
Abstract
Frontotemporal lobar degeneration (FTLD) is a heterogeneous group of disorders characterized by disturbances of behavior and personality and different types of language impairment with or without concomitant features of motor neuron disease or parkinsonism. FTLD is characterized by atrophy of the frontal and anterior temporal brain lobes. Detailed neuropathological studies have elicited proteinopathies defined by inclusions of hyperphosphorylated microtubule-associated protein tau, TAR DNA-binding protein TDP-43, fused-in-sarcoma or yet unidentified proteins in affected brain regions. Rather than the type of proteinopathy, the site of neurodegeneration correlates relatively well with the clinical presentation of FTLD. Molecular genetic studies identified five disease genes, of which the gene encoding the tau protein (MAPT), the growth factor precursor gene granulin (GRN), and C9orf72 with unknown function are most frequently mutated. Rare mutations were also identified in the genes encoding valosin-containing protein (VCP) and charged multivesicular body protein 2B (CHMP2B). These genes are good markers to distinguish underlying neuropathological phenotypes. Due to the complex landscape of FTLD diseases, combined characterization of clinical, imaging, biological and genetic biomarkers is essential to establish a detailed diagnosis. Although major progress has been made in FTLD research in recent years, further studies are needed to completely map out and correlate the clinical, pathological and genetic entities, and to understand the underlying disease mechanisms. In this review, we summarize the current state of the rapidly progressing field of genetic, neuropathological and clinical research of this intriguing condition.
Similar content being viewed by others

Introduction
History and significance
Frontotemporal lobar degeneration (FTLD) is an anatomopathological descriptive term referring to a disorder characterized by the relatively selective atrophy of the frontal and anterior temporal lobes of the brain. Apart from this commonality, FTLD is a clinically, genetically and pathologically heterogeneous group of disorders. Because disease onset occurs before the age of 65 years in 75–80 % of the patients, FTLD is considered a presenile dementia [110, 122]. In the age group between 45 and 65 years, the prevalence of FTLD has been estimated between 10 and 30 per 100,000 [11, 136], but large-scale epidemiological studies are lacking. FTLD is the second most common form of early-onset dementia, after Alzheimer’s disease (AD) [19].
FTLD has a markedly strong familial component: 30–50 % of the FTLD patients report at least one relative with similar symptomatology, and in 10–23 %, the disease segregates in the family with an autosomal dominant inheritance pattern [56, 57]. Five disease genes have been identified to cause FTLD. Further, FTLD is associated with genes that typically cause a clinical picture of amyotrophic lateral sclerosis (ALS), the most common type of motor neuron disease (MND). Common genetic, clinical and pathological characteristics between FTLD and ALS are manifold. In this era of rapidly evolving insights into the pathophysiology of this challenging group of diseases, the present review aims to provide a timely picture of the clinical, neuropathological and genetic findings in FTLD and the correlations between them.
Clinical phenotypes of FTLD and related conditions
FTLD can manifest as two clinically recognized subtypes based on the presenting and predominant features of either behavioral and personality changes, or language disturbances. The behavioral variant of frontotemporal dementia (bvFTD) is characterized by severe changes in behavior and personality such as disinhibition, apathy, loss of empathy, or stereotypic behavior, leading to a loss of social competence [73, 109, 127, 151]. Executive functions are impaired, while at least in the initial stages of the disease, memory and perceptuospatial skills are well preserved. As the differential diagnosis in patients with psychiatric disturbances or AD is not always straightforward, the ‘International Behavioral Variant FTD Criteria Consortium’ developed international consensus criteria for bvFTD. According to these criteria, subclassifications were made in possible bvFTD defined by clinical criteria, probable bvFTD supported by neuro-imaging data, and definite bvFTD confirmation by neuropathological evidence or a pathogenic mutation [127]. bvFTD accounts for more than 50 % of the FTLD patients. The onset of bvFTD is typically before the age of 65 years, with an average onset age of 58 years [71, 73].
If the patient presents with language difficulties, a diagnosis of primary progressive aphasia (PPA) is made. PPA was originally further categorized into progressive nonfluent aphasia (PNFA) and semantic dementia (SD) [73]. However, the clinical picture of a number of PPA patients did not fit either diagnosis, which led to the description of the third variant, logopenic progressive aphasia (LPA). The lack of clear definitions of the three subtypes led in 2011 to new recommendations for the subclassification of PPA into nonfluent/agrammatic variant PPA (the former PNFA), semantic variant PPA (the former SD) and the logopenic variant PPA (also known as LPA) [59]. Nonfluent/agrammatic variant PPA or PNFA is characterized by effortful speech and grammatical error-making, with relatively preserved language comprehension. Apraxia of speech (AOS) or orofacial apraxia is frequently accompanying the aphasia [73]. PNFA is the second most prevalent presentation of FTLD, accounting for a large 25 % [71]. Semantic variant PPA or SD presents with impaired comprehension and conceptual knowledge with concomitant development of anomia, while speech production is spared [59, 109, 151]. SD presents in 20–25 % of the FTLD patients [71]. LPA is mostly associated with a neuropathological diagnosis of AD [121] and is not considered part of the FTLD group of disorders. Based on the evidence supporting the diagnosis of PPA, the label “possible” (clinical features), “probable” (clinical findings in combination with neuro-imaging) and “definite” (after post-mortem examination or when a gene mutation is known) are provided [59, 125].
Overlap between the clinical syndromes of bvFTD, PNFA and SD can occur during the progression of the disease and clinical distinction between them is often complicated in advanced disease stages. The median survival from the onset of symptoms is 6–11 years, independent of age at onset or gender [63, 130]. While some studies show no differences in survival rates among the clinical subtypes of FTLD, other studies suggested shorter survival in bvFTD and longest survival in SD [63, 71, 115, 130]. The gender distribution of FTLD appears to vary by the clinical syndrome, with a male preponderance in the behavioral and personality disorders and a female predominance in the language disorders [71].
The comorbidity of ALS with behavior alterations, cognitive impairment or dementia has been noticed since the early 20th century [91, 119, 129]. FTLD may precede, follow or coincide with the onset of motor symptoms [172]. In one of the largest studies on neuropsychological disturbances in ALS, 47 % of the patients showed some degree of dysfunction in frontal lobe tests, which was in 15 % sufficient for a diagnosis of FTLD [129]. Conversely, motor neuron dysfunction has been described in 40 % of the FTLD patients, and the diagnostic criteria for ALS were met in 15 %, referred to as FTLD-ALS [18, 90]. FTLD-ALS patients have a poor prognosis with a mean survival of 2–3 years from the onset of first symptoms [63, 73]. The reported heritability of FTLD-ALS is high: approximately 50 % is considered familial [56, 132, 141].
Apart from MND, other conditions are closely related to FTLD, including progressive supranuclear palsy (PSP) syndromes, corticobasal syndrome (CBS), FTD with parkinsonism (FTDP) and argyrophilic grain disease (AGD). The most common clinical syndromes are PSP syndromes with a prevalence of 3.1 per 100,000, followed by CBS with a prevalence of less than 1 per 100,000 [41]. PSP was originally described as a clinical syndrome characterized by extrapyramidal symptoms and a progressive dementia. Patients present with a symmetrical, akinetic rigid parkinsonism, severe postural instability and supranuclear ophthalmoplegia. Most patients develop the disease at middle or late age (75 ± 8 years) and death occurs on average 7 years later [35]. In more recent years, the spectrum of PSP syndromes has been extended to not only include classical PSP syndrome, termed the Richardson’s type, but also PSP-parkinsonism, which presents as a more asymmetrical disorder resembling Parkinson’s disease [177] and the syndrome of pure akinesia characterized by freezing of gait and speech dysfluencies as most prominent features [34, 177]. CBS most frequently presents as a combined clinical picture, consisting of a focal cortical deficit (e.g. limb apraxia, aphasia, frontal lobe syndrome, alien limb phenomenon, cortical sensory loss) and a progressive asymmetrical movement disorder (myoclonus, dystonia, tremor). In a later stage of the disease, patients often develop cognitive dysfunctions, sometimes in combination with a frontotemporal behavioral syndrome. The term FTDP-17 was defined in 1997 [47, 153], describing 13 families presenting with a clinical syndrome of autosomal dominant disinhibition, dementia, parkinsonism, and amyotrophy and showing genetic linkage to chromosome 17. Nowadays, we are able to differentiate mutations in the MAPT or to the GRN gene, both located on chromosome 17. The clinical picture resembles bvFTD, while cognitive deficits include anterograde memory dysfunction in an initial stage, later accompanied by progressive deterioration of visuospatial function, orientation and global memory. Eventually, mutism occurs. Motor signs typically include the development of symmetrical bradykinesia without resting tremor, in combination with axial rigidity and postural instability. There is poor or no effect of levodopa therapy. Other motor symptoms include vertical gaze palsy, dystonia, upper and lower motor dysfunction, eye lid apraxia and dysphagia [35]. The onset age can range from the early 20s to the late 70s, with an average of 50 years. The clinical early presentation of AGD is similar to AD but disease progression is less aggressive, with patients having a clinical picture resembling mild cognitive impairment (MCI) for many years [35, 44]. AGD accounts for approximately 5 % of the neurodegenerative dementia cases, with an increasing prevalence with advancing age [35, 44]. As FTLD is a distinctive clinical, genetic and neuropathological entity, it should be noted that an FTD phenotype can also be caused by underlying AD pathology (i.e. frontal variant AD). Similarly, a clinical phenotype of CBS can be caused by AD pathology [61].
Neuropathology of FTLD and cliniconeuropathological correlations
The FTLD brain is by definition characterized by diverse patterns of atrophy of frontal and anterior temporal lobes. Different patterns of atrophy have been described and a strong correlation with clinical phenotypes was found. A relatively symmetrical atrophy of the frontal lobes, insula, anterior cingulate and anterior temporal lobes is associated with bvFTD. An asymmetric atrophy of the left (linguistic dominant) anterior inferior temporal lobe gives rise to SD. Patients with an asymmetrical atrophy of the right anterior temporal lobe (right-sided SD) present with a behavioral syndrome similar to bvFTD. These patients develop emotional bluntness and, in some cases, loss of interest and bizarre affect. As the atrophy progresses, prosopagnosia and associative agnosia are often seen in combination with eating disorders and rigid, compulsive behavior. In patients with PNFA, an asymmetric atrophy involving the anterior perisylvian cortex, mainly of the dominant hemisphere, is seen [59, 122].
On microscopic examination, the neuronal loss and astrocytosis are seen in cortices of atrophied frontal and temporal lobes. In addition, FTLD is a proteinopathy characterized by the presence of abnormal, ubiquitinated protein inclusions in cytoplasm or nuclei of neuronal and glial cells. Adjunctive immunohistochemistry allows subcategorization of these disorders into specific proteinopathies based on the major constituent of the inclusions. From a historic perspective, two pathological categories of FTLD were initially made [20, 95, 103]: in a first group of patients, the disease presented neurons and glial cells containing inclusions of hyperphosphorylated tau protein, therefore referred to as FTLD-tau [97], including Pick’s disease (PiD). However, more than 50 % of the FTLD patients presented with tau-negative ubiquitin staining inclusions at the time of unknown composition, therefore referred to as FTLD-Ubiquitin or FTLD-U [90, 95]. In 80–95 % [94, 131] of this group, inclusions were later found to be composed of transactive response (TAR) DNA-binding protein 43 (TDP-43) [111], thus referred to as FTLD-TDP [95, 112]. A considerable number of TDP-43-negative FTLD-U cases had inclusions of fused-in-sarcoma protein (FUS), thus referred to as FTLD-FUS [96]. However, in a small number of FTLD-U patients, the inclusion protein remains unknown until today. This group is referred to as FTLD-ubiquitin proteasome system (FTLD-UPS) [36, 95].
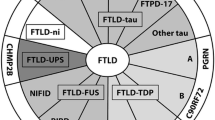
Correlations were noted between the clinical FTLD subtypes and underlying proteinopathies, but a strict one-to-one relationship is lacking. An FTLD-FUS proteinopathy is invariantly associated with a clinical diagnosis of bvFTD, either with or without the signs of MND. However, bvFTD is also associated with FTLD-tau and FTLD-TDP (Fig. 1). Although PNFA and SD are both associated with FTLD-tau and FTLD-TDP, PNFA is commonly associated with tau pathology, especially when AOS or orofacial apraxia is present. On the other hand, SD is, in most cases, linked with TDP-43-immunoreactive pathology, although tau pathology is sometimes observed. When SD is associated with FTLD-tau, patients often present with acalculia [122]. In right-sided SD, the underlying neuropathology is mostly a TDP-43 proteinopathy. Tau pathology was also associated with FTD with parkinsonism, PSP syndromes, CBS and AGD. Similarly, TDP-43 and FUS proteinopathies are also commonly found in MND with or without FTLD.

Diagram illustrating the clinical, genetic and neuropathological correlations in FTLD. The gray background of the genetics box represents the genetically unexplained fraction in FTLD cases overall (as compared to familial cases in Table 2)
Molecular genetics
Positive family history is observed in 40–50 % of the FTLD patients [85] and at least 10–50 % of the patients are associated with an inheritance pattern compatible with a highly penetrant Mendelian mutation [32, 107]. When considering clinical FTLD subtypes, family history is most prominent in bvFTD (45 %), especially when concomitant symptoms of MND are present (60 %), while SD appeared to be the least hereditary FTLD subtype (<20 %) [56]. Molecular genetic studies have identified five genes that, when mutated, cause FTLD: C9orf72, granulin (GRN), the microtubule associated protein tau gene (MAPT), the gene encoding valosin-containing protein (VCP) and the charged multivesicular body protein 2B (CHMP2B) [30]. Mutations in common ALS genes TARDBP and FUS (see ALS review in this issue), in rare cases, present clinically with FTLD [8, 171], even without the observed signs of MND (e.g. [68]). The relative mutation frequencies of these genes vary substantially among different populations due to founder effects resulting in a regionally high occurrence of one or a limited number of specific mutations. Also, reported mutation frequencies vary significantly between studies, depending on the method of patient ascertainment (e.g. population-based vs. hospital-based) and employed inclusion criteria (e.g. clinical vs. pathological diagnosis, familial vs. non-familial or both). However, in general terms, mutations in C9orf72, GRN and MAPT are the most common and together explain at least 17 % of the familial FTLD (Table 2). In two genetically fully documented FTLD series, summed C9orf72, GRN and MAPT mutation frequencies were 32 [33] and 40 % [55]. Mutations in VCP and CHMP2B are rare, each explaining less than 1 % of the familial FTLD.
As the mutant gene is initiating the biological disease processes underlying the neuropathological changes, a relatively strict correlation between the affected gene and associated neuropathology is observed (Fig. 1). However, due to the lack of a tight correlation between the type of pathology and the clinical manifestation thereof, the correlation between the disease gene and associated clinical phenotype is limited.
In addition to the highly penetrant Mendelian FTLD genes, few susceptibility genes are reported. From the Mendelian genes, MAPT appears to also harbor a genetic risk to develop tauopathies [6, 64]. The only systematic genome-wide association study in FTLD reported until today identified TMEM106B at chromosome 7p21 as a risk factor for FTLD-TDP [165].
Autosomal dominant genes
MAPT
The first significant genetic linkage found in FTLD families was with markers at chromosome 17q21 [69, 120, 154]. Linkage was described in 13 families in which a consensus clinical syndrome of autosomal dominant disinhibition, dementia, parkinsonism, and amyotrophy was found, termed FTDP-17 [47]. As tau was implicated in FTLD pathology and the chromosomal location of MAPT coincided with the linkage at chromosome 17, MAPT was the most obvious candidate gene and mutations were identified in FTDP-17 families [69]. Today, 44 different MAPT mutations are reported in 134 FTLD families [30] (Table 2). The mutations are mainly clustered in the five most 3′ exons from 9 to 13, encoding the four microtubule-binding domains of tau. In normal brain, the tau protein occurs as six isoforms of which three contain three microtubule-binding domains (3R tau) and three contain four microtubule-binding domains (4R tau) [41]. A substantial number of mutations were located in the intron 10 splice donor site or intronic splice regulatory elements resulting in aberrant splicing of exon 10. Interestingly, MAPT exon 10 encodes one of the microtubule-binding domains and the mutations affecting the splicing of this exon result in aberrant ratios of 3R and 4R tau [38]. Synonymous and nonsynonymous mutations in exon 10 were shown to locate in exonic splice regulatory elements resulting in similar aberrant splicing effects [39]. In addition, missense mutations are identified that affect the amino acid sequence of the microtubule-binding domains. Mutations affect the binding of tau to tubulin either due to an increased expression of 4R tau relative to 3R tau isoforms, or due to the altered binding properties of mutant tau protein [123]. The mutations thus disturb the subtle equilibrium between cytoskeletal assembly and disassembly affecting neuronal plasticity and axonal transport across the microtubules. In addition, coding MAPT mutations increase the tendency of tau to form neurotoxic aggregates. Mutations in exon 10 lead to 4R tau in both neurons and glia, while mutations outside exon 10 lead to neuronal accumulation of 3R tau and 4R tau [133]. Except for p.P301L and IVS10+16C>T that occur in 32 and 27 families, respectively, MAPT mutations are rare and seen in single families.
FTLD due to MAPT mutations is invariantly of the pathological type FTLD-tau, including PiD. In PiD pathology, the cardinal microscopic features are neuronal intracytoplasmatic spheroid tau-containing Pick bodies in the granular neurons of the dentate gyrus and in pyramidal neurons of the hippocampus and affected neocortical regions (Fig. 2a–c). Pick bodies, mainly localized in cortical layers II and III, and to a lesser extent also in deeper layers [41], specifically accumulate insoluble 3R tau [35, 41, 48, 73]. In addition to FTLD-tau, MAPT mutations have also been associated with other tauopathies including PSP, CBS, FTD with parkinsonism and the rarely seen AGD [73, 79]. The typical tau lesions in PSP are globose neurofibrillary tangles (NFT) found in neurons of the subcortical nuclei [35, 41, 73]. In CBS patients, heterogeneous tau-immunoreactive inclusions are also found in cortical neurons and neurons in substantia nigra and locus coeruleus. Other characteristic hallmarks are astrocytic plaques containing hyperphosphorylated tau and oligodendroglial inclusions [35, 41]. The neuropathological hallmark of FTD with parkinsonism is the combination of tau-immunoreactive aggregates in neurons, astrocytes and oligodendroglial cells, throughout cortex, deep gray and subcortical white matter [35, 41]. Often, these lesions are already extensive in early and intermediate stages of the disease. In AGD, on external macroscopic examination, mild diffuse cortical atrophy is seen, with a predilection of the ambient gyrus [35, 44]. The histopathological hallmark is the presence of argyrophilic grains (ArG). These ArG are present throughout the neuropil of cortical and subcortical structures. Lesions are initially present in the amygdala and temporal allocortex, spreading toward temporal neocortex as disease progresses.

FTLD-tau pathology in brain sections of a Pick disease patient with bvFTD after immunostaining with AT8 antibody. a Frontal cortex (F-cx), b temporal neocortex (T-cx), c hippocampus. Arrowheads indicate the characteristic Pick bodies representing neuronal intracytoplasmatic spheroid tau inclusions
FTLD-tau is mainly associated with bvFTD, however, forms of PPA are also reported [105]. Symptoms of MND are rare. On average, FTLD-tau is characterized by the earliest onset age in the FTLD syndromes, although the range of onset ages is large and strongly overlapping with other types of FTLD. A literature survey of 228 FTLD patients carrying a MAPT mutation demonstrated that the average onset age was 48 ± 10 years with a disease duration of 9 ± 5.5 years [27].
GRN
In some families linked to chromosome 17q21, extensive mutation analyses could not identify a MAPT mutation. It became apparent that, in FTLD families negative for MAPT mutations, the characteristic pathological inclusions were of the tau-negative, ubiquitin-positive types suggesting that mutations in another gene at the same genetic locus were causing this FTLD-U type of disease. Extensive mutation analyses of other positional candidate genes identified mutation in the nearby GRN gene [7, 28]. To date, 69 different GRN mutations have been reported in 231 families [30] (Table 2). GRN mutations are distributed across the complete coding region and splice sites of the gene. They are loss-of-function mutations leading to reduced functional protein and resulting in haplo-insufficiency [52]. Most mutations produce null alleles as a result of mRNA decay mediated by a nonsense or frameshift mutation [7, 28, 50], but other mechanisms including gene deletion [53] and defective protein sorting [143] are described as well. GRN encodes progranulin, a ubiquitously expressed growth factor precursor consisting of 7.5 granulin peptides [1]. Both full-length progranulin and the granulin peptides are implicated in a wide range of biological processes such as inflammation and wound repair, as well as in pathological conditions including tumorigenesis [62]. Although their roles in the CNS are not well established, in vitro and in vivo studies suggest a neurotrophic function involved in neuronal survival and neurite outgrowth [2, 138, 164, 178].
The effect of GRN missense mutations on reduced function and resulting phenotype is not straightforward: some compromise protein stability or cellular sorting, which might result in complete loss of function and lead to haplo-insufficiency, thereby behaving as highly penetrant FTLD mutations. Clear examples of loss-of-function (LOF) missense mutations are those disrupting the signal peptide [104, 143] or affecting the disulfide bonds controlling the characteristic granulin fold [166]. Other missense mutations might only partially compromise function and behave as risk alleles [29]. GRN missense mutations have been implicated in AD risk [15, 150] and survival in ALS [148]. A useful measure of the reduced GRN function, discriminating between pathogenic and neutral missense mutations, is the granulin protein level in cerebrospinal fluid [22, 164], plasma [21, 45] and serum [149], although the correlation between protein expression level and associated disease risk is poorly studied.
All GRN-associated FTLD patients have a FTLD-TDP proteinopathy [4, 117]. The presence of TDP-43 positive inclusions is invariably associated with a decreased physiologically normal nuclear staining [36, 95]. Recent neuropathological studies elicited four FTLD-TDP subtypes A to D based on the cortical distribution, intracellular location and morphology of the inclusions [99] (Table 1; Fig. 3). Based on the large clinicopathological studies, the most common FTLD-TDP subtype is type A accounting for 41–49 % of the cases, followed by type B with 28–34 % and type C with 17–25 %, while type D is rare [19, 73]. All GRN mutations are associated with FTLD-TDP type A (Fig. 3a–c), and in turn mutations in GRN explain about 40 % of these cases [133]. In FTLD-TDP type A, many neuronal cytoplasmic inclusions (NCI), short dystrophic neurites (DN) and some lentiform neuronal intranuclear inclusions (NII) are observed in layer II of the affected neocortex. NCI in the dentate granular gyrus of the hippocampus are variable in number [99].

Different types of TDP-43 pathology in brains of patients with GRN mutation, C9orf72 hexanucleotide repeat expansion and VCP mutation after immunostaining with TDP-43 antibody. a–c TDP-43 type A pathology in a bvFTD patient of family DR8, carrying the GRN IVS0 + 5G>C mutation [14, 28]. Arrowhead, double arrowhead and arrow show moderate NCI, DN and NII load, respectively, in layer II of temporal (a) and frontal (b) neocortex. c Higher magnification of NCI load in frontal cortex. d–f TDP-43 type B pathology in a bvFTD patient with the C9orf72 hexanucleotide repeat expansion [55]. Arrowhead and arrow show NCI and DN, respectively, spread throughout the entire cortical thickness in frontal cortex (d), temporal neocortex (e) and NCI in neostriatum (f, higher magnification). g–i TDP-43 type D pathology in a bvFTD patient carrying the VCP p.Arg159His mutation [168]. Note the extensive NCI (arrowhead), NII (double arrowhead) and DN (arrow) in frontal cortex (g), temporal neocortex (h, i, higher magnification)
Despite the fact that haplo-insufficiency is the common disease mechanism in all patients carrying a GRN mutation, the associated clinical phenotype is variable, including bvFTD and PNFA [88, 125]. Parkinsonian symptoms are often observed, but motor neuron symptoms are rare [23]. Sporadically, clinical diagnoses of related neurodegenerative diseases including AD and parkinsonian disorders have been associated with GRN mutations [14, 21, 22, 88]. A literature survey of 183 patients with GRN-associated FTLD demonstrated that the onset age was on average 61 ± 9 years and 13 years later than in MAPT mutation carriers although as in FTLD-tau, onset ages are highly variable, ranging from 35 to 87 years [29, 30]. A considerable age-dependent penetrance was described with 50–60 % of the mutation carriers being affected by the age of 60 years and 90–95 % by the age of 70 years [50, 125]. Consequently, family history is not always apparent and GRN mutations are found in a significant number of FTLD patients classified as being sporadic [87].
C9orf72
Genetic linkage [12, 54, 89, 93, 102, 162, 163] and association [83, 144, 170] studies in FTLD-TDP, FTLD-ALS and ALS suggested a common genetic defect located at chromosome 9p21 in the complete spectrum of diseases in the FTLD–MND complex [55]. Extensive mutation analyses of positional candidate genes identified an expanded noncoding G4C2 hexanucleotide repeat in C9orf72 explaining linkage and association [33, 55, 128]. In the normal population, the size of the G4C2 repeat ranges from 3 to 25 units, which is expanded to at least 60 units in patients [33, 55, 128]. Accurate size estimations of the expanded repeat in a substantial number of carriers are lacking, as the commonly used detection method based on the repeat-primed PCR does not differentiate repeat sizes >60 units. In one study, Southern blot analyses of a limited number of repeat expansion carriers estimated repeat sizes between 700 and 1,600 units [33]. In an extended study, pathological C9orf72 repeat expansions were observed in 11.4 % of the 1,381 FTLD patients of European origin, rising to 24.8 % in familial patients [101]. The same study reported a C9orf72 repeat expansion frequency of 6.0 % in sporadic FTLD. Whether this high mutation frequency in patients without family history of disease is explained by a high de novo repeat expansion rate remains to be determined, but a preliminary study suggests that this is more likely explained by incomplete penetrance [116].
C9orf72 encodes a ubiquitously expressed protein of unknown function. It is expressed as three major transcripts [33] and the expanded G4C2 repeat is located in the proximal regulatory region of C9orf72 [55], upstream of one and in the first intron of the two other transcripts. Repeat expansion results in near complete loss of expression of the major gene transcripts [33, 55, 128]. In addition, accumulation of transcripts harboring the expanded G4C2 repeat in nuclear RNA foci was described [33] although this was not replicated in another study [146]. Whether haplo-insufficiency due to the loss of transcription, RNA toxicity due to sequestration of RNA-binding proteins in RNA foci, and/or yet unidentified mechanisms are contributing to disease, needs further investigation.
Before the identification of the C9orf72 repeat expansions, FTLD linked to chromosome 9p21 was neuropathologically characterized as being of the TDP-43 proteinopathy type B. The pathological characteristics of FTLD-TDP type B (Fig. 3d–f) include NCI throughout the entire cortical thickness [99]. NCI are seen in the granular cells of the dentate gyrus of the hippocampus. In some patients, NCI can also be found in the motor neurons of the hypoglossal nuclei and ventral horn of the spinal cord. Next to these TDP-43 inclusions, granular “pre”-inclusions are seen in affected cortical regions [36]. Ubiquitin staining of these pre-inclusions is negative. Furthermore, GCI are often seen in brain stem and spinal cord [36]. When the pathological C9orf72 repeat expansion was identified, a larger number patients with neuropathological documentation could be attributed to this mutation, revealing a wider pathological diversity. In a substantial number of patients, the number and distribution of TDP-43 inclusions were more consistent with a TDP-43 type A proteinopathy [107] or intermediate to type B and type A proteinopathy [152]. Alternatively, the TDP-43 lesion load could be relatively limited or even absent resulting in a pathological diagnosis of FTLD-UPS in rare patients [55, 107]. Abundant p62 positive, TDP-43 negative NCI and rare NII were seen in the pyramidal cell layer of the hippocampus and cerebellar granular layer of most patients [3, 13]. Remarkably, similar brain inclusions were also observed in ALS patients without associated cognitive decline [158]. Interestingly, the sequestosome 1 gene (SQSTM1) encoding the p62 protein harbors mutations leading to ALS [42] and Paget disease of bone (PDB) [58]. Nevertheless, p62 immunoreactivity of the TDP-43-negative NCI might indicate general staining of proteins of the ubiquitin–proteasome system (UPS) for protein degradation, and the hallmark protein of these aggregations remains to be identified. The presence of DN and aggregates that were immunoreactive to ubiquilin (UBQLN) in the molecular layer of the hippocampus and throughout the neocortex was shown to be highly specific for the C9orf72 mutation. Further, UBQLN and p62 colocalized in NCI in the dentate gyrus of the hippocampus and the granular layer of the cerebellum [13]. Staining of C9orf72 itself has until today remained inconclusive. Polyclonal antibodies against both protein isoforms showed a punctiform staining of synaptic terminals in the CA4 region of the hippocampus in patients with the C9orf72 mutation, but also in patients with AD and controls [13, 33, 146].
C9orf72 repeat expansions clinically present with a widely variable phenotype including FTLD, ALS or FTLD-ALS [33, 55, 128]. With a mutation frequency of 30 % in FTLD-ALS [55], it is the only known common disease gene in this condition. Independent of concomitant ALS, FTLD is mostly of the behavioral type but patients with PPA have been described [33, 55, 146, 152]. Reported mean onset ages ranged between 55.3 and 58.3 years [33, 55, 146, 152] and are intermediate to onset ages in FTLD associated with MAPT and GRN. Also here, a high variability in onset age was noted, even among patients of the same family [33, 55]. Typical of repeat expansion diseases, genetic anticipation has been suggested with decreasing onset ages in younger generations [5, 24, 43, 156], and a decrease in onset age of 7 years in the younger of two subsequent generations has been suggested for ALS [24]. However, reports correlating sizes of the expanded repeat allele and onset age are lacking.
VCP
VCP mutations were identified by linkage analysis studies in autosomal dominant families with disabling muscle weakness due to inclusion body myopathy (IBM), osteolytic bone lesions consistent with PDB and FTLD (IBMPFD) [174]. Today, 17 different mutations have been identified in 41 independent families [30]. Incomplete penetrance was noted for all three clinical characteristic features and patients may present with classical FTLD.
VCP encodes a ubiquitously expressed member of a family of ATPases associated with a wide range of cellular functions through interactions with different adaptor proteins [175]. All IBMPFD mutations reside at the interface between the D1 ATPase and the N-domain of the CDC48-like protein [175]. The best supported hypotheses of the disease mechanism of VCP mutations are disturbed ubiquitin–proteasome mediated protein degradation [31], autophagy [74], or both [75]. FTLD patients with a VCP mutation are associated with TDP-43 proteinopathy type D [176] characterized by large numbers of NCI, NII and DN in affected neocortical regions [99] (Fig. 3g–i). Some inclusions also stain for VCP protein p97 [140].
Symptoms of FTLD due to a VCP mutation become apparent in the mid-50s in 25–30 % of the IBMPFD patients [76, 78, 174]. The incomplete penetrance of the three clinical characteristics IBM, PDB and FTLD is independent of the underlying mutation [76, 78, 108, 174]. However, in FTLD, VCP mutations are rare and represent less than 1 % of the familial FTLD (Table 2). The most frequently reported FTLD subtypes are bvFTD and SD [176]. Interestingly, a single study reported VCP mutations in 1–2 % of the ALS patients in the absence of dementia symptoms [72, 77].
CHMP2B
Linkage analyses in a large Danish FTLD family identified a mutation in CHMP2B at chromosome 3p11.2 [147]. CHMP2B encodes a component of the heteromeric ESCRT-III complex with functions in the endosomal–lysosomal and the autophagic protein degradation pathway. The gene is expressed in neurons of all major brain regions. Mutations affect the C-terminal end of the protein due to aberrant splicing.
CHMP2B mutations were associated with enlarged vacuoles in cortical neurons in the frontal, temporal, parietal and occipital cortices, due to impaired endosome–lysosome fusion [160], and impairment of autophagy [25]. Ubiquitin-immunoreactive NCI do not stain for tau, TDP-43 or FUS antibodies [65, 66], consistent with a pathological classification of FTLD-UPS [100].
The general clinical diagnoses in patients of the large Danish family corresponded to bvFTD, with early personality changes being the most common feature [60]. In other patients, progressive aphasia involvement was described, although no diagnosis of PNFA, SD or LPA could be made. The aphasia is characterized by a reduction in spontaneous speech sometimes leading to mutism and preserved reading and repetition, most consistent with a dynamic aphasia [70, 167]. The average onset age is 58 years, ranging between 46 and 65 years [60, 70]. The CHMP2B p.Gln206His mutation was reported in two ALS patients [25, 117]. Other missense mutations have been described in patients with FTLD and/or MND; however, their pathogenic nature remains unclear [30].
TARDBP and FUS
Mutations in the genes encoding the TDP-43 (TARDBP) and fused-in-sarcoma (FUS) proteins are typically associated with ALS. TARDBP mutations were initially identified [155] as a direct consequence of the identification of TDP-43-derived protein species as the major constituent of the aggregates found in upper and lower motor neurons of ALS patients without SOD1 mutations and in FTLD-U [4, 117]. Whereas 5 % of the familial ALS patients have a TARDBP mutation, mutations are rarely found in FTLD and FTD-MND [8, 10].
TDP-43 is an RNA-binding protein that forms heterogeneous nuclear ribonucleoprotein complexes (hnRNP) which function in RNA processing activities of several cellular functions, including transcription, RNA splicing and microRNA processing [16, 17, 118]. Missense mutations were found in the C-terminal glycine-rich region involved in protein–protein interactions [118]. Similar to TARDBP, FUS is a member of the hnRNP family. Its location at chromosome 16p11.2, made it an excellent candidate gene to explain previously established genetic linkage to the same chromosomal region in multiple ALS families [137, 139] and mutation analyses of patients in these and other families identified FUS mutations [80]. Although FUS represents an ALS gene, a mutational analysis of the FUS gene in 122 patients with FTLD revealed one novel p.Met254Val mutation in a patient with pure bvFTD. The silico analysis of this missense mutation predicted a pathogenic affect; however, the biologic relevance of this mutation remains elusive [157, 171].
Interestingly, an accumulation of FUS protein in inclusion bodies in neuronal cytoplasm and nucleus was associated with three clinicopathological subtypes of FTLD, defined by specific characteristics and location of NCI and NII [92, 97, 98, 114]. The first type is pathologically characterized by severe atrophy of the caudate nucleus and the frontotemporal cortex. Compact, round to oval kidney-shaped NCI and vermiform NII are localized in neocortex, granule cells of the dentate gyrus, striatum and to a lesser extent in globus pallidus, thalamus and periaqueductal gray matter (Fig. 4a–c). The cerebellar cortex is never affected. NCI and NII are not detected upon hematoxylin–eosin staining and were not immunoreactive to intermediate neurofilament on IHC. This type of FUS pathology is associated with a severe clinical syndrome resembling bvFTD and is referred to as atypical FTLD-U (aFTLD-U) [94]. Onset of disease is early, in 30s or 40s, and disease duration is approximately 7 years [85]. aFTLD-U is the most frequent FTLD-FUS subtype, accounting for 7–20 % of FTLD-U cases [94, 131, 142, 161]. The second FTLD-FUS type is characterized by an asymmetric atrophy of frontotemporal cortex and neostriatum, which is less severe than in aFTLD-U [85]. NCI and NII stain with FUS antibodies and to a lesser extent with antibodies against type IV interfilaments, alpha-internexin and neurofilaments. Therefore, this FTLD-FUS type is referred to as neuronal intermediate filament inclusion disease (NIFID) [94, 113]. Clinically NIFID patients usually develop a rapidly evolving FTD syndrome, mostly bvFTD, in combination with motor disorders such as parkinsonian or motor neuron symptoms. Patients usually develop the disease between 40 and 60 years of age, but earlier onset has been described and disease duration is on average approximately 3 years [85]. Considerable neuropathological similarities exist between aFTLD-U and NIFID; however, distinctive differences are observed. In NIFID, FUS-immunoreactive NCI are more extensive and widespread and are particularly numerous in CA1 and subiculum, but not in the granular layer of the dentate gyrus. The NCI morphology in NIFID patients is very heterogeneous, varying from small round to oval or tangle-like and annular shapes [73, 98, 100]. While less FUS-immunoreactive inclusions are seen in aFTLD-U, microscopic evaluation of aFTLD-U patients often shows sclerosis of the hippocampus and subcortical structures, for example, caudate nucleus, putamen and substantia nigra [85]. Another difference could be the presence of the NII, which are variably found in the hippocampus, but rare in other brain regions of NIFID patients, and are far more numerous in aFTLD-U patients [98, 100]. In the third FTLD-FUS type, referred to as basophilic inclusion body disease (BIBD), basophilic NCI staining with hematoxylin and eosin are seen in the pontine nuclei and to a lesser extent also in cerebral cortex [73, 98, 100, 106]. The basophilic NCI show FUS immunoreactivity in frontal cortex, basal ganglia and brain stem [98, 100]. BIBD presents with a clinical syndrome of early-onset ALS, occasionally accompanied with bvFTD.

aFTLD-U patient with FUS pathology without any known mutation after immunostaining with FUS antibody. The arrowhead shows the kidney-shaped NCI in affected neostriatum (a) and frontal cortex (b, c, higher magnification)
Susceptibility genes and risk loci
Compared with other complex neurodegenerative brain diseases including AD, little is known about susceptibility genes contributing to the risk of developing FTLD. This is mainly due to the fact that the familial component in FTLD is higher, and most research efforts aimed at the identification of Mendelian gene mutations. Also, the clinical and neuropathological heterogeneity of FTLD diseases have hampered large-scale genetic association studies in homogeneous patient series. One exception is the genome-wide association study in FTLD-TDP which has resulted in the identification of TMEM106B at chromosome 7p21 [165]. Nevertheless, other susceptibility genes are to be expected especially in the SD type of FTLD, in which family history is much less pronounced than in bvFTD and PNFA [32]. Consistent with this observation, mutations in the known FTLD genes were not associated with SD apart from a few atypical cases. Interestingly, SD is associated with a distinct type C of TDP-43 pathology suggesting a distinct disease mechanism [73]. FTLD-TDP type C is characterized by the presence of long DN localized in the superficial cortical layers, mainly layer II. NCI and NII are rare or absent compared to the other subtypes. Glial pathology is rare [99]. Together, these observations might suggest that SD is clinically, pathologically and genetically distinct from other types of FTLD and might predominantly be caused by the interaction of multiple yet unknown susceptibility genes.
TMEM106B was the only gene located in a single linkage disequilibrium block at 7p21 in which multiple single nucleotide polymorphisms (SNPs) were significantly associated in a series of 515 FTLD-TDP patients, a finding that was replicated in a second series of 89 FTLD-TDP patients [165]. In subsequent studies, association could be confirmed in one patient series [169], but not in another series [135]. It was found that TMEM106B may also contribute to the risk of developing FTLD in carriers of GRN mutations [165] possibly by modulating the levels of GRN secretion [26], however, other studies were not consistent with these findings [84, 169]. Interestingly, TMEM106B may also be associated with cognitive impairment in ALS [173].
TMEM106B is a type 2 integral membrane protein with unknown function, localizing to late endosomes and lysosomes [84]. The chromosome 7p21 risk haplotype was reported to act through altered TMEM106B gene expression in brain [165], although this was not confirmed in another study [169]. Interestingly, increased levels of TMEM106B were also associated with the inhibition of vacuolar H+-ATPases, a finding which may provide an unexpected biochemical link to GRN, since this protein is also strongly increased by the inhibition of vacuolar H+-ATPases [84].
Apart from the systematic genome-wide association study in FTLD-TDP [165], candidate gene association studies have reported genetic association of FTLD with other genes. In a series of pathologically confirmed FTLD-U patients without GRN mutations, a common genetic variant located in the 3′-untranslated region (UTR) of GRN in a binding site for miR-659 was identified as a major susceptibility factor for FTLD-U [126]. A significant reduction of GRN protein was observed in homozygous T-allele carriers in vivo, suggesting a similar mode of action as heterozygous loss-of-function mutations in GRN [126]. Another variant in the first intron of GRN, potentially affecting its expression, was also reported to be associated with FTLD in another patient series [49]. Nevertheless, other studies could not confirm the genetic risk for FTLD associated with variants in GRN [134, 152].
Besides harboring Mendelian FTLD mutations, MAPT was associated with risk of PSP [6, 64, 67], CBS [37, 67] and PD [40, 159], but inconsistent results were found in FTLD [9, 51, 86]. MAPT is represented in the human population as two genetically distinct haplotypes H1 and H2 due to its genomic location inside an inversion polymorphism [27]. The H1 haplotype is consistently overrepresented in 4R tau disorders, PSP and CBD [6, 67]. In young PSP patients, the risk was in part attributed to an SNP located in the large first intron of MAPT, which potentially modulated tau expression by modifying an LBP-1c/LSF/CP2 binding site. This transcription factor was shown to regulate the expression of genes in other neurodegenerative disorders [124]. A two-staged genome-wide association study in 2,165 PSP patients confirmed and extended these findings, and further implicated STX6, EIF2AK3, and MOBP in PSP [64]. Together, these genes suggested roles of vesicle-membrane fusion at the Golgi–endosomal interface, the endoplasmic reticulum unfolded protein response, and a myelin structure in PSP. The role of these genes in other tauopathies remains to be investigated.
Summary
FTLD is a clinically, neuropathologically and genetically heterogeneous group of disorders with plenty of overlaps between the neurodegenerative mechanism and the clinical expression thereof. As many clinical syndromes belonging to FTLD tend to overlap especially in advanced disease stages, e.g. SD and bvFTD in the TDP-43 proteinopathies, aFTLD-U and NIFID in the FUS proteinopathies, identifying the symptomatology of the patient at an early stage of the disease course is crucial. In that respect, concerted evaluation of clinical and pathological parameters is helpful to obtain a precise diagnosis. The general clinical presentation of FTLD concurs with macroscopic characteristics of brain atrophy including localization, symmetry and degree of atrophy. The underlying neuropathological abnormalities, i.e. the proteinopathies are closely linked with the disease mechanism and are important diagnostic markers. Detailed information on the underlying neuropathology thus provides useful insights for early diagnosis, disease course prognosis and patient counseling and treatment that could not be obtained by clinical evaluation alone. Imaging, biological and genetic biomarkers are valuable parameters to assess the underlying pathological characteristics of the disease. Further research is needed to elicit additional biomarkers to extend the ability to characterize neuropathological causes of disease and to further specify clinicopathological correlations.
Despite immense progress in defining neuropathological abnormalities in FTLD diseases, not all FTLD proteinopathies are fully characterized yet. In the small group of FTLD-UPS patients, the protein accumulating in pathological aggregates remains to be identified. Similarly, not all mutations underlying the proteinopathies have been identified. Especially in the TDP-43 proteinopathies, several yet unknown Mendelian and susceptibility genes are expected to play a role. Specifically for FTLD-TDP type C, no gene has yet been identified. This type of proteinopathy is associated with clinical SD, in which family history was reported to be the least prominent and future genome-wide association studies may provide further insights in the genetics of this disease.
References
Ahmed Z, Mackenzie IR, Hutton ML, Dickson DW (2007) Progranulin in frontotemporal lobar degeneration and neuroinflammation. J Neuroinflam 4:7
Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, Yamanouchi K, Leissring M, Petrucelli L, Nishihara M, Hutton ML, McGowan E, Dickson DW, Lewis J (2010) Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol 177(1):311–324
Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobágyi T, Shaw CE (2011) p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72 linked FTLD and MND/ALS. Acta Neuropathol 122(6):691–702
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351(3):602–611
Arighi A, Fumagalli GG, Jacini F, Fenoglio C, Ghezzi L, Pietroboni AM, De Riz M, Serpente M, Ridolfi E, Bonsi R, Bresolin N, Scarpini E, Galimberti D (2012) Early onset behavioral variant frontotemporal dementia due to the C9ORF72 hexanucleotide repeat expansion: psychiatric clinical presentations. J Alzheimers Dis (Epub ahead of print)
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M (1999) Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet 8(4):711–715
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442(7105):916–919
Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A, French Clinical and Genetic Research Network on Frontotemporal Lobar Degeneration/Frontotemporal Lobar Degeneration with Motoneuron Disease (2009) TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 65(4):470–473
Borroni B, Yancopoulou D, Tsutsui M, Padovani A, Sawcer SJ, Hodges JR, Spillantini MG (2005) Association between tau H2 haplotype and age at onset in frontotemporal dementia. Arch Neurol 62(9):1419–1422
Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, Papetti A, Stuani C, Di Luca M, Gennarelli M, Padovani A (2009) Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat 30(11):E974–E983
Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C, Agosti C, Cosseddu M, Turla M, Di Lorenzo D, Pietro Comi G, Gennarelli M, Padovani A (2010) TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res 13(5):509–517
Boxer AL, Mackenzie IR, Boeve BF et al (2010) Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry 82:196–203
Brettschneider J, Van Deerlin VM, Robinson JL, Kwong L, Lee EB, Ali YO, Safren N, Monteiro MJ, Toledo JB, Elman L, McCluskey L, Irwin DJ, Grossman M, Molina-Porcel L, Lee VM, Trojanowski JQ (2012) Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol 123(6):825–839
Brouwers N, Nuytemans K, van der Zee J, Gijselinck I, Engelborghs S, Theuns J, Kumar-Singh S, Pickut BA, Pals P, Dermaut B, Bogaerts V, De Pooter T, Serneels S, Van den Broeck M, Cuijt I, Mattheijssens M, Peeters K, Sciot R, Martin JJ, Cras P, Santens P, Vandenberghe R, De Deyn PP, Cruts M, Van Broeckhoven C, Sleegers K (2007) Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64(10):1436–1446
Brouwers N, Sleegers K, Engelborghs S, Maurer-Stroh S, Gijselinck I, van der Zee J, Pickut BA, Van den Broeck M, Mattheijssens M, Peeters K, Schymkowitz J, Rousseau F, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C (2008) Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71(9):656–664
Buratti E, Baralle FE (2001) Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276(39):36337–36343
Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F (2010) Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J 277(10):2268–2281
Burrell JR, Kiernan MC, Vucic S, Hodges JR (2011) Motor neuron dysfunction in frontotemporal dementia. Brain 134(Pt 9):2582–2594
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171(1):227–240
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM, Consortium for Frontotemporal Lobar Degeneration (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114(1):5–22
Carecchio M, Fenoglio C, De Riz M, Guidi I, Comi C, Cortini F, Venturelli E, Restelli I, Cantoni C, Bresolin N, Monaco F, Scarpini E, Galimberti D (2009) Progranulin plasma levels as potential biomarker for the identification of GRN deletion carriers. A case with atypical onset as clinical amnestic Mild Cognitive Impairment converted to Alzheimer’s disease. J Neurol Sci 287(1–2):291–293
Carecchio M, Fenoglio C, Cortini F, Comi C, Benussi L, Ghidoni R, Borroni B, De Riz M, Serpente M, Cantoni C, Franceschi M, Albertini V, Monaco F, Rainero I, Binetti G, Padovani A, Bresolin N, Scarpini E, Galimberti D (2011) Cerebrospinal fluid biomarkers in progranulin mutations carriers. J Alzheimers Dis 27(4):781–790
Chen-Plotkin AS, Martinez-Lage M, Sleiman PM, Hu W, Greene R, Wood EM, Bing S, Grossman M, Schellenberg GD, Hatanpaa KJ, Weiner MF, White CL 3rd, Brooks WS, Halliday GM, Kril JJ, Gearing M, Beach TG, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Pickering-Brown SM, Snowden J, van Swieten JC, Heutink P, Seelaar H, Murrell JR, Ghetti B, Spina S, Grafman J, Kaye JA, Woltjer RL, Mesulam M, Bigio E, Lladó A, Miller BL, Alzualde A, Moreno F, Rohrer JD, Mackenzie IR, Feldman HH, Hamilton RL, Cruts M, Engelborghs S, De Deyn PP, Van Broeckhoven C, Bird TD, Cairns NJ, Goate A, Frosch MP, Riederer PF, Bogdanovic N, Lee VM, Trojanowski JQ, Van Deerlin VM (2011) Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol 68(4):488–497
Chiò A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, Pugliatti M, Sotgiu MA, Murru MR, Marrosu MG, Marrosu F, Marinou K, Mandrioli J, Sola P, Caponnetto C, Mancardi G, Mandich P, La Bella V, Spataro R, Conte A, Monsurrò MR, Tedeschi G, Pisano F, Bartolomei I, Salvi F, Lauria Pinter G, Simone I, Logroscino G, Gambardella A, Quattrone A, Lunetta C, Volanti P, Zollino M, Penco S, Battistini S; ITALSGEN consortium, Renton AE, Majounie E, Abramzon Y, Conforti FL, Giannini F, Corbo M, Sabatelli M (2012) Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135(Pt 3):784–793
Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, Hollinger HC, Hartley JA, Brockington A, Burness CE, Morrison KE, Wharton SB, Grierson AJ, Ince PG, Kirby J, Shaw PJ (2010) Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 5(3):e9872
Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, Goate A (2011) Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 68(5):581–586
Cruts M, Rademakers R, Gijselinck I, van der Zee J, Dermaut B, de Pooter T, de Rijk P, Del-Favero J, van Broeckhoven C (2005) Genomic architecture of human 17q21 linked to frontotemporal dementia uncovers a highly homologous family of low-copy repeats in the tau region. Hum Mol Genet 14(13):1753–1762
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442(7105):920–924
Cruts M, Van Broeckhoven C (2008) Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet 24(4):186–194
Cruts M, Theuns J, Van Broeckhoven C (2012) Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. doi:10.1002/humu.22117
Dai RM, Li CC (2001) Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol 3(8):740–744
Davies RR, Hodges JR, Kril JJ, Patterson K, Halliday GM, Xuereb JH (2005) The pathological basis of semantic dementia. Brain 128(Pt 9):1984–1995
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2):245–256
De Letter M, Van Borsel J, Dewulf J, Santens P (2011) Dysfluencies in pure akinesia with gait freezing : two case reports. J Neurolinguist 24:352–356
Dickson D, Weller R (2011) Neurodegeneration: the molecular pathology of dementia and movement disorders, part 3. Wiley-Blackwell, West Sussex, pp 105–190
Dickson D, Weller R (2011) Neurodegeneration: the molecular pathology of dementia and movement disorders, part 7. Wiley-Blackwell, West Sussex, pp 389–436
Di Maria E, Tabaton M, Vigo T, Abbruzzese G, Bellone E, Donati C, Frasson E, Marchese R, Montagna P, Munoz DG, Pramstaller PP, Zanusso G, Ajmar F, Mandich P (2000) Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann Neurol 47(3):374–377
D’Souza I, Schellenberg GD (2000) Determinants of 4-repeat tau expression. Coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J Biol Chem 275(23):17700–17709
D’Souza I, Schellenberg GD (2002) tau Exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5′ and 3′ splice sites. J Biol Chem 277(29):26587–26599
Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Züchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74(2):97–109
Ellison D, Love S, Chimelli L, Harding BN, Lowe J, Vinters HV (2004) A reference text of CNS pathology. In: Chapter 9: neurodegenerative diseases Elsevier Limited. ISBN: 0 7234 3239 2
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud-Driss S, Sufit RL, Heller SL, Deng HX, Siddique T (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68(11):1440–1446
Ferrari R, Mok K, Moreno JH, Cosentino S, Goldman J, Pietrini P, Mayeux R, Tierney MC, Kapogiannis D, Jicha GA, Murrell JR, Ghetti B, Wassermann EM, Grafman J, Hardy J, Huey ED, Momeni P (2012) Screening for C9ORF72 repeat expansion in FTLD. Neurobiol Aging (Epub ahead of print)
Ferrer I, Santpere G, van Leeuwen FW (2008) Argyrophilic grain disease. Brain 31(Pt 6):1416–1432
Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132(Pt 3):583–591
Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu PH, Watts GD, Markesbery WR, Smith CD, Kimonis VE (2006) Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol 65(6):571–581
Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S (1997) Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference Participants. Ann Neurol 41(6):706–715
Fu YJ, Nishihira Y, Kuroda S, Toyoshima Y, Ishihara T, Shinozaki M, Miyashita A, Piao YS, Tan CF, Tani T, Koike R, Iwanaga K, Tsujihata M, Onodera O, Kuwano R, Nishizawa M, Kakita A, Ikeuchi T, Takahashi H (2010) Sporadic four-repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol 120:21–32
Galimberti D, Fenoglio C, Cortini F, Serpente M, Venturelli E, Villa C, Clerici F, Marcone A, Benussi L, Ghidoni R, Gallone S, Scalabrini D, Restelli I, Martinelli Boneschi F, Cappa S, Binetti G, Mariani C, Rainero I, Giordana MT, Bresolin N, Scarpini E (2010) GRN variability contributes to sporadic frontotemporal lobar degeneration. J Alzheimers Dis 19(1):171–177
Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL 3rd, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers R (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15(20):2988–3001
Ghidoni R, Signorini S, Barbiero L, Sina E, Cominelli P, Villa A, Benussi L, Binetti G (2006) The H2 MAPT haplotype is associated with familial frontotemporal dementia. Neurobiol Dis 22(2):357–362
Gijselinck I, Van Broeckhoven C, Cruts M (2008) Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mutat 29(12):1373–1386
Gijselinck I, van der Zee J, Engelborghs S, Goossens D, Peeters K, Mattheijssens M, Corsmit E, Del-Favero J, De Deyn PP, Van Broeckhoven C, Cruts M (2008) Progranulin locus deletion in frontotemporal dementia. Hum Mutat 29(1):53–58
Gijselinck I, Engelborghs S, Maes G et al (2010) Identification of 2 loci atchromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol 67:606–616
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Bäumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin JJ, De Deyn PP, Cruts M, Van Broeckhoven C (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11(1):54–65
Goldman JS, Farmer JM, Wood EM, Johnson JK, Boxer A, Neuhaus J, Lomen-Hoerth C, Wilhelmsen KC, Lee VM, Grossman M, Miller BL (2005) Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 65(11):1817–1819
Goldman JS, Adamson J, Karydas A, Miller BL, Hutton M (2007) New genes, new dilemmas: FTLD genetics and its implications for families. Am J Alzheimers Dis Other Demen 22(6):507–515
Goode A, Layfield R (2010) Recent advances in understanding the molecular basis of Paget disease of bone. J Clin Pathol 63(3):199–203
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014
Gydesen S, Brown JM, Brun A, Chakrabarti L, Gade A, Johannsen P, Rossor M, Thusgaard T, Grove A, Yancopoulou D, Spillantini MG, Fisher EM, Collinge J, Sorensen SA (2002) Chromosome 3 linked frontotemporal dementia (FTD-3). Neurology 59(10):1585–1594
Hassan A, Whitwell JL, Josephs KA (2011) The corticobasal syndrome-Alzheimer’s disease conundrum. Expert Rev Neurother 11(11):1569–1578
He Z, Bateman A (2003) Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (Berl) 81(10):600–612
Hodges JR, Davies R, Xuereb J, Kril J, Halliday G (2003) Survival in frontotemporal dementia. Neurology 61(3):349–354
Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC,Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P; PSP Genetics Study Group, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Müller U, Schellenberg GD (2011) Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 43(7):699–705
Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM (2007) A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol 66(10):884–891
Holm IE, Isaacs AM, Mackenzie IR (2009) Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol 118(5):719–720
Houlden H, Baker M, Morris HR, MacDonald N, Pickering-Brown S, Adamson J, Lees AJ, Rossor MN, Quinn NP, Kertesz A, Khan MN, Hardy J, Lantos PL, St George-Hyslop P, Munoz DG, Mann D, Lang AE, Bergeron C, Bigio EH, Litvan I, Bhatia KP, Dickson D, Wood NW, Hutton M (2001) Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology 56(12):1702–1706
Huey ED, Ferrari R, Moreno JH, Jensen C, Morris CM, Potocnik F, Kalaria RN, Tierney M, Wassermann EM, Hardy J, Grafman J, Momeni P (2011) FUS and TDP43 genetic variability in FTD and CBS. Neurobiol Aging (Epub ahead of print)
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393(6686):702–705
Isaacs AM, Johannsen P, Holm I, Nielsen JE, FReJA consortium (2011) Frontotemporal dementia caused by CHMP2B mutations. Curr Alzheimer Res 8(3):246–251
JohnsonJK Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, Chute DJ, Roberson ED, Pace-Savitsky C, Neumann M, Chow TW, Rosen HJ, Forstl H, Kurz A, Miller B (2005) Frontotemporal lobar degeneration. Demographic characteristics of 353 patients. Arch Neurol 62:925–930
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, Ding J, McCluskey L, Martinez-Lage M, Falcone D, Hernandez DG, Arepalli S, Chong S, Schymick JC, Rothstein J, Landi F, Wang YD, Calvo A, Mora G, Sabatelli M, Monsurrò MR, Battistini S, Salvi F, Spataro R, Sola P, Borghero G; ITALSGEN Consortium, Galassi G, Scholz SW, Taylor JP, Restagno G, Chiò A, Traynor BJ (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68(5):857–864
Josephs K, Hodges J, Snowden J, Mackenzie I, Neumann M, Mann D, Dickson D (2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 122:137–153
Ju JS, Weihl CC (2010) Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum Mol Genet 19(R1):R38–R45
Ju JS, Weihl CC (2010) p97/VCP at the intersection of the autophagy and the ubiquitin proteasome system. Autophagy 6(2):283–285
Kimonis VE, Mehta SG, Fulchiero EC, Thomasova D, Pasquali M, Boycott K, Neilan EG, Kartashov A, Forman MS, Tucker S, Kimonis K, Mumm S, Whyte MP, Smith CD, Watts GD (2008) Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A 146A(6):745–757
Koppers M, van Blitterswijk MM, Vlam L, Rowicka PA, van Vught PW, Groen EJ, Spliet WG, Engelen-Lee J, Schelhaas HJ, de Visser M, van der Kooi AJ, van der Pol WL, Pasterkamp RJ, Veldink JH, van den Berg LH (2011) VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging (Epub ahead of print)
Kovach MJ, Waggoner B, Leal SM, Gelber D, Khardori R, Levenstien MA, Shanks CA, Gregg G, Al-Lozi MT, Miller T, Rakowicz W, Lopate G, Florence J, Glosser G, Simmons Z, Morris JC, Whyte MP, Pestronk A, Kimonis VE (2001) Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab 74(4):458–475
Kovacs GG, Pittman A, Revesz T, Luk C, Lees A, Kiss E, Tariska P, Laszlo L, Molnár K, Molnar MJ, Tolnay M, de Silva R (2008) MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol 116(1):103–118
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH Jr (2009) Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 323(5918):1205–1208
Kumar-Singh S (2011) Progranulin and TDP-43: mechanistic links and future directions. J Mol Neurosci 45(3):561–573
Kwong LK, Uryu K, Trojanowski JQ, Lee VM (2008) TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals 16(1):41–51
Laaksovirta H, Peuralinna T, Schymick JC et al (2010) Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol 9:978–985
Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, Haass C (2012) Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem (Epub ahead of print)
Lashley T, Rohrer JD, Bandopadhyay R, Fry C, Ahmed Z, Isaacs AM, Brelstaff JH, Borroni B, Warren JD, Troakes C, King A, Al-Saraj S, Newcombe J, Quinn N, Ostergaard K, Schrøder HD, Bojsen-Møller M, Braendgaard H, Fox NC, Rossor MN, Lees AJ, Holton JL, Revesz T (2011) A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brain 134(Pt 9):2548–2564
Laws SM, Friedrich P, Diehl-Schmid J, Müller J, Ibach B, Bäuml J, Eisele T, Förstl H, Kurz A, Riemenschneider M (2008) Genetic analysis of MAPT haplotype diversity in frontotemporal dementia. Neurobiol Aging 29(8):1276–1278
Le Ber I, van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M, Laquerrière A, De Pooter T, Camuzat A, Van den Broeck M, Dubois B, Sellal F, Lacomblez L, Vercelletto M, Thomas-Antérion C, Michel BF, Golfier V, Didic M, Salachas F, Duyckaerts C, Cruts M, Verpillat P, Van Broeckhoven C, Brice A, French Research Network on FTD/FTD-MND (2007) Progranulin null mutations in both sporadic and familial frontotemporal dementia. Hum Mutat 28(9):846–855
Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, Hahn-Barma V, van der Zee J, Clot F, Bakchine S, Puel M, Ghanim M, Lacomblez L, Mikol J, Deramecourt V, Lejeune P, de la Sayette V, Belliard S, Vercelletto M, Meyrignac C, Van Broeckhoven C, Lambert JC, Verpillat P, Campion D, Habert MO, Dubois B, Brice A, French research network on FTD/FTD-MND (2008) Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain 131(Pt 3):732–746
Le Ber I, Camuzat A, Berger E et al (2009) Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology 72(The French Research Network on FTD/FTD-MND):1669–1676
Lipton A, White CL III, Bigio EH (2004) Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol 108:379–385
Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B (2003) Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 60(7):1094–1097
Love S, Spillantini MG (2011) Unpicking frontotemporal lobar degeneration. Brain 134:2453–2455
Luty AA, Kwok JB, Thompson EM et al (2008) Pedigree with frontotemporal lobar degeneration—motor neuron disease and TarDNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol 8:32
Mackenzie IR, Foti D, Woulfe J, Hurwitz TA (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131:1282–1293
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117(1):15–18
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119(1):1–4
Mackenzie IR, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9(10):995–1007
Mackenzie I, Munoz DG, Kusaka H, Yokota O, Ishihara K, Roeber S, Kretzschmar HA, Cairns NJ, Neumann M (2011) Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 121:207–218
Mackenzie I, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DMA, Lee VMY (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113
Mackenzie IR, Neumann M, Cairns NJ, Munoz DG, Isaacs AM (2011) Novel types of frontotemporal lobar degeneration: beyond tau and TDP-43. J Mol Neurosci 45(3):402–408
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O; Chromosome 9-ALS/FTD Consortium; French research network on FTLD/FTLD/ALS; ITALSGEN Consortium, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 11(4):323–330
Morita M, Al-Chalabi A, Andersen PM et al (2006) A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 66:839–844
Morris HR, Khan MN, Janssen JC, Brown JM, Perez-Tur J, Baker M, Ozansoy M, Hardy J, Hutton M, Wood NW, Lees AJ, Revesz T, Lantos P, Rossor MN (2001) The genetic and pathological classification of familial frontotemporal dementia. Arch Neurol 58(11):1813–1816
Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S, Behrens MI, Budde J, Hinrichs AL, Norton J, Levitch D, Taylor-Reinwald L, Gitcho M, Tu PH, Tenenholz Grinberg L, Liscic RM, Armendariz J, Morris JC, Goate AM (2006) HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol 60(3):314–322
Munoz DG, Ros R, Fatas M, Bermejo F, de Yebenes JG (2007) Progressive nonfluent aphasia associated with a new mutation V363I in tau gene. Am J Alzheimers Dis Other Demen 22(4):294–299
Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, Kuroda S, Mackenzie IR (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118(5):617–627
Murray ME, Dejesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW (2011) Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol 122(6):673–690
Nalbandian A, Donkervoort S, Dec E, Badadani M, Katheria V, Rana P, Nguyen C, Mukherjee J, Caiozzo V, Martin B, Watts GD, Vesa J, Smith C, Kimonis VE (2011) The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci 45(3):522–531
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51(6):1546–1554
Neary D, Snowden J, Mann D (2005) Frontotemporal dementia. Lancet Neurol 4(11):771–780
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314(5796):130–133
Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM (2007) TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol 64(10):1388–1394
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118(5):605–616
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132(Pt 11):2922–2931
Nunnemann S, Last D, Schuster T, Förstl H, Kurz A, Diehl-Schmid J (2011) Survival in a german population with frontotemporal lobar degeneration. Neuroepidemiology 37(3–4):160–165
Pamphlett R, Cheong PL, Trent RJ, Yu B (2012) Transmission of C9orf72 hexanucleotide repeat expansions in sporadic amyotrophic lateral sclerosis: an Australian trio study. NeuroReport 23(9):556–559
Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, Morrison KE, Pall HS, Hardiman O, Collinge J, Shaw PJ, Fisher EM; MRC Proteomics in ALS Study; FReJA Consortium (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67(6):1074–1077
Pesiridis GS, Lee VM, Trojanowski JQ (2009) Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet 18(R2):R156–R162
Phukan J, Pender NP, Hardiman O (2007) Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 6(11):994–1003
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43(6):815–825
Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, O’Neil JP, Lal RA, Dronkers NF, Miller BL, Gorno-Tempini ML (2008) Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol 64(4):388–401
Rabinovici GD, Miller BL (2010) Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 24(5):375–398
Rademakers R, Cruts M, van Broeckhoven C (2004) The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat 24(4):277–295
Rademakers R, Melquist S, Cruts M, Theuns J, Del-Favero J, Poorkaj P, Baker M, Sleegers K, Crook R, De Pooter T, Bel Kacem S, Adamson J, Van den Bossche D, Van den Broeck M, Gass J, Corsmit E, De Rijk P, Thomas N, Engelborghs S, Heckman M, Litvan I, Crook J, De Deyn PP, Dickson D, Schellenberg GD, Van Broeckhoven C, Hutton ML (2005) High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet 14(21):3281–3292
Rademakers R, Baker M, Gass J, Adamson J, Huey ED, Momeni P, Spina S, Coppola G, Karydas AM, Stewart H, Johnson N, Hsiung GY, Kelley B, Kuntz K, Steinbart E, Wood EM, Yu CE, Josephs K, Sorenson E, Womack KB, Weintraub S, Pickering-Brown SM, Schofield PR, Brooks WS, Van Deerlin VM, Snowden J, Clark CM, Kertesz A, Boylan K, Ghetti B, Neary D, Schellenberg GD, Beach TG, Mesulam M, Mann D, Grafman J, Mackenzie IR, Feldman H, Bird T, Petersen R, Knopman D, Boeve B, Geschwind DH, Miller B, Wszolek Z, Lippa C, Bigio EH, Dickson D, Graff-Radford N, Hutton M (2007) Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C-->T (Arg493X) mutation: an international initiative. Lancet Neurol 6(10):857–868
Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Caselli RJ, Wszolek ZK, Uitti RJ, Feldman H, Hutton ML, Mackenzie IR, Graff-Radford NR, Dickson DW (2008) Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet 17(23):3631–3642
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134(Pt 9):2456–2477
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M; ITALSGEN Consortium, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72(2):257–268
Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65(4):586–590
Roberson ED, Hesse JH, Rose KD, Slama H, Johnson JK, Yaffe K, Forman MS, Miller CA, Trojanowski JQ, Kramer JH, Miller BL (2005) Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology 65(5):719–725
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116:147–157
Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, Mackenzie IR, Warren JD, de Silva R, Holton J, Revesz T, Hardy J, Mead S, Rossor MN (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73(18):1451–1456
Rohrer JD, Warren JD (2011) Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol 24(6):542–549
Rollinson S, Rohrer JD, van der Zee J, Sleegers K, Mead S, Engelborghs S, Collinge J, De Deyn PP, Mann DM, Van Broeckhoven C, Pickering-Brown SM (2011) No association of PGRN 3′UTR rs5848 in frontotemporal lobar degeneration. Neurobiol Aging 32(4):754–755
Rollinson S, Mead S, Snowden J, Richardson A, Rohrer J, Halliwell N, Usher S, Neary D, Mann D, Hardy J, Pickering-Brown S (2011) Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol Aging 32(4):758.e1–758.e7
Rosso SM, Kamphorst W, de Graaf B, Willemsen R, Ravid R, Niermeijer MF, Spillantini MG, Heutink P, van Swieten JC (2001) Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q21-22. Brain 124(Pt 10):1948–1957
Ruddy DM, Parton MJ, Al-Chalabi A, Lewis CM, Vance C, Smith BN, Leigh PN, Powell JF, Siddique T, Meyjes EP, Baas F, de Jong V, Shaw CE (2003) Two families with familial amyotrophic lateral sclerosis are linked to a novel locus on chromosome 16q. Am J Hum Genet 73(2):390–396
Ryan CL, Baranowski DC, Chitramuthu BP, Malik S, Li Z, Cao M, Minotti S, Durham HD, Kay DG, Shaw CA, Bennett HP, Bateman A (2009) Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci 27(10):130
Sapp PC, Hosler BA, McKenna-Yasek D, Chin W, Gann A, Genise H, Gorenstein J, Huang M, Sailer W, Scheffler M, Valesky M, Haines JL, Pericak-Vance M, Siddique T, Horvitz HR, Brown RH Jr (2003) Identification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosis. Am J Hum Genet 73(2):397–403
Schröder R, Watts GD, Mehta SG, Evert BO, Broich P, Fliessbach K, Pauls K, Hans VH, Kimonis V, Thal DR (2005) Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann Neurol 57(3):457–461
Seelaar H, Schelhaas HJ, Azmani A, Küsters B, Rosso S, Majoor-Krakauer D, de Rijk MC, Rizzu P, ten Brummelhuis M, van Doorn PA, Kamphorst W, Willemsen R, van Swieten JC (2007) TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without progranulin mutations. Brain 130(Pt 5):1375–1385
Seelaar H, Klijnsma KY, de Koning I et al (2010) Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol 257:747–753
Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C (2008) Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem 283(3):1744–1753
Shatunov A, Mok K, Newhouse S et al (2010) Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol 9:986–994
Simón-Sánchez J, Seelaar H, Bochdanovits Z, Deeg DJ, van Swieten JC, Heutink P (2009) Variation at GRN 3′-UTR rs5848 is not associated with a risk of frontotemporal lobar degeneration in Dutch population. PLoS ONE 4(10):e7494
Simón-Sánchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, van den Berg LH, Schelhaas HJ, De Die-Smulders CE, Majoor-Krakauer D, Rozemuller AJ, Willemsen R, Pijnenburg YA, Heutink P, van Swieten JC (2012) The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135(Pt 3):723–735
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sørensen SA, Gydesen S, Fisher EM, Collinge J (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37(8):806–808
Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, van Vught PW, van der Zee J, Serneels S, De Pooter T, Van den Broeck M, Cruts M, Schymkowitz J, De Jonghe P, Rousseau F, van den Berg LH, Robberecht W, Van Broeckhoven C (2008) Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71(4):253–259
Sleegers K, Brouwers N, Van Damme P, Engelborghs S, Gijselinck I, van der Zee J, Peeters K, Mattheijssens M, Cruts M, Vandenberghe R, De Deyn PP, Robberecht W, Van Broeckhoven C (2009) Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 65(5):603–609
Sleegers K, Brouwers N, Van Broeckhoven C (2010) Role of progranulin as a biomarker for Alzheimer’s disease. Biomark Med 4(1):37–50
Snowden JS, Thompson JC, Stopford CL, Richardson AM, Gerhard A, Neary D, Mann DM (2011) The clinical diagnosis of early-onset dementias: diagnostic accuracy and clinicopathological relationships. Brain 134(Pt 9):2478–2492
Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, DuPlessis D, Neary D, Mann DM, Pickering-Brown SM (2012) Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 135(Pt 3):693–708
Spillantini MG, Bird TD, Ghetti B (1998) Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol 8(2):387–402
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319(5870):1668–1672
Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, DeJesus-Hernandez M, Eisen A, Rademakers R, Mackenzie IR (2012) Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 123(3):409–417
Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, Taroni F, Kwiatkowski TJ Jr, McKenna-Yasek DM, Sapp PC, Brown RH Jr, Landers JE (2009) Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73(15):1180–1185
Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklós L, Bell C, Smith B, Newhouse S, Vance C, Johnson L, Hortobágyi T, Shatunov A, Al-Chalabi A, Leigh N, Shaw CE, King A, Al-Sarraj S (2011) An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. doi:10.1111/j.1440-1789.2011.01286.x
UK Parkinson’s Disease Consortium; Wellcome Trust Case Control Consortium 2, Spencer CC, Plagnol V, Strange A, Gardner M, Paisan-Ruiz C, Band G, Barker RA, Bellenguez C, Bhatia K, Blackburn H, Blackwell JM, Bramon E, Brown MA, Brown MA, Burn D, Casas JP, Chinnery PF, Clarke CE, Corvin A, Craddock N, Deloukas P, Edkins S, Evans J, Freeman C, Gray E, Hardy J, Hudson G,Hunt S, Jankowski J, Langford C, Lees AJ, Markus HS, Mathew CG, McCarthy MI, Morrison KE, Palmer CN, Pearson JP, Peltonen L,Pirinen M, Plomin R, Potter S, Rautanen A, Sawcer SJ, Su Z, Trembath RC, Viswanathan AC, Williams NW, Morris HR, Donnelly P, Wood NW (2011) Dissection of the genetics of Parkinson’s disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 20(2):345–353
Urwin H, Authier A, Nielsen JE, Metcalf D, Powell C, Froud K, Malcolm DS, Holm I, Johannsen P, Brown J, Fisher EM, van der Zee J, Bruyland M; FReJA Consortium, Van Broeckhoven C, Collinge J, Brandner S, Futter C, Isaacs AM (2010) Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet 19(11):2228–2238
Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, Seelaar H, Van Swieten JC, Brown JM, Johannsen P, Nielsen JE, Holm IE; FReJA Consortium, Dickson DW, Rademakers R, Graff-Radford NR, Parisi JE, Petersen RC, Hatanpaa KJ, White CL 3rd, Weiner MF, Geser F, Van Deerlin VM, Trojanowski JQ, Miller BL, Seeley WW, van der Zee J, Kumar-Singh S, Engelborghs S, De Deyn PP, Van Broeckhoven C, Bigio EH, Deng HX, Halliday GM, Kril JJ, Munoz DG, Mann DM, Pickering-Brown SM, Doodeman V, Adamson G, Ghazi-Noori S, Fisher EM, Holton JL, Revesz T, Rossor MN, Collinge J, Mead S, Isaacs AM (2010) FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 120(1):33–41
Valdmanis PN, Dupre N, Bouchard JP et al (2007) Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol 64:240–245
Vance C, Al-Chalabi A, Ruddy D et al (2006) Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain 129:868–876
Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W (2008) Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol 181(1):37–41
Van Deerlin VM et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42(3):234–239
van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I, Camuzat A, Brouwers N, Vandenberghe R, Sleegers K, Hannequin D, Dermaut B, Schymkowitz J, Campion D, Santens P, Martin JJ, Lacomblez L, De Pooter T, Peeters K, Mattheijssens M, Vercelletto M, Van den Broeck M, Cruts M, De Deyn PP, Rousseau F, Brice A, Van Broeckhoven C (2007) Mutations other than null mutations producing a pathogenic loss of progranulin in frontotemporal dementia. Hum Mutat 28(4):416
van der Zee J, Urwin H, Engelborghs S, Bruyland M, Vandenberghe R, Dermaut B, De Pooter T, Peeters K, Santens P, De Deyn PP, Fisher EM, Collinge J, Isaacs AM, Van Broeckhoven C (2008) CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum Mol Genet 17(2):313–322
van der Zee J, Pirici D, Van Langenhove T, Engelborghs S, Vandenberghe R, Hoffmann M, Pusswald G, Van den Broeck M, Peeters K, Mattheijssens M, Martin JJ, De Deyn PP, Cruts M, Haubenberger D, Kumar-Singh S, Zimprich A, Van Broeckhoven C (2009) Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 73(8):626–632
van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, Santens P, Van den Broeck M, Joris G, Brys J, Mattheijssens M, Peeters K, Cras P, De Deyn PP, Cruts M, Van Broeckhoven C (2011) TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134(Pt 3):808–815
van Es MA, Veldink JH, Saris CG et al (2009) Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 41:1083–1087
Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijsens M, Peeters K, De Deyn PP, Cruts M, Van Broeckhoven C (2010) Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74:366–371
van Langenhove T, van der Zee J, van Broeckhoven C (2012) The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med (Epub ahead of print)
Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, Elman L, McCluskey L, Lee VM, Van Deerlin VM, Trojanowski JQ, Chen-Plotkin AS (2012) Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 121(3):373–380
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36(4):377–381
Weihl CC, Pestronk A, Kimonis VE (2009) Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul Disord 19(5):308–315
Weihl CC (2011) Valosin-containing protein associated fronto-temporal lobar degeneration: clinical presentation, pathologic features and pathogenesis. Curr Alzheimer Res 8(3):252–260
Williams DR, Pittman AM, Resesz T, Lees AJ, de Silva R (2007) Genetic variation at the tau locus and clinical syndromes associated with progressive supranuclear palsy. Mov Disord 22(6):895–897
Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A (2010) Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med 207(1):117–128
Acknowledgments
The authors are grateful to the participants to this study for their kind cooperation. We acknowledge the contribution of the lab technician Karen Sterck and photographer Inge Bats. We further thank the medical doctors in collaboration Dr. Adrian Ivanoiu, Dr. Alex Michotte, Dr. Jan Versijpt, Dr. Olivier Deryck, Dr. Chris Willems, Dr. Boudewijn Michielsens, Dr.Alexandra Keersmaekers, Dr. Dirk Ververken, Dr. Jen Maes, Dr. Philippe Maere, Dr. Marc Bruyland who recruited patients to this study and provided clinical data. We further thank the personnel of the Genetic Service Facility of VIB (http://www.vibgeneticservicefacility.be), the Antwerp Biobank of the Institute Born-Bunge and the neurological departments of the participating hospitals. None of the funding agencies listed below had a role in writing of the manuscript or the decision to submit for publication. All funding sources are listed and were from public or private research organizations. The research was in part funded by the European initiative on Centers of Excellence in Neurodegeneration (CoEN), the Special Research Fund of the University of Antwerp, the Research Foundation—Flanders (FWO) and the Agency for Innovation by Science and Technology—Flanders (IWT), the Methusalem excellence grant of the Flemish Government, the Interuniversity Attraction Poles program (IUAP) P7/16 of the Belgian Science Policy Office, the Stichting Alzheimer Onderzoek (SAO-FRMA) and the Queen Elisabeth Medical Foundation (QEMF). The FWO provided a clinical investigator mandate to A.S. and the IWT a PhD fellowship to T.V.L.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sieben, A., Van Langenhove, T., Engelborghs, S. et al. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 124, 353–372 (2012). https://doi.org/10.1007/s00401-012-1029-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-1029-x