
Abstract
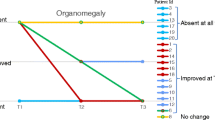
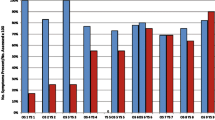
The current paper describes the natural history and management of mucopolysaccharidosis VI (MPS VI) in all patients currently diagnosed with the disease in Italy. Nine patients (5.5–14.4 years) were included in the data review in March 2008. Gestational and perinatal data were normal for all patients. Median age at diagnosis was 1.9 years. During the course of the disease, all patients developed coarsened facial features, short stature, heart valve disease, eye problems, musculoskeletal problems, hepatosplenomegaly and neurological abnormalities. All patients received rhASB enzyme replacement therapy (ERT) and showed improvement or stabilisation in clinical manifestations after onset of therapy. The most frequently reported improvements were increased joint mobility and reduced hepatosplenomegaly. No relevant safety issues of ERT were reported. In conclusion, patients in Italy with MPS VI are diagnosed early in life. All patients have access to ERT and appear to benefit from this therapy.

Similar content being viewed by others

References
Auclair D, Hopwood JJ, Brooks DA et al (2003) Replacement therapy in mucopolysaccharidosis type VI: advantages of early onset of therapy. Mol Genet Metab 78:163–174
Byers S, Nuttall JD, Crawley AC et al (1997) Effect of enzyme replacement therapy on bone formation in a feline model of mucopolysaccharidosis type VI. Bone 21:425–431
Crawley AC, Niedzielski KH, Isaac EL et al (1997) Enzyme replacement therapy from birth in a feline model of mucopolysaccharidosis type VI. J Clin Invest 99:651–662
Giugliani R, Harmatz P, Wraith JE (2007) Management guidelines for mucopolysaccharidosis VI. Pediatrics 120:405–418
Harmatz P, Giugliani R, Schwartz I et al (2006) Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr 148:533–539
Harmatz P, Giugliani R, Schwartz IVD et al (2008) Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab 94:469–475
Harmatz P, Ketteridge D, Giugliani R et al (2005) Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux–Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 115:e681–e689
Harmatz P, Whitley CB, Waber L et al (2004) Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). J Pediatr 144:574–580
Acknowledgments
The authors are grateful to Ismar Healthcare NV for their assistance in writing of the manuscript. They also thank the Italian MPS Society and the Fondazione Pierfranco e Luisa Mariani (Milano) for their contribution.
Conflict of interest
This study was supported by BioMarin Europe Ltd. All authors have received travel expenses for scientific meetings by BioMarin.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Scarpa, M., Barone, R., Fiumara, A. et al. Mucopolysaccharidosis VI: the Italian experience. Eur J Pediatr 168, 1203–1206 (2009). https://doi.org/10.1007/s00431-008-0910-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-008-0910-z