec® [prescribing information] (2008) Distributed by Novartis Pharmaceuticals Corporation, East Hanover, New Jersey
Preudhomme C, Guilhot J, Nicolini FE, Guerci-Bresler A, Rigal-Huguet F, Maloisel F et al (2010) Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N Engl J Med 363:2511–2521
Acknowledgments
National Cancer Institute Grants CA016672, U01-CA70172-01 and N0-CM-17003 to University of Texas MD Anderson Cancer Center; National Cancer Institute Award P30CA042014 to Huntsman Cancer Institute; Public Health Services research grants UL1-RR025764 and National Center for Research Resources C06-RR11234 to University of Utah. We thank Schering/Merck for supplying peginterferon α-2b and partial funding; Katie Thomas, RN and Grace Noda, PA-C, for excellent patient care; Cherry Vanover for superb clinical trial assistance; Aida B. Narvios, MD and Regan Butterfield for assistance in leukapheresis and radiology.
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Chen, L.L., Chen, X., Choi, H. et al. Exploiting antitumor immunity to overcome relapse and improve remission duration. Cancer Immunol Immunother 61, 1113–1124 (2012). https://doi.org/10.1007/s00262-011-1185-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-011-1185-1