
Abstract
A rapid subtraction hybridization approach was used to isolate genes differentially expressed during mycelial contact between Trichoderma harzianum (Hypocrea lixii) and Rhizoctonia solani, and could serve as marker genes for selection of superior biocontrol strains. Putatively positive clones were evaluated by transcription analysis during mycelial contact with R. solani versus growth on glucose, and for their differential transcription between two strains with either strong or poor biocontrol capability before, at, and after contact with R. solani. Besides four clones, which had similarity to putative but as yet uncharacterized proteins, they comprised ribosomal proteins, proteins involved in transcriptional switch and regulation, amino acid and energy catabolism, multidrug resistance, and degradation of proteins and glucans. Transcription of three clones was evaluated in five T. harzianum strains under confrontation conditions with R. solani. Two clones—acetyl-xylane esterase AXE1 and endoglucanase Cel61b—showed significant upregulation during in vivo confrontation of a T. harzianum strain that successively demonstrated a very high antagonistic capability towards R. solani, while expression was progressively lower in a series of T. harzianum strains with intermediate to poor antagonistic activity. These clones are promising candidates for use as markers in the screening of improved T. harzianum biocontrol strains.
Similar content being viewed by others
Introduction
Several taxa from the filamentous fungal genus Trichoderma became well known as biocontrol agents against aerial and soil-borne plant pathogens (Samuels 1996; Lorito et al. 1996b; Yedidia et al. 1999; Harman et al. 2004; Brunner et al. 2005). Successful application of a given strain depends on the interaction of a number of physiological properties, such as mycoparasitic activity, rapid growth rate, capability to utilize various substrates, resistance to noxious chemicals, ability to induce a systemic resistance response in the plant to be protected, and production of antibiotics and extracellular hydrolytic enzymes necessary for inhibition of the plant pathogen (Carsolio et al. 1994; Lorito et al. 1996a; Yedidia et al. 1999; Kullnig et al. 2000; Harman et al. 2004).
Over the last decade, the agricultural market has increased its demand for biological control formulates that work at least with similar or even better efficacy than their chemical counterparts and that can be applied for a wide range of pathogens and plants (Gielen et al. 2004). Consequently, the search for Trichoderma isolates with a high antagonistic potential against plant pathogens has become more and more attractive for the development of environment-friendly agricultural practices in the last years.
Despite the promising scenario, the use of Trichoderma spp. as biocontrol agents is still limited by the insufficient understanding of the biochemical and genetic basis of biocontrol. This also impedes the straightforward selection of good biocontrol genes. Most of the available literature has dealt with the potential role of hydrolytic enzymes (chitinases, glucanases, proteases) and antibiotics in mycoparasitism (Inbar and Chet 1995; Lorito et al. 1996a; Carsolio et al. 1999; Ramot et al. 2000; Kullnig et al. 2000; Donzelli and Harman 2001; Prabavathy et al. 2006). Other factors have only scarcely been dealt with. To this end, genome-wide screening techniques—such as subtractive hybridization (Wieland et al. 1990; Lopez-Fernandez and del Mazo 1993; Jiang et al. 2000) or EST sequencing (Velculescu et al. 1995; Adams 1996)—bear clear advantages. In fact, both approaches have recently been applied to T. harzianum (Liu and Yang 2005; Vizcaino et al. 2006), T. hamatum (Carpenter et al. 2005) and T. asperellum, T. longibrachiatum, T. cf. viride and T. virens (Vizcaino et al. 2007), but the obtained clones were not thoroughly characterized. In addition, in the majority of these studies, the fungi were grown in submerged culture on cell wall of potential plant pathogens as carbon source, and the physiological relevance of this approach to biocontrol is debatable.
The aim of this work was to clone genes which are specifically expressed by Trichoderma during the antagonistic interaction with the common plant pathogenic fungus Rhizoctonia solani Kühn, using the rapid subtraction hybridization (RaSH) approach (Jiang et al. 2000; Schmoll et al. 2004), and to test whether any of them could be used to specifically screen for superior biocontrol activity. To this end, we first selected a Trichoderma strain with high biocontrol ability to protect cotton seedlings against R. solani as a model. We then adopted enforced mycoparasitic conditions, in which pregrown biomass of a mycoparasitic strain of Trichoderma was placed in contact with preformed biomass of R. solani, for the tester cDNA. Evaluation of the so obtained genes under confrontation conditions allowed us to detect two clones which are significantly more strongly expressed in superior Trichoderma strains than in strains displaying comparatively moderate or low biocontrol performance.
Materials and methods
Strains and growth conditions
Five Sardinian strains of T. harzianum—T4.26, T4.67, T4.57, T4.31, and T17.25—were used in the present work. These were isolated during a larger strain selection programme (Migheli et al. 2008), and unambiguously identified as T. harzianum by the TrichOKey barcode (Druzhinia et al. 2005). In addition, T. atroviride P1 (ATCC 74058; Lorito et al. 1996a), T. harzianum T22 (Harman 2000) and R. solani RT-10 (anastomosis group AG-4, kindly provided by Dr. E. Lahoz) were used in the biocontrol experiments. Monoconidial cultures of T. harzianum and clonal cultures of R. solani were grown in potato dextrose broth (PDB, Sigma-Aldrich, Steinheim, Germany) or on potato dextrose agar (PDA, Difco, Franklin Lakes, NJ) supplemented with streptomycin and tetracycline (50 μg l−1 each; Sigma-Aldrich) to prevent bacterial contamination. Long-term storage of colonized PDA plugs was in 50% glycerol at −80°C.
Escherichia coli DH5α or JM109 were grown at 37°C on Luria-Bertani (LB) medium as described by Sambrook et al. (1989).
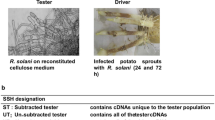
For the construction of the subtractive library under enforced contact conditions, T. harzianum T4.67 (an effective biocontrol strain) and R. solani were pregrown as liquid cultures in PDB for 3 days at 25°C. Mycelia were harvested by gentle filtration, washed thrice with sterile water and the mycelia of T. harzianum then transferred on (1) agar plates of synthetic medium (Yedidia et al. 1999) containing glucose [SM + glucose, consisting of (g l−1): (NH4)2NO3, 1.0, MgSO4·7H2O, 0.2, K2HPO4, 0.9, KCl, 0.15, FeSO4·7H2O, 0.01, ZnSO4·7H2O, 0.009, MnSO4·H2O, 0.007, glucose, 30] or (2) agar plates of synthetic medium with no glucose but with a homogenous layer of 0.65 ± 3 g l−1 (wet weight) of pre-grown washed R. solani mycelium (SM + Rh). Mycelia of T. harzianum were separated from the residual medium and from the R. solani mycelium by a sterile sheet of cellophane (Model 583 Gel Dryer, Bio-Rad, CA, USA) that allowed assimilation of nutrients and interaction between the two fungi. These cultures were further incubated for 1–24 h at 25°C.
Plate confrontation tests
Circular plaques of 5 mm diameter were cut from mycelium of 5-day-old cultures of T. harzianum strains T17.25, T4.31, T4.57, T4.67, T.4.26 and of R. solani grown on malt-extract (MEX, Becton Dickinson and Co., MD, USA) plates. These plaques served as inoculum to confront the different T. harzianum strains with R. solani in vivo, hence they were placed in the respective combinations of each T. harzianum isolate with R. solani at a distance of 7.5 cm of each other on fresh MEX plates covered with cellophane. Plate confrontation experiments were performed in the dark at 25°C, and mycelia were harvested during the precontact, contact, and after-contact phase.
Isolation of RNA
For northern blot hybridization experiments mycelia of T. harzianum strains T4.67, T4.31 or R. solani were harvested, ground to a fine powder with liquid nitrogen and total RNAs were isolated using either the RNeasy Maxi Kit (No. 75162, Qiagen, Hilden, Germany) following the manufacturer’s instructions, or according to the guanidiniumthiocyanate/phenol method (Chomczynski and Sacchi 1987; Schmoll et al. 2004).
Extraction of total RNA applied in quantitative real-time RT-PCR was carried out from mycelium of T. harzianum strains T4.26, T4.67, T4.57, T4.31 and T17.25 sampled at different phases (pre-contact, contact, after-contact) during in vivo confrontation with R. solani (see “Plate confrontation tests”). Mycelia were harvested with a spatula, flash-frozen in liquid N2 and ground to a fine powder, and RNA extraction was carried out with the Pure Link™ Micro-to-Midi Kit (No. 12183-018, Invitrogen, Carlsbad, CA, USA). For genomic DNA removal, an on-column DNase digestion with the RQ1 RNase-Free DNase (Promega, WI, USA) set was carried out according to manufacturer’s instructions.
Rapid subtraction hybridization
The procedure of RaSH was essentially the same as described by Schmoll et al. (2004). RNAs of T. harzianum T4.67 (grown on SM + glucose) were used as driver, and RNAs from the same strain grown on SM interacting with the mycelium of R. solani, were used as tester. For the latter, RNAs were isolated after 1, 2, 3, 4, 6, 8, 12 and 24 h of incubation, pooled, respectively, and an aliquot of 1 μg was transcribed into cDNA using the CreatorSMART library construction Kit (BD Biosciences Clontech, CA, USA). cDNAs were digested with EcoRII (New England Biolabs, Beverly, MA, USA) at 37°C for 3 h. The oligonucleotides XE-14, XEA-13, and XET-13 (Table 1) were used as adapters. After incubation at 55°C for 3 min, the mix was slowly cooled down to 8°C, and ligation of adapters was performed at 4°C overnight. To amplify the digested cDNA, PCR was performed with primers XEA-18 and XET-18 (Table 1) that correspond to the adapter sequences. The PCR was carried out under the following conditions: GoTaq polymerase reaction buffer (Promega), MgCl2 (2.5 mM), dNTP (0.2 mM), XEA-18 (1 μM), XET-18 (1 μM), GoTaq polymerase (Promega) and 1 μl of cDNA; the PCR-programme was set as follows: 72°C, 5 min; 25 cycles of 1 min, 94°C denaturation; 1 min, 55°C annealing; and 1 min, 72°C extension with a final extension step of 10 min at 72°C.
PCR products were purified using the Qiaquick PCR Purification Kit (No. 28104; Qiagen). 1 μg of amplified tester cDNA was digested by XhoI (New England Biolabs) at 37°C for 4 h, and then applied to the hybridization reaction. 100 ng of tester cDNA were mixed with 3 μg of the driver cDNA (ratio 1:30, μg/μg) in 10 μl of hybridization solution (0.5 M NaCl, 50 mM Tris-HCl pH 7.5, 0.2% (w/v) SDS, 40% (v/v) formamide), overlaid with a drop of mineral oil and, after boiling for 5 min, incubated at 42°C for 48 h. The hybridization mix was diluted with sterile water to 100 μl, purified using the Qiaquick PCR Purification Kit and adjusted to a final volume of 30 μl. Eight microlitres of this mixture was ligated with XhoI-digested and dephosphorylated (Shrimp alkaline phosphatase, Fermentas, Vilnius, Lithuania) pBlueScript SK (+/−) vector at 4°C overnight and transformed into E. coli DH5α or JM109, respectively.
Reverse northern hybridization
Bacterial colonies were picked randomly, grown in 100 μl LB medium, supplemented with ampicillin to a final concentration of 100 μg ml−1, for 2 h and 1 μl of the culture was used for insert screening PCR using primers RaSH-F and RaSH-R (Table 1), which bind within pBluescript SK. The PCR products were first analysed by agarose gel electrophoresis and then blotted onto Hybond-N membrane (Amersham Biosciences, Freiburg, Germany) using 0.4 N NaOH. The hybridization step was performed using 2.5 μg of PCR amplified, EcoRII digested and subsequently fluorescein (F1-dUTP; Gene Images Random Prime Labelling Module, Amersham Biosciences), random-labelled cDNA from tester or driver, respectively, as probes. The candidates for a more detailed analysis were chosen by visual inspection first, and then this decision was cross-checked by quantitative measurements using the Bio-Rad GelDoc Imaging system and Bio-Rad Quantity One software, both for three different exposures of the blot.
Northern blot analysis
Gene expression of selected clones was analyzed in a first set of northern blots with RNA used in the RaSH experiments, to be then confirmed in a second set of northern blots with RNA deriving from in vivo confrontation of T. harzianum and R. solani in plate. Standard methods (Sambrook et al. 1989; Schmoll et al. 2004) were used for electrophoresis, blotting and hybridization of RNA, using α32P-dCTP random-labelled plasmid inserts of the respective clones as a probe.
Quantitative real-time RT-PCR
Primers used in quantitative real-time RT-PCR (qPCR) (Table 1) were designed using the Primer3 software (Rozen and Skaletsky 1998) and were synthesized by Invitrogen. Primers for target genes were based on subtractive library sequences of clone 22 (NADH-dehydrogenase, 22-For and 22-Rev), clone 115 (acetyl xylan esterase, 115-For and 115-Rev) and clone 140 (endoglucanase Cel61b, 140-For and 140-Rev) of T. harzianum. Reference gene primers were designed for the glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH), the beta-actin gene (ACTB), and the tubulin gene (TUB) of T. harzianum (gapdh-For, gapdh-Rev, actb-For, actb-Rev, tub-For, tub-Rev). Product size and specificity were confirmed by agarose gel electrophoresis.
PCR efficiency was determined for each primer pair on fivefold serial dilutions of cDNA and calculated according to Rasmussen (2001). Three potential reference genes were tested (GAPDH, ACTB, TUB), however, only the GAPDH gene met qualifications of stability. A total of 500 ng of RNA of the pre-contact, contact and after-contact phase of the T. harzianum strains (T4.26, T4.67, T4.57, T4.31 and T17.25) grown in plate confrontation with R. solani were reverse-transcribed into cDNA using the iScript® cDNA Sythesis Kit (No. 170-8890, Bio-Rad, CA, USA) following the manufacturer’s protocol. The qPCR was performed using the SYBR® Green I dye. Optimal PCR conditions in a 25 μl reaction volume were 1 μl of 1:5 diluted cDNA template, 12.5 μl of 2X iQ SYBR® Green Supermix (Bio-Rad), 400 nM of Forward primer and 400 nM of Reverse primer. PCR was performed in triplicate in 96-well optical plates on a MyiQ Single Colour Real-Time Detection System (Bio-Rad) The PCR programme was as follows: 95°C for 3 min, 40 cycles of 95°C for 10 s and 60°C for 30 s, followed by the measurement of fluorescence during a melting curve in which the temperature raised from 55 to 95°C in sequential steps of 0.5°C for 30 s. This ensured the detection of one gene-specific peak and the absence of primer-dimer peaks. cDNA of sample “T4.67 after-contact” was chosen as calibrator sample and was therefore tested for the reference gene and the respective target gene in each qPCR experiment. Samples of two independent cultivations were analyzed and yielded consistent results.
Biological control of R. solani infection of cotton
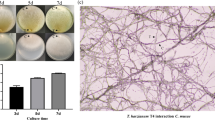
Two biocontrol experiments were carried out on cotton (Gossypium barbadense L.) seedlings (cv Gedera-236, Zeraim Gedera Co., Israel). Using a sterile cork borer, plugs of 15 mm in diameter were cut from 7-day-old cultures of R. solani, T. harzianum and T. atroviride grown at 25°C on PDA. One plug of Trichoderma was paired with one plug of R. solani, by placing the mycelia in direct contact. Plug pairs were incubated in the dark at 25°C for 24 h, then transferred to the centre of plastic sowing pots (4.5 cm in diameter, 55 ml capacity, one plug per pot), and covered by a 2-cm layer of sterilized (121°C for 60 min on two successive days) potting mix (Humin-Substrat N17, Neuhaus, Germany). For each treatment, three replicates (10 seeds for each replicate) were incubated on a bench in a glasshouse for 7 days before cotton seeds (one seed per pot) were added. Pots were watered daily and the average temperature was 20–25°C (min 10–15°C, max 25–35°C). Seedling emergence was checked weekly after 7–21 days. After the last survey, The severity of disease was calculated using the McKinney index (McKinney 1923), which expresses the percentage of the maximum severity of disease (i.e. 100) according to the formula: I = [Σ (c × f)/n × N] × 100, where c = disease class; f = frequency; n = number of observations; N = the highest value of an empirical scale adopted, estimating the severity of symptoms on the stem. Five classes were set: class 0 = healthy stem; class 1 = mild browning on the stem; class 2 = browning on one half of the stem; class 3 = complete browning of the stem; class 4 = plant death after emergence or plant not emerged in comparison with the emergence of the uninoculated control treatment.
Bioinformatic analysis of RaSH clones
To identify individual clones from their nt-sequence, these were first trimmed from vector sequences, and then subjected to a BLASTN search against the JGI TRIRE database (http://genome.jgi-psf.org/Trire2/Trire2.home.html). This was done in order to by-pass the drawback that the genome sequence of T. harzianum is not available, assuming that the sequences of its clones would show highest similarity to the gene sequences of another Trichoderma species. In case of hits showing high nt-similarity (above 80%), the annotated full-length sequences of the respective H. jecorina hypothetical proteins were then used to perform a BLASTX search of the NCBI database (http://www.ncbi.nlm.nih.gov) to identify the protein on the basis of its closest orthologue. In case a clone did not yield any hit in the JGI TRIRE database, this was nevertheless given a try at NCBI in order to identify genes which might have arisen by horizontal transfer from other more distant organisms. In silico analysis of the predicted proteins was done with PROTPARAM (http://www.expasy.org/tools/protparam.html), and signal peptide prediction was performed with SignalP (http://www.cbs.dtu.dk/services/SignalP/) (version 3.0; Bendtsen et al. 2004), both available at Expasy-Tools (http://www.expasy.org/tools).
Statistical methods
Data from biocontrol experiments were analyzed by the analysis of variance (ANOVA) after transforming the original data (expressed as percentage, %) using the arcsin√x function, where x is the relative proportion. Means separation was done by the Dunnett test by using Minitab® for Windows release 12.1 software.
Validation of two separate qPCR assays and data handling were performed using the Bio-Rad iQ5 2.0 standard edition software where baseline and cycle threshold (C T) were set automatically for each assay. The standard curves were generated by plotting the DNA amount (nanogram of cDNA) against the C T value exported from the MyiQ Single Colour Real-Time Detection System (Fig. 3).
Results
RaSH-cloning of genes specifically expressed in T. harzianum during forced antagonism with R. solani
Using the RaSH protocol, described above, we isolated several hundred bacterial colonies comprising plasmids with apparently differentially expressed tester sequence tags as inserts. In order to select those which are truly upregulated, 400 of these clones were randomly picked and analysed by reverse northern blotting, using the total cDNA of T. harzianum grown on SM, and also total cDNA from growth on SM medium plus R. solani mycelium. This analysis strongly reduced the number of putatively positives, resulting in 50 clones which showed a significantly more intense hybridization signal after hybridization with the cDNA from the latter conditions (i.e. antagonistic interaction). These clones were sequenced and used for further analysis.
Due to the expected properties of the genes for which we obtained these EST-fragments they were denominated aag (antagonism associated gene). We first looked for orthologues in the T. reesei genome sequence (http://genome.jgi-psf.org/Trire2/Trire2.home.html). T. reesei orthologues were found for 36 of these clones. The remaining 14 genes (aag13, aag17, aag19, aag34, aag44, aag49, aag54, aag57, aag74, aag75, aag83, aag91-2, aag92, aag135; sequence data not shown) did not produce any hits in BLASTN search against the T. reesei, T. atroviride (http://genome.jgi-psf.org/Triat1/Triat1.home.html), T. virens (http://genome.jgi-psf.org/Trive1/Trive1.home.html) genome sequences, and the NCBI database (both within the nr database and the EST-database). BLASTX search on the NCBI database produced hits without significant e values for 9 of the 14 clones: a mitochondrial precursor of a choline dehydrogenase (aag17: 2.0E−21), a conserved hypothetical protein with GMC oxidoreductase activity (aag49: 2.0E−9, aag74: 4.0E−7, aag75: 9.0E−9), hypothetical proteins (aag83: 2.0E−6, aag91-2: 2.0E−10, aag57: 4.0E−13, aag135: 2.0E−7), and a putative methionine sulfoxide reductase SelR (aag92: 7.6). These results suggest that the sequences may represent unique T. harzianum genes.
Retrieving the full length nt-sequences of the corresponding genes from T. reesei and aligning them with that of the respective T. harzianum clone identified some of them actually to be different ESTs from the same gene, e.g. aag91 = aag21 = aag22 = aag29; aag10 = aag7; aag33 = aag86 = aag127; aag288 = aag141; aag69 = aag87 = aag95 = aag100 = aag109 = aag110, thus corroborating that these genes are more strongly expressed under the target conditions. Therefore, the 36 clones actually represented only 25 different genes (Table 2).
Our identifications of the 25 putative genes are listed in Table 2: 4 of them (aag14, aag76, aag91, aag48) encoded unknown or hypothetical proteins. A search of T. harzianum EST sequences within NCBI GenBank did not yield any result. However, they all contained orthologues in F. graminearum and M. grisea, which among all fungi whose genome sequence is accessible in GenBank are phylogenetically the closest members to Trichoderma, indicating that these genes encode a protein whose structure, and thus function, is conserved at least among Sordariomycetes. One of them—aag14—encoded a protein characterized by a signal peptide and four transmembrane helices, respectively, suggesting that it represents a membrane-spanning protein. One unknown clone (aag91) yielded an intriguing result, as it showed similarity to an untranscribed area 5′ of the epl1 gene encoding a protein of the ceratoplatanin-family, which was recently shown to be upregulated under mycoparasitic conditions in H. atroviridis (Seidl et al. 2006). Apart from this interesting, albeit contingent correlation, no identification of function of this clone was possible. However, the possibility that this clone was an artefact is unlikely because it was isolated four times (aag91, aag21, aag22, aag29).
All the other genes could reliably be identified: although their potential functions are widespread, most of them can be grouped into two major clusters, i.e. those representing genes/proteins involved in transcription and protein synthesis/turnover, and those encoding extracellular hydrolases. Members of the former were: a high mobility box protein (aag51); ribosomal protein S10 (aag90); 40S ribosomal protein 25 (aag20); an ARM-repeat protein involved in Puf-RNA folding (Andrade et al. 2001; aag32); an U2-associated snRNP A protein (aag34); and polyubiquitin (aag107). The extracellular hydrolases comprised two proteases (a metalloprotease, aag63; and an S28 peptidase, aag97), a triacylglycerol lipase (whose clones were most abundant: aag69, aag87, aag95, aag100, aag109, aag110), and an acid phosphatase (aag288). An orthologue of H. jecorina acetyl xylan esterase I (aag115; Axe1), and an orthologue of the H. jecorina GH family 61 endoglucanase Cel61b (aag140; Foreman et al. 2003), which corresponds to the previously published EGIV (Saloheimo et al. 1997) but lacks a cellulose-binding domain. Besides these two major groups, most of the other clones represented enzymes involved in transport, metabolism and energy production: clones aag22 and aag115a, encoding NADH-dehydrogenase 51 kDa subunit and succinate dehydrogenase, respectively, represented oxidative energy metabolism; a 12-membrane hexose permease (aag33, aag86, aag128) and another protein of the major facilitator subfamily (aag10, aag71) representing transport proteins; and tryptophan synthase (aag74), acetate kinase (aag75) and saccharopine reductase (aag81) representing metabolic enzymes. Finally, two clones fell outside of all other categories: a multicopper oxidase (aag58) and an adenosyl-methionine-cyclopropan fatty acyl phospholipid synthase (aag45).
Expression pattern of selected aag genes during in vitro confrontation with R. solani
Based on the results described above, selected clones (aag51, aag63, aag90, aag10, aag22, aag33, aag74, aag75, aag81, aag97, aag109, aag115, aag140) were used as probes to investigate the expression of these genes during the antagonistic interaction between T. harzianum and R. solani, separated by a cellophane sheet, under the same conditions as used for the initial RaSH experiment. Only 4 of these 13 clones showed the difference in the expression patterns that would have been anticipated from the reverse northern blot experiments (Fig. 1): NADH-dehydrogenase (aag22) and Cel61b (aag140) exhibited only a very low level of expression under both conditions, but their expression increased dramatically after 12–24 h of antagonistic interaction. A similar expression pattern was shown by axe1 (aag115), although this gene exhibited a higher basal level of expression. Only the transcript of the hexose transporter (aag33) exhibited a significantly higher abundance under antagonistic conditions. Expression of aag51 and aag63 is also shown as example for false positive genes. Interestingly, all genes but the hexose transporter displayed two mRNA transcripts of different size, that could be due to different lengths of the 5′ and 3′ untranslated regions or alternative splicing.

Northern blot analysis of selected subtracted library clones. RNA was prepared after 1, 2, 3, 4, 6, 8, 12, and 24 h from Trichoderma harzianum antagonist isolate T4.67 grown on minimal medium containing glucose as sole carbon source (driver), as well as from T4.67 during interaction with Rhizoctonia solani (tester), the two mycelia being separated by a cellophane sheet, and loaded in the given order (20 μg per lane). Hybridizations were performed with [α-32P]-labelled sequence tags as obtained by the RaSH experiment. 18S rRNA was used as hybridization control
Expression pattern of selected aag genes during in vivo antagonistic interaction between T. harzianum and R. solani (plate confrontation assay)
In order to learn whether the four genes, identified above as being up-regulated under antagonistic conditions, would also be expressed during in vivo antagonistic interaction of T. harzianum and R. solani, hence by direct contact of their mycelia, we used clones aag22, aag33, aag115, and aag140 as probes in Northern analysis of mRNA prepared from T. harzianum in confrontation experiments on plates during pre-contact, contact and after-contact phases. Two strains of T. harzianum—T4.67 (a biocontrol strain), and T4.31 (a weakly antagonizing strain not capable of biocontrol activity against R. solani) were used in these confrontations with R. solani. Five other genes (aag51, aag63, aag288, aag97, aag45 and aag14), which were not expected to be upregulated during mycoparasitism, were included as a control. NADH-dehydrogenase (aag22), endoglucanase Cel61b (aag140) and the S28 peptidase (aag97, originally not recognized as biocontrol-related) were most strongly expressed (Fig. 2). Only the acid phosphatase (aag288) and the cyclopropan fatty acyl phospholipid synthase (aag45) failed to show a specific difference in the expression pattern, whereas all the other genes were significantly overexpressed in the biocontrol strain T4.67 (Fig. 2).

Expression pattern of genes during in vivo antagonistic interaction with Rhizoctonia solani. 20 μg of total RNA were applied in each lane, obtained from Trichoderma harzianum isolates T4.67 (antagonist) and T4.31 (non-antagonist) before, during, and after contact with Rhizoctonia solani in constant darkness and from pure cultures of the three fungi under otherwise equal conditions. Hybridization was performed with the [α-32P]-labelled clones aag22, aag33, aag115, aag140, aag51, aag63, aag288, aag97, and aag45; 18S rRNA was used as hybridization control
Expression pattern of potential biocontrol marker genes in T. harzianum strains
The above described expression patterns obtained from in vivo interaction between T. harzianum T4.67 and T4.31 with R. solani identified three promising gene clones (aag22, aag115, aag140) that could potentially be used as indicators for selection of biocontrol strains. In order to test this hypothesis, an on-plate confrontation assay was performed with a wider range of T. harzianum isolates (T17.25, T4.31, T4.57, T4.67 and T4.26) obtained from a large field study in Sardinia (Migheli et al. 2008). The expression of the NADH-dehydrogenase (aag22), acetyl xylan esterase (aag115) and endoglucanase Cel61b (aag140) in the absence of confrontation and during pre-contact, contact, and after-contact with R. solani was monitored by quantitative Real-time RT-PCR (qPCR). The almost “yes-no” expression for T4.67 and T4.31 in the northern blot was confirmed for all three genes but was not the rule for the other isolates. Instead, the strains showed an increasing expression of the acetyl xylan esterase and of the Cel61b endoglucanase, from isolates T17.25 and T4.31 (displaying the lowest expression level), through isolates T4.57 and T4.67 (having an intermediate level of expression), to isolate T4.26, which displayed the maximum level of expression (Fig. 3b, c, respectively). The expression of the NADH-dehydrogenase was, with the exception of isolate T4.31, only marginally regulated for all the isolates (Fig. 3a).

Real-time qPCR analysis of expression of genes encoding NADH-dehydrogenase (a), acetyl xylan esterase (b) and endoglucanase Cel61b (c) during in vivo confrontation of five Trichoderma harzianum isolates with Rhizoctonia solani. Expression was monitored in the absence of confrontation, before, during, and after contact with Rhizoctonia solani in constant darkness. Data are relative expression ratios (average values ± SE between biological replicates from two separate experiments) normalised against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels and adjusted for PCR efficiency
Validation of the marker-assisted selection of a superior T. harzianum strain capable of antagonizing R. solani on cotton seedlings in vivo
The five isolates of T. harzianum T17.25, T4.31, T4.57, T4.67 and T4.26, were further tested for their ability to antagonize R. solani on cotton seedlings. Table 3 shows the results of two separate biocontrol assays: under a very high disease pressure (McKinney index ranging from 95.8 to 100), the most effective isolate was T. harzianum T4.26, which consistently reduced the disease incidence in both experiments. T. harzianum T4.67 displayed a significant biocontrol potential, which was comparable to that presented by the well-known antagonistic strains T. harzianum T22 and T. atroviride P1 (Table 3). On the contrary, T. harzianum isolates T17.25, T4.31, and T4.57 had no significant effect on plant emergence nor on the disease index when compared to the R. solani-inoculated control.
Discussion
The advent of techniques allowing identification of differentially expressed genes has strongly advanced the characterization of genes which play key roles in physiological processes. Despite the widespread use of this principle with industrially applied and plant pathogenic fungi, it has not yet yielded unambiguous results with respect to genes required for the physiological changes accompanying mycoparasitism and biocontrol. While this study was completed, three papers were published, two of which dealt with the collection of ESTs (Vizcaino et al. 2006, 2007), whereas a third one used subtractive hybridization in T. hamatum (Carpenter et al. 2005). However, in the first two cases mRNAs were isolated from Trichoderma spp. grown on cell wall of plant pathogenic fungi, a condition which is only remotely related to the situation which Trichoderma faces during in vivo biocontrol or mycoparasitism, because it ignores the interaction with the host, which likely provokes a relevant response by the biocontrol organism. In addition, the collection of ESTs has the general drawback that they contain a high proportion of housekeeping genes, which are strongly expressed under all conditions and are consequently not specific for mycoparasitism.
In the study carried out by Carpenter et al. (2005), the authors identified their differentially expressed clones by directly subjecting the sequences of the obtained EST-fragments to a NCBI BLAST search. In our experience, this approach has serious pitfalls: when the gene fragment encodes a part of the protein which is only poorly conserved, the result of such an analysis often does not reflect the actual characteristics and thus the identity of the encoded gene. Such “problem cases” become evident when best hits are obtained with proteins from other kingdoms, e.g. bacteria, plants or even mammals, which makes it very probable that the identification is wrong. In order to avoid such flaws, we first searched for full-length orthologues of our clones in a closely related species from the same genus, i.e. H. jecorina. This strategy proved that for all of the clones for which a H. jecorina orthologue could be identified there were also closest neighbours present in other fungal species, and the similarity was highest with closest neighbours from GenBank (G. zeae and M. grisea). On the other hand, of the 14 clones which could not be identified, we could also not find any protein with significant similarity in any other organism from non-fungal kingdoms. This is consistent with the fact that—while cases of apparently horizontal transfer have been found in Trichoderma and other fungi (El-Bondkly 2006) - the genome annotation of several filamentous fungi has provided only limited evidence for cases of such horizontal transfer, and in most cases from bacteria (Braun et al. 2000). We therefore emphasize that caution should be applied when interpreting results from EST or proteomic studies in fungi where “best hits” are found with genes/proteins from evolutionary distant organisms such as insects, amphibia and vertebrates (Marra et al. 2006).
The genes identified in this study roughly fell into three groups: proteins involved in protein synthesis and turn-over; proteins involved in metabolism; and proteins involved in the hydrolysis of macromolecules. As a general picture, this reflects the necessity to degrade the cell wall of the mycoparasitic host, to take up and metabolize its constituents, but also to provide the energy needed for an induced synthesis, modification and excretion of the hydrolases and eventually to compete with the metabolic rate of the host. Such an interpretation is also supported by the findings that most of the genes tested were more highly expressed in the biocontrol strain, thereby indicating that the major advantage of this strain may be its more rapid and efficient reaction to the presence of the host.
The isolation of a cellulase CEL61b and the cellulose-induced acetyl xylan esterase AXE1 was intriguing, because R. solani does not contain cellulose in its cell walls. The isolation of these two genes is probably due to their induction by another component from the R. solani cell wall. We should like to note that—although we used dialysis membranes composed of modified cellulose in these experiments—this is unlikely the reason for their isolation, because the transcript analyses clearly showed their up-regulation also in the absence of the dialysis membranes. Among the other hydrolase-encoding genes that were found, two encoded proteases which is in agreement with the fact that several proteases have been shown to be involved in the mycoparasitic response in different Trichoderma strains (Viterbo et al. 2002; Olmedo-Monfil et al. 2002; Pozo et al. 2004). However, a triacylglycerol lipase has not yet been shown to be important for mycoparasitism, and the present identification of it makes further studies with this gene worthwhile.
The main purpose of this study was to isolate genes which could be used as markers for the pre-identification of biocontrol strains and their monitoring after application in the field. The expression pattern of the acetyl xylan esterase and the endoglucanase Cel61b suggests the possibility to differentiate very effective biocontrol isolates (such as T4.26) from intermediate biocontrol strains or from strains with no biocontrol capability. Although in theory these need not to be necessarily identical with genes which are critical or essential for mycoparasitism and biocontrol, they turned out to be more strongly expressed in the most effective biocontrol strain, thus opening the possibility to adopt a marker-assisted selection strategy for this fungal antagonist.
References
Adams MD (1996) Serial analysis of gene expression: ESTs get smaller. Bioassays 18:261–262
Andrade MA, Petosa C, O’Donoqhue SI, Muller CW, Bork P (2001) Comparison of ARM and HEAT protein repeats. J Mol Biol 309:1–18
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795
Braun EL, Halpern AL, Nelson MA, Natvig DO (2000) Large-scale comparison of fungal sequence information: mechanisms of innovation in Neurospora crassa and gene loss in Saccharomyces cervisiae. Genome Res 10:416–430
Brunner K, Zeilinger S, Ciliento R, Woo SL, Lorito M, Kubicek CP, Mach RL (2005) Improvement of the fungal biocontrol agent Trichoderma atroviride to enhance both antagonism and induction of plant systemic disease resistance. Appl Environ Microbiol 3959–3965
Carpenter MA, Stewart A, Ridgway HJ (2005) Identification of novel Trichoderma hamatum genes expressed during mycoparasitism using subtractive hybridisation. FEMS Microbiol Lett 251:105–112
Carsolio C, Gutiérrez A, Jiménez B, Montagu MV, Herrera-Estrella A (1994) Characterization of ech-42, a Trichoderma harzianum endochitinase gene expressed during mycoparasitism. Proc Natl Acad USA 91:10903–10907
Carsolio C, Benhamou N, Haran S, Cortes C, Gutierrez A, Chet I, Herrera-Estrella A (1999) Role of the Trichoderma harzianum endochitinase gene, ech42, in mycoparasitism. Appl Environ Microbiol 65:929–935
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:56–59
Donzelli BGG, Harman GE (2001) Interaction of ammonium, glucose, and chitin regulates the expression of cell wall-degrading enzymes in Trichoderma atroviride strain P1. Appl Environ Microbiol 67:5643–5647
Druzhinia IS, Kopchinskiy AG, Komon M, Bisset J, Szakacs G, Kubicek CP (2005) An oligonucleotide barcode for species identification in Trichoderma and Hypocrea. Fungal Genet Biol 42:813–828
El-Bondkly AM (2006) Gene transfer between different Trichoderma species and Aspergillus niger through intergeneric protoplast fusion to convert ground rice straw to citric acid and cellulases. Appl Biochem Biotechnol 135:117–132
Foreman PK, Brown D, Dankmeyer L, Dean R, Diener S, Dunn-Colemann NS, Goedegebuur F, Houfek TD, England GJ, Kelley AS, Meerman HJ, Mitchell T, Mitchinson C, Olivares HA, Teunissen PJ, Yao J, Ward M (2003) Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J Biol Chem 278:31988–31997
Gielen S, Aerts R, Seels B (2004) Different products for biological control of Botrytis cinerea examined on wounded stem tissue of tomato plants. Commun Agric Appl Biol Sci 69:641–647
Harman GE (2000) Myths and dogmas of biocontrol. Changes perceptions derived from research on Trichoderma harzianum T22. Plant Dis 84:377–393
Harman GE, Howell CR, Viterbo A, Chet I, Lorito M (2004) Trichoderma species—opportunistic, avirulent plant symbionts. Nat Rev Microbiol 2:43–56
Inbar J, Chet I (1995) The role of recognition in the induction of specific chitinases during mycoparasitism by Trichoderma harzianum. Microbiology 141:2823–2829
Jiang H, Kang DC, Alexandre D, Fisher PB (2000) RaSH, a rapid subtraction hybridisation approach for identifying and cloning differentially expressed genes. Proc Natl Acad Sci USA 97:12684–12689
Kullnig C, Mach RL, Lorito M, Kubicek CP (2000) Enzyme diffusion from Trichoderma atroviride (=T. harzianum P1) to Rhizoctonia solani is a prerequisite for triggering of Trichoderma ech42 gene expression before mycoparasitic contact. Appl Environ Microbiol 66(5):2232–2234
Liu P, Yang Q (2005) Identification of genes with a biocontrol function in Trichoderma harzianum mycelium using the expressed sequence tag approach. Res Microbiol 156:416–423
Lopez-Fernandez LA, del Mazo J (1993) Construction of subtractive cDNA libraries from limited amounts of mRNA and multiple cycles of subtraction. Biotechniques 15:654–656 658–659
Lorito M, Mach RL, Sposato P, Strauss J, Peterbauer CK, Kubicek CP (1996a) Mycoparasitic interaction relieves binding of the Cre1 carbon catabolite repressor protein to promoter sequences of the ech42 (endochitinase-encoding) gene in Trichoderma harzianum. Proc Natl Acad Sci USA 93:14868–14872
Lorito M, Farkas V, Rebuffat S, Bodo B, Kubicek CP (1996b) Cell wall synthesis is a major target of mycoparasitic antagonism by Trichoderma harzianum. J Bacteriol 178(21):6382–6385
Marra R, Ambrosino P, Carbone V, Vinale F, Woo SL, Ruocco M, Ciliento R, Lanzuise S, Ferraioli S, Soriente I, Gigante S, Turra D, Fogliano V, Scala F, Lorito M (2006) Study of the three-way interaction between Trichoderma atroviride, plant and fungal pathogens by using a proteomic approach. Curr Genet 50:307–321
McKinney HH (1923) Influence of soil temperature and moisture on infection of wheat seedlings by Helminthosporium sativum. J Agric Res 26:195–217
Migheli Q, Balmas V, Komoń-Zelazowska M, Scherm B, Fiori S, Kopchinskiy A, Kubicek CP, Druzhinina I (2008) Soils of a Mediterranean hotspot of biodiversity and endemism (Sardinia, Tyrrhenian Islands) are inhabited by pan-European invasive species of Hypocrea/Trichoderma. Env Microbiol. doi:10.1111/j.1462-2920.2008.01736.x
Olmedo-Monfil V, Mendoza-Mendoza A, Gomez I, Cortes C, Herrera-Estrella A (2002) Multiple environmental signals determine the transcriptional activation of the mycoparasitism related gene prb1 in Trichoderma atroviride. Mol Genet Genomics 267:703–712
Pozo MJ, Baek JM, García JM, Kenerley CM (2004) Functional analysis of tvsp1, a serine protease-encoding gene in the biocontrol agent Trichoderma virens. Fungal Genet Biol 41:336–348
Prabavathy VR, Mathivanan N, Sagadevan E, Murugesan K, Lalithakumari D (2006) Self-fusion of protoplasts enhances chitinase production and biocontrol activity in Trichoderma harzianum. Bioresour Technol 97:2330–2334
Ramot O, Cohen-Kubiec R, Chet I (2000) Regulation of β-1, 3-glucansase by carbon starvation in the mycoparasite Trichoderma harzianum. Mycol Res 104:415–420
Rasmussen R (2001) Quantification on the LightCycler. In: Meuer S, Wittwer C, Nakagawara K (eds) Rapid cycle real-time PCR, methods and applications. Springer, Heidelberg, pp 21–34
Rozen S, Skaletsky H (1998) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Saloheimo M, Nakari-Setala T, Tenkanem M, Pentilla M (1997) cDNA cloning of a Trichoderma reesei cellulase and demonstration of endoglucanase activity by expression in yeast. Eur J Biochem 249:584–591
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual, 2nd edn. Cold Spring Harbour, NY
Samuels GJ (1996) Trichoderma: a review of biology and systematics of the genus. Mycol Res 100:923–935
Schmoll M, Zeilinger S, Mach RL, Kubicek CP (2004) Cloning of genes expressed early during cellulase induction in Hypocrea jecorina by a rapid subtraction hybridisation approach. Fungal Genet Biol 41:877–887
Seidl V, Marchetti M, Schandl R, Allmaier G, Kubicek CP (2006) Epl1, the major secreted protein of Hypocrea atroviridis on glucose, is a member of a strongly conserved protein family comprising plant defense response elicitors. FEBS J 273:4346–4359
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial analysis of gene expression. Science 270:484–487
Viterbo A, Ramot O, Chernin L, Chet I (2002) Significance of lytic enzymes from Trichoderma spp. in the biocontrol of fungal plant pathogens. Anton Leeuw Int J G 81:549–556
Vizcaino JA, Gonzalez FJ, Suarez MB, Redondo J, Heinrich J, Delgado-Jarana J, Hermosa R, Gutierrez S, Monte E, Llobell A, Rey M (2006) Generation, annotation, analysis of ESTs from Trichoderma harzianum CECT 2413. BMC Genomics 7:193
Vizcaino JA, Redondo J, Suarez MB, Cardoza RE, Hermosa R, Gonzales FJ, Rey M, Monte E (2007) Generation, annotation and analysis of ESTs from four different Trichoderma strains grown under conditions related to biocontrol. Appl Microbiol Biotechnol 75:853–862
Wieland I, Bolger G, Asouline G, Wigler M (1990) A method for difference cloning: gene amplification following subtractive hybridization. Proc Natl Acad Sci USA 87:2720–2724
Yedidia I, Benhamou N, Chet I (1999) Induction of defense responses in cucumber plants (Cucumis sativus L.) by the biocontrol agent Trichoderma harzianum. Appl Environ Microbiol 65:1061–1070
Acknowledgments
This work was carried out with the financial support of: Fondazione Banco di Sardegna, Italian Ministry of Foreign Affairs, Büro für Akademische Kooperation und Mobilität des Österreichischen Austauschdienstes, University of Sassari. Work in the laboratory of CPK and MS was supported by a grant from the Austrian Science Foundation (P17325-B17), which is gratefully acknowledged. MS is recipient of an APART-fellowship of the Austrian Academy of Sciences at the Institute of Chemical Engineering, Vienna University of Technology.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by U. Kück.
B. Scherm and M. Schmoll have equally contributed to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Scherm, B., Schmoll, M., Balmas, V. et al. Identification of potential marker genes for Trichoderma harzianum strains with high antagonistic potential against Rhizoctonia solani by a rapid subtraction hybridization approach. Curr Genet 55, 81–91 (2009). https://doi.org/10.1007/s00294-008-0226-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-008-0226-6