
Abstract
Whilst randomised control trials are undoubtedly the best way to demonstrate whether plasma exchange or infusion alone is the best first-line treatment for patients with atypical haemolytic uremic syndrome (aHUS), individual case reports can provide valuable information. To that effect, we have had the unique opportunity to follow over a 10-year period three sisters with aHUS associated with a factor H mutation (CFH). Two of the sisters are monozygotic twins. A similar natural evolution and response to treatment would be expected for the three patients, as they all presented with the same at-risk polymorphisms for CFH and CD46 and no identifiable mutation in either CD46 or CFI. Our report of different modalities of treatment of the initial episode and of three transplantations and relapses in the transplant in two of them, strongly suggest that intensive plasma exchange, both acutely and prophylactically, can maintain the long-term function of both native kidneys and allografts. In our experience, the success of plasma therapy is dependent on the use of plasma exchange as opposed to plasma infusion alone, the prolongation of daily plasma exchange after normalisation of haematological parameters followed by prophylactic plasma exchange, the use of prophylactic plasma exchange prior to transplantation and the use of prophylactic plasma exchange at least once a week posttransplant with immediate intensification of treatment if there are any signs of recurrence.
Similar content being viewed by others


Introduction
In ∼60% of patients with atypical haemolytic uremic syndrome (aHUS), mutations have been detected in complement components, including factor H (FH), membrane cofactor protein, factor I, factor B and C3 [1–5]. Factor H abnormalities are the most frequent, being found in ∼30% of patients. The prognosis in this group is poor, with most patients developing end-stage renal failure (ESRF) and a recurrence rate posttransplant of ∼80% [1, 6]. Although the use of plasma therapy is recommended, there is little consensus either with regard to efficacy or the optimal regimen to be used. To date, more than 100 FH mutations have been described (https://doi.org/www.fh-hus.org). The penetrance of these mutations is ∼50%, suggesting that other genetic and environmental factors act as modifiers [4]. Retrospective analysis of the response to plasma therapy in patients with FH mutations is supportive of a beneficial effect [2, 3]. To that effect, we have had a unique opportunity to follow over a 10-year period a family of three sisters with aHUS associated with a FH mutation (c.3572C>T, Ser1191Leu). The response to treatment has been of particular interest, as two of the sisters are monozygotic twins, and all three patients presented with the same at-risk polymorphisms for CFH and CD46. Therefore, they would be expected to present with a similar natural evolution of the disease and treatment response. Individual members of this family have been reported previously [7–10]. However, analysis of the whole family enables assessment of the effect of plasma therapy on the natural history of the disease in both native kidneys and allografts.
Case report
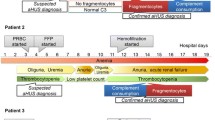
We summarise below our experience with plasma therapy in three sisters, two of whom are monozygotic twins, with atypical HUS associated with a FH mutation (c.3572C>T, Ser1191Leu) (Table 1).
In all three sisters, serum FH concentration has always been normal, and no evidence of systemic complement activation (such as a low C3) has ever been detected. Mutation screening of CFI and CD46 revealled no abnormalities. Genotyping of the CD46 single nucleotide polymorphism (SNP) *4074T>C (rs7144) showed that all three sisters were heterozygous for the at-risk C allele. Genotyping of CFH SNPs showed that all three sisters were homozygous for the at-risk T allele of -332C>T (rs3753394), heterozygous for the at-risk G allele of c.2016A>G, Q672Q (rs3753396) and heterozygous for the at-risk T allele of c.2808G>T, E936D (rs1065489). Using multiplex ligation-dependent probe amplification (MLPA), all three sisters were found to have two copies of CFHR1 and CFHR3.
The other family members (father, mother, an older brother and an older sister) were asymptomatic. The disease manifested first in the older sister when she was 3 years old and resulted immediately in ESRF. Her first renal transplant at the age of 5 years was lost to recurrent HUS within weeks of surgery. Plasma therapy was not given at that time.
One year later, one of the twins (twin 1) presented with the features of HUS. Plasma exchange (PE) was initiated, and ten daily sessions of PE (40 ml/kg per session) undertaken. Platelet count normalised, the evidence of haemolysis resolved but plasma creatinine decreased only slightly from 166 to 137 μmol/l. She received thereafter three sessions of five plasma infusions (10 ml/kg) in response to recurrent thrombocytopaenia and haemolysis with good effect. However, plasma creatinine continued to increase, and ESRF was reached 4 months after presentation.
Six months later, her twin sister (twin 2) presented with the disease. Because of the dramatic evolution of the two other sisters and the good response to plasma therapy in twin 1, an intensified plasma therapy protocol was implemented. Daily PE was performed until normalisation of plasma creatinine after 3 weeks (132 μmol/l to 61 μmol/l). Thereafter, PE was undertaken once every 2 weeks. Three and 19 months later, thrombocytopaenia and haemolysis in association with a respiratory tract infection prompted a temporary increase in the frequency of PE to daily. Following the second relapse, weekly PE was undertaken for 6 months, followed by a frequency of once every 2 weeks indefinitely. There were no further relapses, and renal function was normal six years after presentation.
This response to intensive PE prompted us to undertake transplantation with prophylactic plasma therapy in her two sisters. The older sister was transplanted for the second time at the age of 12 years with a cadaver kidney. Immunosuppression comprised prednisone, cyclosporine (maximum trough level 150 μg/ml), anti-interleukin-2-receptor (IL-2-R) antibodies and mycophenolate mofetil. PE was undertaken immediately prior to transplantation, daily for the first postoperative week, and then progressively tapered to once every 2 weeks by 2 months posttransplant. The graft functioned well (plasma creatinine 80 μmol/l) until 2 months posttransplant, when plasma creatinine increased to 200 μmol/l, without either evidence of thromboctyopaenia or significant haemolysis. A clinical diagnosis of acute rejection was made, and treatment commenced with pulse methylprednisolone. A renal biopsy performed 5 days later following a further deterioration in transplant function showed HUS, and despite an increase in the frequency of PE, the graft did not recover.
Twin 1 was the second to be transplanted, also with a cadaver kidney. We used an identical immunosuppressive regimen. PE was undertaken as in her older sister, but the frequency of PE was never decreased to less than once a week. There was immediate graft function, and plasma creatinine fell to 80 μmol/l within days. Ten months posttransplant, she presented with biopsy-proven recurrent HUS associated with primary cytomegalovirus (CMV) infection. The plasma creatinine was 167 μmol/l. After 4 weeks treatment with daily PE and ganciclovir, plasma creatinine stabilised at 90 μmol/l. When ganciclovir was stopped 6 months later, there was a further episode of recurrent HUS, and plasma creatinine increased to 187 μmol/l. Again, intensification of PE and the use of ganciclovir resulted in recovery of graft function. Continuous CMV prophylaxis with acyclovir was instituted, and no further episodes of either CMV reactivation or recurrent HUS were seen. Five years after transplantation, plasma creatinine was 127 μmol/l, and transplant biopsy showed allograft nephropathy but no HUS activity.
PE was performed using plasma filtration technique with a Gambro filter PF 1000 N and a Gambro device AK 200. Reactions to plasma were rarely observed and were always reversible using steroids and antihistaminic treatment.
Discussion
None of the affected members of this family showed evidence of systemic complement activation. This is in keeping with functional studies that show that mutations in the C-terminal domains of FH lead to reduced binding to surface-bound C3b and heparin, whereas the capacity to bind fluid-phase C3b is maintained [11]. The FH mutation (S1191L) found in the three affected sibs in this family has been reported previously in another HUS patient [12]. Although the functional significance of this mutation has not been examined, other missense mutations (W1183L, V1197A, R1210C, R1215G) in the same complement-control protein module (CCP 20) of FH have been shown to result in impaired protection of host surfaces against complement activation [11, 13]. It has been recently shown that multiple additive factors, including both mutations and susceptibility factors, in other complement genes are probably responsible for the ×50% penetrance seen in aHUS [4]. In these three patients, no mutations in either CD46 or CFI were detected, and there was no evidence of deletion of either CFHR1 or CFHR3, as previously described in association with aHUS [14]. However, all three presented with the same at-risk polymorphisms of CFH and CD46.
The efficacy of plasma therapy is difficult to assess in recent large series, because treatment modalities are poorly described [2, 3]. The general impression is that plasma therapy is beneficial in patients known to have either a CFH or CFI mutation, whereas it does not appear to influence the outcome in patients with an MCP mutation [2, 3].
It is possible that the improvement seen in twin 2 after her initial presentation was not related to PE, as spontaneous recovery has been reported previously [2]. This is unlikely because of the severity of the disease in her two sisters and the similar profile of CFH and CD46 susceptibility factors in the three patients. Our observation confirms previous reports on the role of plasma therapy in aHUS associated with FH deficiency [2, 3, 15–18]. It confirms also that plasma infusions do not always prevent deterioration of renal function or relapses, even when given prophylactically [18]. A possible cause of the failure of plasma infusion treatment reported in the latter study as well in twin 1 in this study may be that the volume of plasma allowed by infusion alone (10 ml/kg) is four times less than the amount used in PE. Another possible cause may be the type of mutation and the resulting functional defect. In the report of Nathanson and coworkers, a child with total FH deficiency due to a homozygous CFH mutation was successfully treated with plasma infusions [17]. In this patient, 13.5 ml/kg of fresh frozen plasma infused every 2 weeks was associated with a relapse-free period of several years. A similar successful short-term outcome was seen in another patient with complete FH deficiency by using plasma infusions (20 ml/kg) both during relapses and prophylactically [16]. The duration of the daily PE treatment may also be important, as the initial episode in twin 1 was treated daily for 10 days compared with 3 weeks for twin 2. The degree of renal failure at presentation does not seem to explain the different responses to PE, as the difference between plasma creatinine at treatment initiation in both twins was minimal (166 vs. 132 μmol/l).
The risk of recurrence of aHUS in allografts is at least 20% [1] and is higher in patients known to have an FH mutation [1]. The risk of early graft loss after recurrence in some series of familial HUS is close to 100% despite treatment [19]. For this reason, it has been suggested by some that transplantation is contraindicated in familial HUS [20]. A particularly high risk of relapse was observed in this family, as the illness recurred in the three transplants performed in two patients. However, PE profoundly influenced the natural history of the disease both in native kidney and transplants. The first transplant in the oldest sister was performed without prophylactic PE and was associated with immediate relapse and transplant loss. In the same patient, the second transplant performed with prophylactic PE had good function until 2 months posttransplant. At that time, PE frequency was reduced to once every 2 weeks, and at that time, there was a deterioration in transplant function. This was initially felt to be secondary to rejection and intensification of PE was delayed. Subsequently, the graft was lost. The third transplant, performed in twin 1 under PE prophylaxis, resulted in satisfactory long-term function (plasma creatinine 127 μmol/l after 5 years) despite several relapses associated with CMV infection.
In conclusion, in this family with atypical HUS associated with a FH mutation, we have shown that intensive and prophylactic PE has enabled maintenance of native and transplant renal function. The success of plasma therapy was associated with the use of PE and not plasma infusion, prolongation of daily PE after normalization of haemolysis followed by prophylactic PE, the use of prophylactic PE prior to transplantation and the use of a minimum long-term PE frequency of once a week and immediate intensification of PE frequency in case of relapse.
As mutations of FH are heterogeneous, our strategy of intensive and prophylactic PE may not be necessary in all patients with FH mutations. Indeed, several authors have reported success with plasma infusions [16, 17]. The strategy of prophylactic PE is expensive, necessitates creation of an arteriovenous fistula and implies life-long regular treatment in hospital. It is likely that purified FH will become available within the next few years, and this may provide sufficient FH in a small volume to replace PE. However, PE is not only a means to provide high volumes of plasma but also removes mutant FH. Recently, success has been obtained by combined kidney–liver transplantation with prophylactic PE immediately prior to transplantation [21]. Treatment choice should take into account the benefit versus risk as well as the local feasibility and financial aspects.
References
Kavanagh D, Goodship TH, Richards A (2006) Atypical haemolytic uraemic syndrome. Br Med Bull 77:1–18
Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G (2006) Genetics of HUS: the impact of MCP, CFH and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108:1267–1279
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, Boudaillez B, Bouissou F, Deschesnes G, Gie S, Tsimaratos M, Fischbach M, Morin D, Nivet H, Alberti C, Loirat C, French Society of Pediatric Nephrology (2007) Differential impact of complement mutations on clinical characteristics in atypical HUS. J Am Soc Nephrol 18:2392–2400
Saunders RE, Abarrategui-Garrido C, Frémeaux-Bacchi V, Goicoechea de Jorge E, Goodship THJ, Lopez Trascasa M, Noris M, Ponce Castro IM, Remuzzi G, Rodriguez de Cordoba S, Sanchez-Corral P, Skerka C, Zipfel PF, Perkins SJ (2007) The interactive Factor H-atypical hemolytic uremic syndrome mutation database and Website: Update and integration of membrane cofactor protein and factor I mutations with structural models. Hum Mutat 28:222–234
Geelen J, van den Dries K, Roos A, van de Kar N, de Kat Angelino K, Klasen I, Monnens L, van den Heuvel L (2007) A missense mutation in factor I (IF) predisposes to atypical haemolytic uraemic syndrome. Pediatr Nephrol 22:371–375
Zimmerhackl LB, Scheiring J, Prüfer F, Taylor M, Loirat C (2007) Renal transplantation in HUS patients with disorders of complement regulation. Pediatr Nephrol 22:10–16
Davin JC, Gruppen M, Bouts AHM, Groothoff JW, Florquin S, Weening JJ (1999) Relapse of atypical haemolytic uraemic syndrome after kidney transplantation: role of ATG and failure of mycophenolate mofetil as rescue therapy. Nephrol Dial Transplant 14:984–987
Olie KH, Florquin S, Groothoff JW, Verlaak R Weening JJ, Strain L, Goodship TH, Davin JC (2004) Atypical relapse of hemolytic uremic syndrome after transplantation. Pediatr Nephrol 19:1173–1176
Olie KH, Goodship TH, Verlaak R, Florquin S, Groothoff JW, Strain L, Weening JJ, Davin JC (2005) Posttransplantation cytomegalovirus-induced recurrence of atypical hemolytic uremic syndrome associated with a factor H mutation: successful treatment with intensive plasma exchanges and ganciclovir. Am J Kidney Dis 45:e12–15
Davin JC, Olie KH, Verlaak R, Florquin S, Weening JJ, Groothoff JW, Strain L, Goodship TH (2006) Familial hemolytic uremic syndrome associated with factor H mutation: prevention of chronic renal failure by continuous prophylactic plasma exchange. Am J Kidney Dis 47:e27–30
Sanchez-Corral P, Perez-Caballero D, Huarte O, Simckes AM, Goicoechea E, Lopez-Trascasa M, Rodriguez De Cordoba S (2002) Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet 71:1285–1295
Richards A, Buddles MR, Donne RL, Kaplan BS, Kirk E, Venning MC, Tielemans CL, Goodship JA, Goodship THJ (2001) Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet 68:485–490
Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, Jozsi M, Neumann HP, Remuzzi G, Zipfel PF (2003) Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest 111:1181–1190
Zipfel PF, Edey M, Heinen S, Jozsi M, Richter H, Misselwitz J, Hoppe B, Routledge D, Strain L, Hughes AE, Goodship JA, Licht C, Goodship AJ, Skerka C (2007) Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. Plos Genet 3:388–392
Gerber A, Kirchhoff-Moradpour AH, Obieglo S, Brandis M, Kirschfink M, Zipfel PF, Goodship JA, Zimmerhackl LB (2003) Successful (?) therapy of hemolytic uremic syndrome with factor H abnormality. Pediatr Nephrol 18:952–955
Cho HY, Lee BS, Moon KC, Ha IS, Cheong HI, Choi Y (2007) Complete factor H deficiency-associated atypical hemolytic uremic syndrome in a neonate. Pediatr Nephrol 22:874–880
Nathanson S, Frémeaux-Bacchi V, Deschênes (2001) Successful therapy in the haemolytic uremic syndrome with factor H deficiency. Pediatr Nephol 16:554–556
Ohali M, Shalev H, Schlesinger M, Katz Y, Kachko L, Carmi R, Sofer S, Landau D (1998) Hypocomplementemic autosomal recessive haemolytic uremic syndrome with decreased factor H. Pediatr Nephrol 12:619–624
Kaplan BS, Papadimitrion M, Brelin TH, Tomlanovitch JJ, Zulkharian J (1997) Renal transplantation in adults with autosomal recessive inheritance of haemolytic uremic syndrome. Am J Kidney Dis 30:760–765
Ruggenenti P (2002) Post-transplant haemolytic uremic syndrome. Kidney Int 62:1093–1104
Saland JM, Emre SH, Shneider BL, Benchimol C, Ames S, Bromberg JS, Remuzzi G, Strain L, Goodship TH (2006) Favorable long-term outcome after liver-kidney transplant for recurrent hemolytic uremic syndrome associated with a factor H mutation. Am J Transplant 6:1948–1952
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Davin, J.C., Strain, L. & Goodship, T.H.J. Plasma therapy in atypical haemolytic uremic syndrome: lessons from a family with a factor H mutation. Pediatr Nephrol 23, 1517–1521 (2008). https://doi.org/10.1007/s00467-008-0833-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-008-0833-y