
Abstract
Intragenic homozygous deletions in the Large gene are associated with a severe neuromuscular phenotype in the myodystrophy (myd) mouse. These mutations result in a virtual lack of glycosylation of α-dystroglycan. Compound heterozygous LARGE mutations have been reported in a single human patient, manifesting with mild congenital muscular dystrophy (CMD) and severe mental retardation. These mutations are likely to retain some residual LARGE glycosyltransferase activity as indicated by residual α-dystroglycan glycosylation in patient cells. We hypothesized that more severe LARGE mutations are associated with a more severe CMD phenotype in humans. Here we report a 63-kb intragenic LARGE deletion in a family with Walker-Warburg syndrome (WWS), which is characterized by CMD, and severe structural brain and eye malformations. This finding demonstrates that LARGE gene mutations can give rise to a wide clinical spectrum, similar as for other genes that have a role in the post-translational modification of the α-dystroglycan protein.
Similar content being viewed by others


Introduction
Abnormal O-linked glycosylation of α-dystroglycan is the common pathogenic mechanism in a group of patients with a clinical spectrum ranging from severe congenital muscular dystrophy (CMD), structural brain, and eye abnormalities [Walker-Warburg syndrome (WWS), MIM 236670] to a relative mild form of limb-girdle muscular dystrophy (LGMD2I, MIM 607155) (van Reeuwijk et al. 2005a). In muscle tissue, α-dystroglycan acts as a bridge between the extracellular matrix laminin and the actin cytoskeleton through the transmembranal β-dystroglycan. The interaction of α-dystroglycan with the extracellular matrix proteins is mediated by its O-glycosylated moiety. Mature α-dystroglycan in brain binds to laminin and neurexin, which is important for neuronal migration in the neocortex and the integrity of the glia limitans (Barresi and Campbell 2006).
Mutations that result in hypoglycosylation of α-dystroglycan have been identified in known and putative glycosyltransferase genes: FCMD, FKRP, LARGE, POMGnT1, POMT1, and POMT2 (Freeze 2006). Four of these genes, FCMD, FKRP, POMT1, and POMT2, have been implicated in WWS. These genes explain approximately one-third of the WWS patients in our cohort. Interestingly, different mutations in these genes are known to cause phenotypic variability ranging from WWS to limb-girdle muscular dystrophy. Thus, it appears that mutations in genes that affect the glycosylation of α-dystroglycan can give rise to a number of related disorders, thereby blurring the boundaries between these clinically defined ‘dystroglycanopathies’ (Freeze 2006; Mercuri et al. 2006; van Reeuwijk et al. 2006; van Reeuwijk et al. 2005a). Here, we have investigated whether allelic variability does also occur for the LARGE gene.
The LARGE protein is a putative glycosyltransferase, required for addition of as yet unknown glycans onto the α-dystroglycan protein. Interestingly, overexpression of LARGE can by-pass the glycosylation defects in cells from patients with WWS or muscle-eye–brain disease (MEB, MIM 253280) (Barresi et al. 2004). Mutations in LARGE have been identified in a patient with relatively mild CMD and severe mental retardation (MDC1D: MIM 608840). This patient is compound heterozygous for p.E509K and p.C667fs. These mutations do not seem to abolish the activity of the LARGE protein completely, as Western blot analysis of skeletal muscle from this patient indicates residual functional glycosylation and laminin binding activity for α-dystroglycan (Longman et al. 2003). Interestingly, myd mice that carry a spontaneous deletion in Large (Largemyd), resemble the more severe clinical Fukuyama congenital muscular dystrophy (FCMD, MIM 253800), and MEB (Grewal et al. 2001; Longman et al. 2003; Michele et al. 2002).
Based on linkage studies we expect that there are multiple other genes that give rise to a WWS phenotype when mutated (van Reeuwijk et al. 2005b, and unpublished data). Here we report a homozygous 63-kb intragenic deletion in LARGE, in a patient who had familial classical WWS characteristics. This result establishes LARGE as the fifth WWS gene.
Patients and methods
Case reports
Patient 1 is a Saudi female and was delivered normally at term. Parents were second-degree cousins. Mother was gravida 5 para 4 with uneventful pregnancy, no polyhydramnios, or reduced fetal movements. Apgar scores were 5 and 8 at 1 and 5 min, respectively. Birth weight 2,880 g (25th centile), length 50 cm (50th centile), head circumference 37 cm (>90th centile). Physical examination (Fig. 1a) showed no dysmorphic features, severe generalized hypotonia with very little spontaneous movements of the limbs and valgus deformity of the feet. She had poor respiratory effort. Anterior fontanel was wide with separated sutures, deep tendon reflexes were absent, and sensations were intact. Ophthalmic examination revealed dense bilateral congenital cataract in the left eye and mild lens opacity with pigmentary degeneration of the retina and optic atrophy in the right eye. She was discharged from the Neonatal Intensive Care Unit (NICU) at the age of 3 months. At the age of 4 months her weight was on the 25th centile, length 10th centile and head circumference above 95th centile. She was developmentally delayed and blind. She died at the age of 6 months.

Phenotype and brain CT scans of patient 1 (a–c) and patient 2 (d–f). Patient 1 showed no dysmorphic features, severe generalized hypotonia with very little spontaneous movements of the limbs, and valgus deformity of the feet (a). Brain CT of patient 1 (b, c), and patient 2 (e, f) show absence of the inferior cerebellar vermis, a hypoplastic cerebellum, and marked dilatation of the lateral ventricles. Patient 2 also shows severe hydrocephalus with wide fontanel and separated sutures and cysts (d–f)
Laboratory investigations showed remarkably high-creatine kinase (CK) of 28,600 U/l (N 24–170) on the third day of life and 1,086 U/l at the age of 1 month. Liver function tests (LFT), metabolic screen, TORCH panel for congenital infections were all normal. Chromosome analysis was normal female karyotype. Brain CT (Fig. 1b, c) showed marked dilatation of the lateral ventricles with moderate dilatation of the third ventricle, there was a striking decrease in attenuation surrounding the dilated ventricles, the inferior cerebellar vermis was absent, and the cerebellum was hypoplastic. EEG showed frequent right tempro-occipital burst of sharp and slow activity. Brain auditory evoked responses (BAER) using monoaural click stimulation was normal. Muscle biopsy showed features of dystrophy. A diagnosis of WWS was made based on the CNS, eye, and muscle involvements. No DNA or tissue sample was available for this study.
Patient 2 is the younger sibling of patient 1. Antenatal ultrasound showed hydrocephalus and he was delivered by emergency cesarean section. Apgar scores were 6 and 9 at 1 and 5 min, respectively. Physical examination (Fig. 1d) showed severe hydrocephalus with wide fontanel and separated sutures, and head circumference was 46 cm, above the 97th centile. He had generalized hypotonia, absent deep tendon reflexes but no dysmorphic features. Ophthalmic examination showed bilateral leukocornia, retinal dysplasia, and posterior synechia. Brain CT (Fig. 1e, f) revealed severe hydrocephalus with Dandy-Walker malformation and minimal brain tissue, absent inferior cerebellar vermis and hypoplastic cerebellum. CK was elevated at 18,000 U/l (N 24–195). Lactate dehydrogenase was high at 1,690 U/l (N 230–460). Liver enzymes were mildly elevated, alanine aminotransferase (ALT) 53 U/l (N 10–50), and aspartate aminotransferase (AST) 142 U/l (N 10–45 U/l). Tandem mass spectrometry (MS) for metabolic screen was unremarkable; TORCH for congenital infections was negative. Chromosome analysis showed normal male karyotype. Muscle biopsy was done at the age of 11 days and revealed dystrophic features in the form of myofiber necrosis, basophilic fibers and interstitial endomysial, and perimysial fibrosis. There was no specific fiber type atrophy or grouping. A limited number of immunostains were done and included dystrophin and α-sarcoglycan. Both immunostains were normally positive.
A ventriculoperitoneal shunt was inserted. He was weaned off the ventilatory support and died at the age of 2 months. A diagnosis of WWS was made.
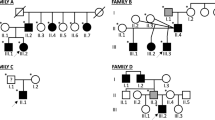
Nine other siblings were born healthy and DNA was obtained from seven, as well as the parents. A pedigree of the family is given in Fig. 2.

Family pedigree. Females are represented by circles, males by squares. Open symbols represent the unaffected family members, the solid black symbols the WWS affected siblings
Linkage analysis and mutation analysis
DNA was extracted from blood lymphocytes using standard procedures. Linkage to the LARGE locus was assessed by genotyping two microsatellite markers flanking the gene (D22S281 and D22S529) and two intragenic markers (D22S1162 and D22S1172).
Primer3 (http://www.frodo.wi.mit.edu) was used to design specific primers (supplementary Table 1) for PCR amplification and direct sequencing of the 14 coding exons (including intron–exon boundaries) of LARGE (NM_004737.3).
Copy number detection
Copy number detection of the 16 LARGE exons was performed by Multiplex Amplifiable Probe Hybridization (MAPH). A series of probes for the 16 LARGE exons were generated by PCR and cloned into pZERO (Invitrogen, Carlsbad, CA, USA). MAPH was carried out and probe ratios analyzed essentially as previously described (Armour et al. 2000; Hollox et al. 2002).
Multiplex Ligation-dependent Probe Amplification (MLPA) was used to further restrict the deletion breakpoints. MLPA probes were designed within exons 8–11 and intron 8 and 10 of the LARGE gene (supplementary Table 2). A protocol for designing these probes and hybridization, ligation, and amplification of these probes is provided by MRC-Holland (http://www.mlpa.com). Product separation was performed using capillary electrophoresis on an ABI 3730 or 3100 sequencer (Applied Biosystems, Foster City, CA). For quantitative analysis, trace data were retrieved using Genemapper software following the manufacturer’s protocol (Applied Biosystems).
Long-range PCR method and sequence analysis of breakpoints
The breakpoint-spanning region was amplified by long-range PCR amplification with specific primers (supplementary Table 3) and LA TaqTM (TaKaRa Bio Inc., Shiga, Japan) using PCR conditions recommended by the manufacturer. We then used different combinations of primers to further restrict the breakpoint-spanning region and identified the breakpoint by direct sequencing.
Results
Linkage to the LARGE locus and mutation analysis
We hypothesized that severe LARGE mutations give rise to WWS. To assess this hypothesis we selected intragenic and LARGE flanking genetic markers to test for homozygosity in 30 WWS patients from consanguineous parents. Seven patients from six unrelated families showed homozygosity for at least two intragenic markers (D22S1162 and D22S1172) and one marker (D22S281 or D22S529) in close proximity of LARGE.
We performed mutation analysis for all 14 coding exons including intron–exon boundaries by direct sequence analysis. No mutations were found. However, difficulties during the PCR amplification of some of the exons suggested a possible deletion of these exons in one of the families. Two affected siblings in this family manifest typical WWS features as described in the patients and methods section.
Identification of a submicroscopic deletion by copy number detection
Using MAPH we screened all 16 exons for copy number changes. Exon 9 and 10 showed copy number changes in one family, indicative for a deletion of these exons in patient 2 (homozygous) and in the parents (hemizygous). This result was confirmed by MLPA analysis of probes specific to these exons and surrounding intronic sequences, which also further defined the size of the deletion to 61.1–66.6 kb (Fig. 3).

MLPA analysis, showing a deletion of two exonic probes (EX9 and EX10) and two flanking intronic probes (IN8-2 and IN10) in the patient (diamonds), and in the unaffected carriers (black circles, triangles, and squares). No intronic probes were tested for the patient due to limited availability of DNA. Open circles, triangles, and squares depict control individuals
Analysis of breakpoints
By long-range PCR amplification we obtained a deletion-spanning fragment of ∼3.5 kb from patient 2. Sequence analysis of this fragment revealed the 5′ and 3′ breakpoints, and a deletion size of 63.1 kb (Fig. 4). This genomic deletion predicts a 239 bp deletion in the transcript resulting in a shift of the open reading frame within the first predicted catalytic domain, coding for 29 alternative codons followed by a premature stop codon, which most likely results in non-sense-mediated decay of the transcript. Unfortunately no cell-line or tissue sample was available to test this hypothesis. Only the affected individual was homozygous for the deletion, the parents and five of the seven analyzed unaffected sibs were heterozygous carriers of the deletion. No significant match between the 5′ and 3′ junction sequences is present, therefore the LARGE deletion is likely the result of non-homologous end-joining as reported also for the Large deletion in myd mice (Browning et al. 2005).

Schematic overview of LARGE exons 8–11 of wild-type sequence (a) and patient sequence (b), showing the exons depicted by black boxes and the MLPA probes depicted by black bars. MLPA analysis revealed deletion of four MLPA probes (IN8-2, EX9, EX10, and IN10). Sequence analysis of the breakpoint region in the patient revealed the exact position of the 63.1 kb deletion (c)
Discussion
We previously reported mutations in POMT1, POMT2, FCMD, and FKRP in approximately one-third of the WWS patients in our cohort (van Reeuwijk et al. 2005b). Here we report a homozygous 63-kb intragenic deletion in LARGE in a patient with WWS. The clinical features of the patients in this family do not diverge from the typical manifestations of other WWS patients. Hence, none of the five WWS genes that are known to date are associated with discriminating clinical features. The deletion described in this report is likely a loss of function mutation due to a predicted frameshift of the open reading frame within the first predicted catalytic domain. Mice carrying a similar disruptive defect in the Large gene display a severe muscle, eye, and brain phenotype, and have a shortened life span. With regard to the brain defects, these mice have severe neuronal migration defects resulting in a lissencephalic phenotype (Holzfeind et al. 2002; Lee et al. 2005; Mathews et al. 1995; Michele et al. 2002). The only previously known human LARGE mutations (p.E509K and p.C667fs) were identified in a patient with CMD, subtle structural brain abnormalities and severe mental retardation (MDC1D). The less severe clinical phenotype of this patient could be explained by residual activity of the LARGE protein. By an overlay assay, the authors demonstrated that residual α-dystroglycan present in a skeletal muscle biopsy in the patient retained laminin-binding, whereas this binding is lost in the myd mice (Holzfeind et al. 2002; Longman et al. 2003; Michele et al. 2002).
The existence of phenotypic variability for different mutations is also reported for other WWS genes. Mutations in the POMT1/2 genes were initially identified in WWS (Beltrán-Valero de Bernabé et al. 2002; van Reeuwijk et al. 2005b), but subsequently also in milder conditions including limb-girdle muscular dystrophy subtype 2K (LGMD2K, MIM 609308) (Balci et al. 2005; Mercuri et al. 2006; van Reeuwijk et al. 2006). Conversely, FKRP mutations are a common cause of LGMD, denoted subtype LGMD2I, but rare mutations are also found in severe conditions such as MEB and WWS, two similar disorders with CMD and severe brain and eye malformations (van Reeuwijk et al. 2005a). A common hypomorphic mutation in the FCMD gene causes FCMD in the Japanese population (Kobayashi et al. 1998). However, loss-of-function FCMD mutations are found in more severe conditions, including WWS (Beltrán-Valero de Bernabé et al. 2003). Finally, different mutations in POMGnT1 cause phenotypic variability within the MEB disease spectrum (Taniguchi et al. 2003; Yoshida et al. 2001).
LARGE is localized to the Golgi apparatus but the exact function of LARGE is unknown (Brockington et al. 2005; Grewal et al. 2005). It contains two putative catalytic domains, one related to a bacterial glycosyltransferase, and one related to a human glycosyltransferase (Grewal et al. 2001). In addition, LARGE interacts with the N-terminal domain of α-dystroglycan, which is essential for normal glycosylation of this protein (Kanagawa et al. 2004). Another remarkable finding is the therapeutic potential of LARGE, demonstrated by the recovery of dystroglycan processing and functioning in WWS/MEB fibroblasts by overexpression of the LARGE gene (Barresi et al. 2004).
Our finding demonstrates the existence of phenotypic variability, especially with regard to the brain, caused by different mutations in LARGE. We identified a mutation in this gene in 1 of 30 families, indicating that this gene is causal for only a small percentage of WWS patients. However, two spontaneous mouse mutants for Large, both due to intragenic deletions, have been reported in addition to the WWS patient described here (Grewal et al. 2001; Lee et al. 2005). The genomic size of LARGE may predispose this gene for genomic deletions. To exclude LARGE from genetic involvement in LGMD, or CMD with or without brain involvement it will be important to examine patients for genomic deletions in this gene.
References
Armour JA, Sismani C, Patsalis PC, Cross G (2000) Measurement of locus copy number by hybridisation with amplifiable probes. Nucleic Acids Res 28:605–609
Balci B, Uyanik G, Dincer P, Gross C, Willer T, Talim B, Haliloglu G, Kale G, Hehr U, Winkler J, Topaloglu H (2005) An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul Disord 15:271–275
Barresi R, Campbell KP (2006) Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci 119:199–207
Barresi R, Michele DE, Kanagawa M, Harper HA, Dovico SA, Satz JS, Moore SA, Zhang W, Schachter H, Dumanski JP, Cohn RD, Nishino I, Campbell KP (2004) LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med 10:696–703
Beltrán-Valero de Bernabé D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der ZB, Kayserili H, Merlini L, Chitayat D, Dobyns WB, Cormand B, Lehesjoki AE, Cruces J, Voit T, Walsh CA, van Bokhoven H, Brunner HG (2002) Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet 71:1033–1043
Beltrán-Valero de Bernabé D, van Bokhoven H, van Beusekom E, van den Akker W, Kant S, Dobyns WB, Cormand B, Currier S, Hamel B, Talim B, Topaloglu H, Brunner HG (2003) A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Genet 40:845–848
Brockington M, Torelli S, Prandini P, Boito C, Dolatshad NF, Longman C, Brown SC, Muntoni F (2005) Localization and functional analysis of the LARGE family of glycosyltransferases: significance for muscular dystrophy. Hum Mol Genet 14:657–665
Browning CA, Grewal PK, Moore CJ, Hewitt JE (2005) A rapid PCR method for genotyping the Large(myd) mouse, a model of glycosylation-deficient congenital muscular dystrophy. Neuromuscul Disord 15:331–335
Freeze HH (2006) Genetic defects in the human glycome. Nat Rev Genet 7:537–551
Grewal PK, Holzfeind PJ, Bittner RE, Hewitt JE (2001) Mutant glycosyltransferase and altered glycosylation of alpha-dystroglycan in the myodystrophy mouse. Nat Genet 28:151–154
Grewal PK, McLaughlan JM, Moore CJ, Browning CA, Hewitt JE (2005) Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 15:912–923
Hollox EJ, Atia T, Cross G, Parkin T, Armour JA (2002) High throughput screening of human subtelomeric DNA for copy number changes using multiplex amplifiable probe hybridisation (MAPH). J Med Genet 39:790–795
Holzfeind PJ, Grewal PK, Reitsamer HA, Kechvar J, Lassmann H, Hoeger H, Hewitt JE, Bittner RE (2002) Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Large(myd) mouse defines a natural model for glycosylation-deficient muscle-eye-brain disorders. Hum Mol Genet 11:2673–2687
Kanagawa M, Saito F, Kunz S, Yoshida-Moriguchi T, Barresi R, Kobayashi YM, Muschler J, Dumanski JP, Michele DE, Oldstone MB, Campbell KP (2004) Molecular recognition by LARGE is essential for expression of functional dystroglycan. Cell 117:953–964
Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T (1998) An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 394:388–392
Lee Y, Kameya S, Cox GA, Hsu J, Hicks W, Maddatu TP, Smith RS, Naggert JK, Peachey NS, Nishina PM (2005) Ocular abnormalities in Large(myd) and Large(vls) mice, spontaneous models for muscle, eye, and brain diseases. Mol Cell Neurosci 30:160–172
Longman C, Brockington M, Torelli S, Jimenez-Mallebrera C, Kennedy C, Khalil N, Feng L, Saran RK, Voit T, Merlini L, Sewry CA, Brown SC, Muntoni F (2003) Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet 12:2853–2861
Mathews KD, Rapisarda D, Bailey HL, Murray JC, Schelper RL, Smith R (1995) Phenotypic and pathologic evaluation of the myd mouse. A candidate model for facioscapulohumeral dystrophy. J Neuropathol Exp Neurol 54:601–606
Mercuri E, D’Amico A, Tessa A, Berardinelli A, Pane M, Messina S, van RJ, Bertini E, Muntoni F, Santorelli FM (2006) POMT2 mutation in a patient with ‘MEB-like’ phenotype. Neuromuscul Disord 16:446–448
Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP (2002) Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418:417–422
Taniguchi K, Kobayashi K, Saito K, Yamanouchi H, Ohnuma A, Hayashi YK, Manya H, Jin DK, Lee M, Parano E, Falsaperla R, Pavone P, Van Coster R, Talim B, Steinbrecher A, Straub V, Nishino I, Topaloglu H, Voit T, Endo T, Toda T (2003) Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum Mol Genet 12:527–534
van Reeuwijk J, Brunner HG, van Bokhoven H (2005a) Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet 67:281–289
van Reeuwijk J, Janssen M, van den Elzen C, Beltrán-Valero de Bernabé D, Sabatelli P, Merlini L, Boon M, Scheffer H, Brockington M, Muntoni F, Huynen MA, Verrips A, Walsh CA, Barth PG, Brunner HG, van Bokhoven H (2005b) POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet 42:907–912
van Reeuwijk J, Maugenre S, van den Elzen C, Verrips A, Bertini E, Muntoni F, Merlini L, Scheffer H, Brunner HG, Guicheney P, van Bokhoven H (2006) The expanding phenotype of POMT1 mutations: from Walker-Warburg syndrome to congenital muscular dystrophy, microcephaly, and mental retardation. Hum Mutat 27:453–459
Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell 1:717–724
Acknowledgments
We thank the families for contributing material for this study. This work was supported by grants from the ‘Prinses Beatrix Fonds’ and ‘Stichting Spieren voor Spieren’ (MAR02-226), and the ‘Hersenstichting Nederland’ (11F503.21). JEH thanks John Armour and Jess Tyson for advice on MAPH. JEH is a BBSRC Research Development Fellow. Funding from MDA USA, The Wellcome Trust and The BBSRC to JEH.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Reeuwijk, J., Grewal, P.K., Salih, M.A.M. et al. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet 121, 685–690 (2007). https://doi.org/10.1007/s00439-007-0362-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-007-0362-y