
Abstract
Recent developments in methods to study virus internalisation are providing clearer insights into mechanisms used by viruses to enter host cells. The use of dominant negative constructs, specific inhibitory drugs and RNAi to selectively prevent entry through particular pathways has provided evidence for the clathrin-mediated entry of hepatitis C virus (HCV) as well as the caveolar entry of Simian Virus 40. Moreover, the ability to image and track fluorescent-labelled virus particles in real-time has begun to challenge the classical plasma membrane entry mechanisms described for poliovirus and human immunodeficiency virus. This review will cover both well-documented entry mechanisms as well as more recent discoveries in the entry pathways of enveloped and non-enveloped viruses. This will include viruses which enter the cytosol directly at the plasma membrane and those which enter via endocytosis and traversal of internal membrane barrier(s). Recent developments in imaging and inhibition of entry pathways have provided insights into the ill-defined entry mechanism of HCV, bringing it to the forefront of viral entry research. Finally, as high-affinity receptors often define viral internalisation pathways, and tropism in vivo, host membrane proteins to which viral particles specifically bind will be discussed throughout.
Similar content being viewed by others


Introduction
Viruses are obligate intracellular pathogens and entry into the host cell is a critical step in the viral life cycle. Cellular membranes present a barrier between the viral particle and intracellular site(s) of replication in the cytosol or nucleus. While enveloped viruses are bound by a lipid bilayer, non-enveloped viruses are surrounded by a proteinaceous capsid. Both enveloped viruses and non-enveloped viruses have evolved complex and often poorly defined mechanisms to enter cells. Identification of host cell receptors and their trafficking pathways provide tools to study viral entry. Although this generally involves the endocytic processes of the host cell, the exact entry mechanisms of many medically important viruses, such as hepatitis C virus (HCV), have yet to be fully elucidated.
The entry of both enveloped and non-enveloped viral particles requires specific interactions between host cell molecules, or receptors, and viral encoded envelope or capsid proteins. One key result of this is to bring the virus into close contact with the plasma membrane and in some documented cases, to initiate a cascade of signalling events important to the viral life cycle (Dangoria et al. 1996). In addition to primary receptors critical for virus attachment to the cell surface (e.g. CD4 for human immunodeficiency virus (HIV)), important co-receptors have been identified (e.g. chemokine receptors CXCR4 or CCR5 for HIV). It is now becoming apparent that a wide variety of host cell molecules are important for virus internalisation in the absence of any direct association with the virus particle. This is leading to the notion of “entry factors”, for example the tight junction proteins claudin-1 and occludin appear to have an indirect role in HCV entry (Ploss et al. 2009).
Following attachment to the host cell surface, virus entry at the plasma membrane has been described for many viruses such as HIV and Poliovirus (PV; Stein et al. 1987; Dunnebacke et al. 1969). Enveloped viruses can fuse directly with the plasma membrane, releasing the capsid directly into the cytosol, whilst non-enveloped viruses disrupt or form pore(s) in the plasma membrane to gain entry. In contrast to fusion at the plasma membrane, many viruses (such as SV40 and Influenza A) utilise intracellular trafficking pathways to fuse with internal membranes in order to release their genomic material into the cytosol (Anderson et al. 1996; Lakadamyali et al. 2004). The biophysical mechanisms of endosomal entry have been well documented (Cohen et al. 1984; Sarkar et al. 1989; Lee 2010).
Host cells offer a variety of internalisation mechanisms for virus entry, including: macropinocytosis (cell drinking), a process where substances present in extracellular fluid are internalised in a non-specific manner, and receptor-mediated endocytosis, including the well-described clathrin- and caveolar-dependent pathways, in addition to the more recently described lipid-raft-based endocytic mechanisms that are clathrin and caveolin independent. Receptor trafficking pathways often define particle internalisation routes and viruses typically enter the cell by a single defined pathway, although examples have been reported where viruses utilise multiple pathways in diverse cell types (Meier et al. 2002; Damm et al 2005; Patel et al 2009).
The majority of host cell membrane proteins internalise through clathrin-mediated endocytosis. Whilst the clathrin-mediated pathway is ubiquitous, caveolae have only been demonstrated in a limited number of cell types. Nevertheless, caveolar entry pathways have been described for a selected number of viruses including SV40 and human coronavirus 229E (Anderson et al. 1996; Nomura et al. 2004; Kawase et al. 2009). However, SV40 has also been reported to infect cells devoid of caveolae (Damm et al. 2005).
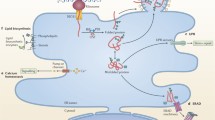
Non-enveloped and enveloped viruses enter the cytosol directly at the plasma membrane or via host cell endocytic pathways (Fig. 1). The mechanisms employed by non-enveloped and enveloped viruses to cross membrane barriers differ significantly, most likely as a consequence of the biophysical constraints imposed by the viral envelope (Cohen et al. 1984). The goal of this review is to summarise the different entry strategies employed by specific viruses and to highlight research areas where classical understanding of viral entry mechanisms have been challenged by more recent analyses. Finally, we will identify specific pharmacological and genetic manipulations which have been demonstrated to inhibit viral entry with medically relevant implications.

Conventional understanding of entry pathways for particular viruses. Both HIV and Polio virus have been considered able to directly penetrate the host cell plasma membrane. SV40 and coronavirus 229E have been shown to enter through caveolae, while adenovirus2/5, influenza and HCV can all infect through clathrin-mediated endocytosis. However, as described in the text, recent observations are challenging some of these conclusions
Non-enveloped viruses
Non-enveloped viruses can enter the cytosol by directly penetrating the plasma membrane, as well as through a variety of endocytic mechanisms leading to penetration of internal membrane(s). Internal membranes crossed by non-enveloped viruses include the endosomal membrane (e.g. adenovirus; Wickham et al. 1993), the Golgi (e.g. papillomavirus; Day et al. 2003) and the endoplasmic reticulum (e.g. SV40; Pelkmans et al. 2001; Schelhaas et al. 2007). Strategies to disrupt or traverse host cell membranes must be included in the mechanisms of non-enveloped virus entry. However, the precise molecular and biophysical means by which non-enveloped viruses gain entry to the cytosol have not been clearly defined in all cases. The entry mechanisms of the non-enveloped viruses PV, AdV 2/5 and SV40 will be discussed below.
Poliovirus
Early electron micrographs of PV-infected cells demonstrated virus particles adjacent to and directly penetrating the plasma membrane (Dunnebacke et al. 1969). Such evidence gave rise to a long-standing belief that PV entry occurs directly at the cell surface. However, use of drugs such as the ionophore monensin to dissipate cellular proton gradients suggested a pH-dependent route of PV entry (Madshus et al. 1984a, b), contradicting the earlier electron microscopic observations. It was known that under low pH, substances such as diphtheria toxin exposed a hydrophobic region which could insert into the membrane (Sandvig and Olsnes 1980). To investigate such a model for PV entry, Madshus and colleagues measured PV entry into the detergent phase of Triton-X114 under different pH conditions (Madshus et al. 1984a, b). The results indicated that PV was hydrophilic at neutral pH but became amphipathic at low pH, suggesting the exposure of hydrophobic regions or ‘fusion domains’. These data suggest PV association with cellular receptors induced the exposure of hydrophobic regions at low pH. Although it is possible that micro-regions of low pH occur at the cell surface, apart from specialised tissues such as the stomach, extracellular fluid is typically neutral, suggesting that PV enters cells via an acidic intracellular compartment.
Recent developments allowing the imaging of fluorescent PV in live cells have supported an endocytic internalisation route (Brandenburg et al. 2007). Furthermore, the involvement of actin in the entry process was implicated by inhibition of infection following incubation with cytochalasin D. Use of siRNA and inhibitors of clathrin-, caveolin- and flotillin-dependent pathways as well as macropinocytosis suggest that PV entry is independent of all these cellular pathways (Brandenburg et al. 2007). This study also used total internal reflection fluorescence microscopy to demonstrate that genome release into the cytosol occurs from recently internalised vesicles, close to the cell surface. Taken together, recent studies of PV entry highlight an endocytic event that is independent of the classical clathrin- and caveolin-mediated pathways, followed by low pH-mediated exposure of hydrophobic residues in an early endocytic vesicle. Recent reports of clathrin- and caveolin-independent routes of endocytosis await further clarification (reviewed in Kirkham and Parton 2005), and it is likely that studies of PV entry will provide important information about alternative endocytic mechanisms operating in host cells.
SV40
Although SV40 represents a virus with a particularly well-studied entry pathway, recent evidence suggests that this well-characterised route is not as clear cut as it may initially have seemed. SV40 became of particular interest to virologists and cell biologists alike as being the first virus shown to enter cells via a clathrin-independent endocytic pathway (Anderson et al. 1996). In fact it was, at least in part, the study of SV40 entry which led to the discovery of what we know as caveolar endocytosis and the caveosome, and as such, labelled SV40 is often used as a tool to study intracellular trafficking (Fig. 2).

Caveolar endocytosis of SV40. Fluorescence imaging of red-labelled SV40 (Texas Red) and green-labelled caveolae (Caveolin1-GFP) demonstrates a caveolar entry route for SV40 (adapted from Pelkmans et al. 2001; reprinted by permission from Macmillan Publishers Ltd., copyright 2001)
Early studies on SV40 entry found the internalisation pathway to differ in several ways from the classical clathrin-mediated endocytic process described for many viruses. Firstly, electron micrographs of SV40-infected cells showed the majority of particles in small tight-fitting vesicles, which appeared to be uncoated (Hummeler et al. 1970). These vesicles were able to fuse to generate larger compartments within the cell (which we now term caveosomes; Maul et al. 1978). Secondly, these vesicles could be tracked to the ER, rather than the endosomal/lysosomal compartment, which is the fate of most endocytosed viruses (Maul et al. 1978; Pelkmans et al. 2001; Norkin et al. 2002). Moreover, the ability of SV40 to internalise into different endocytic compartments was dependent on the capacity to be enveloped by cellular membranes. Subsequently, the caveolar entry pathway of SV40 was confirmed by the use of drugs found to disrupt caveolae, such as nystatin, which inhibited SV40 entry (Anderson et al. 1996).
However, as relatively stable membrane domains, it was unclear how caveolae could offer a productive entry pathway for SV40. The related murine polyoma virus has also been reported to internalise via caveolae (Richterová et al. 2001; Gilbert et al. 2003; Liebl et al. 2006), but, while SV40 employs the class I Major Histocompatability Complex as a receptor, murine polyoma virus interacts with sialyloligosaccharide receptors (Breau et al. 1992; Stehle and Harrison 1996). Furthermore, murine polyoma virus binding to host cells induces an up-regulation of primary and early response genes that regulate particle internalisation (Zullo et al. 1987; Glenn and Eckhart 1990). SV40–host cell interactions induce an intracellular signalling event that up-regulates the expression of primary response genes c-myc, c-jun, c-cis within 30 min and JE, a PDGF-inducible gene encoding monocyte chemoattractant MCP-1, within 90 min (Dangoria et al. 1996). SV40 up-regulation of both c-myc and c-jun was blocked by the tyrosine kinase inhibitor genistein. Furthermore, genistein was shown to block SV40 infection in a reversible manner, resulting in a model where SV40 binding induces a signalling pathway that primes particle internalisation. More recently, labelling of the virus and use of live-cell imaging have demonstrated a role for actin in SV40 internalisation and trafficking (Pelkmans et al. 2002). Upon virus binding, a transient breakdown of actin-stress fibres was observed. After leaving the caveosome, labelled SV40 trafficked in tubular, caveolin-free vesicles, which move along microtubules to fuse with smooth ER organelles (Pelkmans et al. 2001; Schelhaas et al. 2007).
Recent data showing SV40 entry into cells lacking detectable caveolae, including the human hepatoma Huh7 cell line and embryonic fibroblasts from a Cav1 knockout mouse, suggest alternative pathways may be in operation (Damm et al. 2005). The same authors noted caveolin-independent SV40 internalisation in wild-type embryonic fibroblasts with an active caveolar pathway (Damm et al. 2005). In all of the aforementioned cells, viruses were seen to internalise in small, tight-fitting vesicles similar to those seen in the first EM images of SV40-infected cells. SV40 was subsequently transported to pH-neutral organelles, which resembled caveosomes despite being devoid of both Cav1 and Cav2 (Damm et al. 2005). Importantly, expression of SV40 encoded T-antigen suggests these caveolin-independent pathway(s) represent productive infection. The observation that this entry mechanism was observed in cells with detectable Cav1, indicates that this alternative pathway is active even in the presence of Cav1. This could represent further evidence of host cell endocytic mechanisms acting independently of clathrin and caveolin, similar to that utilised by Adenovirus 2/5 (AdV 2/5).
Adenovirus 2/5
AdV 2 and 5 are non-oncogenic adenoviruses that infect the upper respiratory tract. Ad2 and Ad5 bind to coxsackie virus B Ad receptor (CAR; Leon et al. 1998). CAR is expressed at different levels on different tissues and is the primary determinant for susceptibility to Ad infection (Bergelson et al. 1997; Meier and Greber 2004). In addition to a requirement for CAR binding, activation of αv-integrin is necessary for virus entry into cells (Wickham et al. 1993; Greber et al 1993; Wang et al. 1998).
The first electron micrographs depicting AdV 2 entry suggested a role for coated vesicles early in the entry process (Brown and Burlingham 1973; Svensson 1985). Furthermore, the inhibitory effects of lysosomotropic agents on infection indicated that this was followed by escape from acidic endosomes (Svensson 1985). The use of drugs known to selectively inhibit receptor-mediated endocytosis reduced the extent of virus internalisation similarly to effects on transferrin entry (a marker for clathrin-mediated endocytosis; Varga 1991). Despite the mounting evidence for a clathrin-mediated entry pathway (Fig. 3), other studies reported direct penetration at the plasma membrane in addition to the proposed receptor-mediated endocytic pathway (Svensson 1985; Meier et al. 2002).

Although the use of dominant negative constructs of Eps15 (required for clathrin-mediated endocytosis) and dynamin (required for vesicle fission) seemed to confirm the clathrin-mediated entry pathway, AdV was reported to stimulate the uptake of fluid phase markers, largely via macropinocytosis (Wang et al. 1998; Meier et al. 2002). Whilst this could result from virus disruption of general membrane integrity, the process was shown to be dependent on αv-integrin, protein kinase C, F-actin and the amiloride-sensitive sodium/proton exchanger, all of which are involved in the AdV endosomal escape mechanism (Meier et al. 2002). Taken together, these results suggest a model whereby AdV 2/5 binds to CAR and activates integrin co-receptors leading to the clathrin-mediated endocytosis of the virus. This is followed by trafficking to the early endosome, where low pH mediates endosomal escape (Gastaldelli et al. 2008). Alongside endosomal escape, macropinocytosis is stimulated leading to the non-specific internalisation of virus particles from the extracellular environment. It is unclear whether this is a coincidental side effect of adenovirus infection, or whether this represents a manipulation of host cell machinery to ensure the uptake of more virions.
As the name implies, CAR also acts as the receptor for coxsackie B group viruses (CVB), which are known to infect polarised epithelia (Coyne and Bergelson 2005). As CAR is a component of tight junctions, it is inaccessible to virus on the apical side of the epithelium, for example, viral particles in the intestinal lumen. Many isolates of CVB interact with decay accelerating factor (DAF or CD55), a GPI-anchored protein, which is expressed on the apical surface, and is accessible to virus. Virus attachment to DAF on the apical surface is thought to trigger signalling events culminating in actin rearrangement and virus movement to the tight junction (Coyne and Bergelson 2006; Coyne et al. 2007). Signalling events induced by virus interaction with DAF are thought to induce caveolar endocytosis of virus at the tight junction (Coyne et al. 2007; Coyne et al. 2009). Moreover, although not involved in CVB binding, particle internalisation from the tight junction is dependent on occludin (Coyne et al. 2007). Cell polarity and viral receptors localised to the tight junction are also important factors in hepatitis C virus entry, which will be discussed below.
Enveloped viruses
Infection with an enveloped virus requires the fusion of the viral envelope with a cellular membrane. In some cases, this can occur at the plasma membrane, as reported for HIV, where binding to plasma membrane-expressed forms of CD4 and chemokine receptors induce changes in the viral envelope glycoprotein that are thought to mediate membrane fusion under neutral pH conditions. Fusion of other enveloped viruses occurs within the low-pH environment of an acidic endosomal compartment. Enveloped viruses typically reach the endosomal compartment via trafficking in clathrin-coated vesicles, although a caveolar route of entry has been reported for human coronavirus 229E (Nomura et al. 2004; Kawase et al. 2009). Examples of enveloped viruses with entry mechanisms of particular interest are HCV, influenza A and HIV and will be discussed in more detail below.
HCV
Hepatitis C virus is a hepatotropic enveloped virus associated with liver disease, fibrosis, cirrhosis and hepatocellular carcinoma. The HCV particle comprises a single-stranded positive-sense RNA surrounded by an icosahedral capsid and envelope derived from a host cell lipid bilayer (Bartosch and Cosset 2006). HCV encodes two envelope glycoproteins E1 and E2 which play a critical role in binding host cell surface receptors and membrane fusion (Keck et al. 2004; Op De Beeck et al. 2004; Helle and Dubuisson 2008). Recombinant forms of HCV E2 were used to identify interaction(s) with host cell proteins, tetraspanin CD81 (Pileri et al. 1998; Bartosch et al. 2003a; Cormier et al. 2004; Flint et al. 2006) and scavenger receptor B type I (SR-BI; Scarselli et al. 2002; Grove et al. 2007; Catanese et al. 2010). The recent development of infectious lentiviral pseudoparticles bearing HCV glycoproteins (HCVpp; Bartosch et al. 2003b; Hsu et al. 2003; Drummer et al. 2003), and native HCV capable of replication in cell culture (HCVcc; Lindenbach et al. 2005; Wakita et al. 2005; Zhong et al. 2005) have enabled studies to validate the role of CD81 and SR-BI in HCV entry. Following development of these experimental systems, the tight junction proteins Claudin-1 and Occludin were recently reported to be essential for virus internalisation (Evans et al. 2007; Zheng et al. 2007; Yang et al. 2008; Liu et al. 2009; Ploss et al. 2009). HCVpp bearing diverse glycoproteins of all major genotypes show a marked preference for infecting liver-derived cells suggesting that receptor-dependent entry events may in part define hepatotropism (Bartosch et al. 2003c; McKeating et al. 2004).
Hepatocytes in the liver are highly polarised with bile canaliculi, surrounded by tight junctions, running between adjacent cells at the apical membrane. HCV enters the liver via the sinusoidal blood and is likely to encounter the basolateral surface of hepatocytes. The observation that CD81 and SR-BI localise at basolateral hepatocellular membranes (Reynolds et al. 2008) is consistent with particle attachment occurring at this site. Recent data from our laboratory demonstrate a role for CD81–claudin complexes in HCV entry (Harris et al. 2008; Harris et al. 2010 JBC submitted). The involvement of tight junction proteins has raised many questions about HCV entry. For example, does the virus need to locate to tight junctions to internalise? Current data demonstrate that HCV E2 engagement of CD81 promotes clathrin-mediated endocytosis and there is limited evidence to support a role for CD81-induced movement of the virus to tight junctions in polarised hepatoma cells (Farquhar, personal communication).
Soon after the development of HCVpp and HCVcc, the use of drugs such as Bafilomycin A1 and concanamycin A (which inhibit the vacuolar ATPase, dissipating membrane proton gradients) demonstrated the pH-dependence of HCV entry, implicating the involvement of receptor-mediated endocytosis and fusion in an acidic endosomal compartment (Hsu et al. 2003; Blanchard et al. 2006; Tscherne et al. 2006). Furthermore, the use of dominant negative constructs of Eps15 and dynamin demonstrated a clathrin-mediated endocytic entry process (Meertens et al. 2006). A recent siRNA study confirmed the involvement of several genes involved in clathrin-mediated endocytosis and actin polymerisation in the viral entry process and used time-lapse imaging to observe viral entry in live unpolarised liver-derived cells (Coller et al. 2009). The authors demonstrated an association between labelled HCV and clathrin, suggesting that virions bind to filopodia and traffic towards the cell body, where endocytosis occurs. However, analysis of viral particles in polarised cells will be necessary before the exact entry pathway of the virus is clear.
Influenza A
The entry process of Influenza A is a much-studied topic and differs from the simple membrane fusion processes described for the majority of enveloped viruses. The first electron micrographs of influenza-infected cells demonstrated particles inside cytoplasmic compartments. Subsequently, the particles were seen to penetrate the membrane of this compartment followed by genome release in the cytosol (Dourmashkin and Tyrrell 1974). While this evidence pointed towards an endocytic mechanism of entry, the images failed to show the fusion of viral and host cell membranes that is believed to play a critical role in escaping the low pH environment of the endosome.
Lysosomotropic agents such as ammonium chloride (Matlin et al. 1981) and chloroquine (Yoshimura et al. 1982) inhibited influenza infection, suggesting an endocytic entry pathway. Many studies have demonstrated the pH-dependence of the Influenza entry mechanism (Guinea and Cerrasco 1994; Guinea and Cerrasco 1995), where fusion with the plasma membrane occurs at pH 5.5 or below but not in less acidic environments (Matlin et al. 1981). Further evidence for an endocytic entry mechanism includes dynamin dependence and the involvement of the Rab GTPases Rab5 and Rab7 (Roy et al. 2000; Sieczkarski and Whittaker 2003). Taken together, the data point towards endocytosis in coated pits followed by trafficking to an acidic internal compartment where low pH mediates membrane fusion and the release of the viral genetic material into the cytosol.
Influenza binds to cell surface sialic acid residues via interaction with the viral hemagglutinin (HA) glycoprotein (Weiss et al. 1988). Interestingly, strain-specific viral tropism is dependent on the specific types of sialic acids expressed on the target cell surface (Carroll et al. 1981). HA is critical for influenza fusion with host cells, which does not passively occur in the presence of low pH but is triggered in an HA-dependent process. HA is a trimeric spike glycoprotein which undergoes an irreversible conformational change at low pH which is necessary for endosomal escape of the virus. Evidence suggests that this process involves the insertion of the coiled coil of the HA trimer into the membrane, bringing the viral envelope and endosomal membranes into close contact and facilitating membrane fusion (Doms and Helenius 1986; Chambers et al. 1990; Yu et al. 1994).
Recent observations demonstrate influenza A entry into cells expressing dominant negative Eps15. Furthermore, inhibitors of caveolar endocytosis, such as nystatin, genistein and dominant negative caveolin-1 had minimal effect(s) on viral entry (Sieczkarski and Whittaker 2003). Other studies, including pharmacological manipulations and fluorescence imaging of live cells suggest that Influenza can enter via diverse pathways, including clathrin-dependent (Fig. 4) and clathrin- and caveolin-independent endocytic mechanisms (Nunes-Correia et al. 2004; Rust et al. 2004).

Clathrin-mediated endocytosis of Influenza. Fluorescence imaging of red-labelled virus (DiD) and YFP-tagged clathrin (shown in green) demonstrates a clathrin-mediated entry route for Influenza (adapted from Rust et al 2004; reprinted by permission from Macmillan Publishers Ltd., copyright 2004)
Various aspects of the influenza A entry process represent targets of a new class of antivirals, entry inhibitors (Voss et al. 2008; Saong et al. 2009; Yamaya et al. 2010). Whilst Influenza entry inhibitors are not currently clinically available, one recent study found clarithromycin, the macrolide antibiotic clinically used to treat pharyngitis and tonsillitis could inhibit two steps in the influenza virus entry process (Yamaya et al. 2010). The drug was found to reduce expression of sialic acid residues on the surface of airway epithelial cells, reducing virus binding, as well as reducing the number of acidic endosomes in the cell, inhibiting endosomal escape.
HIV-1
Despite being extensively studied, the entry process of HIV, the causative agent of AIDS remains a debated issue. At a relatively early stage in the study of HIV infection, CD4 was identified as an essential receptor molecule defining HIV infection (Dalgleish et al. 1984). However, it soon became clear that CD4 was not the sole determinant of HIV entry and the chemokine receptors, CCR5 and CXCR-4, were implicated as co-receptors (Broder and Dimitrov 1996; Feng et al. 1996; Deng et al. 1996; Dragic et al. 1996). The importance of HIV co-receptors was evident in the observation that humans with mutant CCR-5 are resistant to HIV infection (Samson et al. 1996). Our current understanding of the role of these HIV receptors in viral fusion supports an interaction with the primary receptor CD4 that leads to conformational change(s) that primes the virus to interact with CCR5. It is this receptor complex formation which alters the conformation of gp160 and leads to pH-independent gp41 fusion (Chan et al. 1997; Tan et al. 1997; Weissenhorn et al. 1997).
It has long been thought that productive entry of HIV occurs via direct fusion of the viral envelope with the host cell plasma membrane. One key piece of evidence for an endocytic-independent entry is the observation that infection is insensitive to neutralisation of endosomal pH (Stein et al. 1987). In fact, blocking endosome acidification was in some cases seen to augment viral infection, perhaps by sparing particles from lysosomal degredation (Fredericksen et al. 2002; Schaeffer et al. 2004; Wei et al. 2005). Despite this long-standing belief, EM studies have shown internalised virions in membrane-bound vesicles (Pauza and Price 1988; Grewe et al. 1990). Moreover, the rates of entry and uncoating of radiolabelled virions in the human T-lymphoid cell line CEM were consistent with a receptor-mediated mechanism of entry (Pauza et al. 1988). However, CD4 endocytosis was not required for HIV entry, suggesting that endocytosed particles were degraded by host cell lysosomes (Maddon et al. 1988).
Whilst the clathrin-mediated entry pathway has been documented for almost as long as the classical plasma membrane entry mechanism, it has only recently been confirmed that this route offers a productive entry pathway for HIV (Daecke et al. 2005). Recent data may even suggest that clathrin-mediated HIV entry is the only productive pathway for infection (Miyauchi et al. 2009). In fact, live cell imaging has demonstrated that HIV fusion at the plasma membrane did not proceed further than mixing of lipids and could therefore not lead to productive infection.
Individual steps in the HIV entry process have become the target for the development of antiviral entry inhibitors (for review see Kuritzkes 2009). These include inhibitors of the CD4–gp120 interaction (Guo et al. 2003; Lin et al. 2003), post-attachment inhibitors such as ibalizumab, which is thought to decrease CD4 flexibility, inhibiting access of CD4–gp120 to chemokine receptors (Burkly et al. 1992; Burkly et al. 1995; Kuritzkes et al. 2004) and inhibitors of the fusion process such as Enfuvirtide (Wild et al. 1994; Lalezari et al. 2003). Fusion inhibitors have perhaps been the most successful of these inhibitors to date, with Enfuvirtide approved for clinical use following phase 3 clinical trials (Clotet et al. 2007; Hicks et al. 2006).
Conclusions
The use of specific drugs, dominant negative constructs and siRNA to inhibit specific internalisation pathways have provided insights into viral entry mechanisms, providing evidence for clathrin-mediated entry of HCV as well as the caveolar-dependent entry of SV40. Recent developments in live-cell imaging have allowed the real-time tracking of fluorescent virus particles and have advanced our understanding of HCV entry in addition to challenging the classical plasma membrane pathways of two well-studied viruses, HIV and PV. However, the more information gained through these new techniques, the more complicated viral entry mechanisms seem to become, with AdV and influenza A seeming to enter via two distinct mechanisms. Further studies involving single-particle tracking of fluorescent-labelled virus particles in real-time will help elucidate the mechanisms of entry of many viruses and provide a greater understanding of the endocytic mechanisms active within the host cell which these viruses exploit.
References
Anderson HA et al (1996) Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol Biol Cell 7(11):1825–1834
Bartosch B et al (2003a) Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem 278:41624–41630
Bartosch B et al (2003b) Infectious hepatitis C virus Pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197(5):633–642
Bartosch B et al (2003c) In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc Natl Acad Sci U S A 100(24):14199–14204
Bartosch B, Cosset FL (2006) Cell entry of hepatitis C. Virology 348(1):1–12
Bergelson JM et al (1997) Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275(5304):1320
Blanchard E et al (2006) Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol 80(14):6964–6972
Brandenburg B et al (2007) Imaging poliovirus entry in live cells. PLoS Biol 5(7):1543–1555
Breau WC et al (1992) Class I major histocompatibility proteins are an essential component of the simian virus 40 receptor. J Virol 66(4):2037–2045
Broder C, Dimitrov DS (1996) HIV and the 7-transmembrane domain receptors. Pathobiology 64(4):171–179
Brown T, Burlingham BT (1973) Penetration of host cell membranes by adenovirus 2. D J Virol 12(2):386–396
Burkly L et al (1992) Inhibition of HIV infection by a novel CD4 domain 2-specific monoclonal antibody. Dissecting the basis for its inhibitory effect on HIV-induced cell fusion. J Immunol 149:1779–1787
Burkly L et al (1995) Synergistic inhibition of human immunodeficiency virus type 1 envelope glycoprotein-mediated cell fusion and infection by an antibody to CD4 domain 2 in combination with anti-gp120 antibodies. J Virol 69:4267–4273
Carroll SM et al (1981) Different cell-surface receptor determinants of antigenically similar influenza virus hemagglutinins. J Biol Chem 256(16):8357–8363
Catanese MT et al (2010) Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol 84:34–43
Chambers P et al (1990) Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J Gen Virol 71(12):3075–3080
Chan DC et al (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell 89(2):263–273
Clotet B et al (2007) Efficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trials. Lancet 369:1169–1178
Cohen FS et al (1984) Parameters affecting the fusion of unilamellar phospholipid vesicles with planar bilayer membranes. J Cell Biol 98(3):1054–1062
Coller KE et al (2009) RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathogens 5(12):e1000702
Cormier EG et al (2004) CD81 is an entry coreceptor for hepatitis C virus. Proc Natl Acad Sci U S A 101:7270–7274
Coyne CB, Bergelson JM (2005) CAR: a virus receptor within a tight junction. Adv Drug Deliv Rev 57(6):869–882
Coyne CB, Bergelson JM (2006) Virusinduced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124(1):119–131
Coyne CB et al (2007) Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2(3):181–192
Coyne CB et al (2009) The distinct roles of JAM-A in reovirus pathogenesis. Cell Host Microbe 5(1):3–5
Daecke J et al (2005) Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J Virol 79(3):1581–1594
Dalgleish AG et al (1984) The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312(5996):763–767
Damm E-M et al (2005) Clathrin and Caveolin-1- independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. JCB 168(3):477–488
Dangoria NS et al (1996) Extracellular simian virus 40 induces and ERK/MAP kinase-independent signalling pathway that activates primary response genes and promotes virus entry. J General Virol 77:2173–2182
Day PM et al (2003) Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 307(1):1–11
Deng H et al (1996) Identification of a major co-receptor for primary isolates of HIV-1. Nature 381(6584):661–666
Doms RW, Helenius A (1986) Quarternary structure of influenza virus hemagglutinin after acid treatment. J Virol 60(3):833–839
Dourmashkin RR, Tyrrell TA (1974) Electron microscopic observations on the entry process of influenza virus into susceptible cells. J Gen Virol 24(1):129–141
Dragic T et al (1996) HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381(6584):667–673
Drummer HE et al (2003) Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett 546(2–3):385–390
Dunnebacke TH et al (1969) Entry and release of poliovirus as observed by electron microscopy of cultured cells. J Virol 4:505–513
Evans MJ et al (2007) Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805
Feng Y et al (1996) HIV-1 entry co-factor: functional cDNA cloning of a seven-transmembrane G-protein coupled receptor. Science 272(5263):872–877
Flint M et al (2006) Diverse CD81 proteins support hepatitis C virus infection. J Virol 80:11331–11342
Fredericksen BL et al (2002) Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus. J Virol 76(22):11440–11446
Gastaldelli M et al (2008) Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 9:2265–2278
Gilbert JM et al (2003) Cell penetration and trafficking of polyomavirus. J Virol 77(4):2615–2622
Glenn GM, Eckhart W (1990) Transcriptional regulation of early-response genes during polyomavirus infection. J Virol 64(5):2193–2201
Grewe C et al (1990) HIV: early virus-cell interactions. J Acquir Immune Defic Syndr 3(10):965–974
Greber UF et al (1993) Stepwise dismantling of adenovirus 2 during entry into cells. Cell 75(3):477–486
Grove J et al (2007) Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J Virol 81:3162–3169
Guinea R, Cerrasco L (1994) Concanamycin a blocks influenza virus entry into cells under acidic conditions. FEBBS Lett 349(3):327–330
Guinea R, Cerrasco L (1995) Requirement for vacuolar proton-atpase activity during entry of influenza virus into cells. J Virol 69(4):2306–2312
Guo Q et al (2003) (2003) Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J Virol 77:10528–10536
Harris HJ et al (2008) CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol 82(10):5007–5020
Helle F, Dubuisson J (2008) Hepatitis C virus entry into host cells. Cell Mol Life Sci 65(1):100–112
Hicks CB et al (2006) Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatment-experienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 368:466–475
Hsu M et al (2003) Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 100(12):7271–7276
Hummeler K et al (1970) Morphological aspects of the uptake of simian virus 40 by permissive cells. J Virol 6(1):87–93
Kawase M et al (2009) Protease-mediated entry via the endosome of human coronavirus 229E. J Virol 83(2):712–721
Keck ZY et al (2004) Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J Virol 78(13):7257–7263
Kirkham M, Parton RG (2005) Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta 1745(3):273–286
Kuritzkes DR et al (2004) Antiretroviral activity of the anti-CD4 monoclonal antibody TNX-355 in patients infected with human immunodeficiency virus type 1. J Infect Dis 189:286–291
Kuritzkes DR (2009) HIV-1 entry inhibitors: an overview. Curr Opin HIV AIDS 4(2):82–87
Lakadamyali M et al (2004) Endocytosis of influenza viruses. Microbes Infect 6:929–936
Lalezari JP et al (2003) Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 348:2175–2185
Lee KK (2010) Architecture of a nascent viral fusion pore. EMBO J 29(7):1299–1311
Leon RP et al (1998) Adenoviral-mediated gene transfer in lymphocytes. Proc Natl Acad Sci U S A 95(22):13159–13164
Liebl D et al (2006) Mouse polyomavirus enters early endosomes, requires their acidic pH for productive infection, and meets transferrin cargo in Rab11-positive endosomes. J Virol 80(9):4610–4622
Lin PF et al (2003) A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A 100:11013–11018
Lindenbach BD et al (2005) Complete replication of hepatitis C virus in cell culture. Science 309(5734):623–626
Liu S et al (2009) Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83:2011–2014
Maddon PJ et al (1988) HIV Infection does not require endocytosis of its receptor, CD4. Cell 54(6):865–874
Madshus IH et al (1984a) Requirements for entry of poliovirus RNA into cells at low pH. EMBO J 3(9):1945–1950
Madshus IH et al (1984b) Mechanism of entry into the cytosol of poliovirus type 1: requirement for low pH. J Cell Biol 98(4):1194–1200
Matlin KS et al (1981) Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 91(3pt1):601–613
Maul GG et al (1978) Membrane fusion as a mechanism of simian virus 40 entry into different cellular compartments. J Virol 28(3):936–944
McKeating JA et al (2004) Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J Virol 78(16):8496–8505
Meertens L et al (2006) Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol 80(23):11571–11578
Meier O et al (2002) Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J Cell Biol 158(6):1119–1131
Meier O, Greber UF (2004) Adenovirus endocytosis. J Gene Med 6(Suppl 1):S152–S163
Miyauchi K et al (2009) HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 137:433–444
Nomura R et al (2004) Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J Virol 78(16):8701–8708
Norkin LC et al (2002) Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J Virol 76(10):5156–5166
Nunes-Correia I et al (2004) Caveolae as an additional route for influenza virus endocytosis in MDCK cells. Cell Mol Biol Lett 9(1):47–60
Op De Beeck A et al (2004) Characterisation of functional hepatitis C virus envelope glycoproteins. J Virol 78(6):2994–3002
Patel KP et al (2009) Dynamin- and lipid raft-dependent entry of decay-accelerating factor (DAF)-binding and non-DAF-binding coxsackieviruses into nonpolarized cells. J Virol 83(21):11064–11077
Pauza CD, Price TM (1988) Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis. J Cell Biol 107(3):959–968
Pauza CD et al (1988) Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis. J Cell Biol 107(3):959–968
Pelkmans L et al (2001) Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular- transport pathway to the ER. Nat Cell Biol 3:473–483
Pelkmans L et al (2002) Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296:535–538
Pileri P et al (1998) Binding of hepatitis C virus to CD81. Science 282:938–941
Ploss A et al (2009) Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886
Reynolds GM et al (2008) Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 47(2):418–427
Richterová Z et al (2001) Caveolae are involved in the trafficking of mouse polyomavirus virions and artificial VP1 pseudocapsids toward cell nuclei. J Virol 75(22):10880–10891
Roy AM et al (2000) Early stages of influenza virus entry into MV-1 lung cells: involvement of dynamin. Virology 267(1):1728
Rust MJ et al (2004) Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 11(6):567–573
Samson M et al (1996) Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382(6593):722–725
Sandvig K, Olsnes S (1980) Diphtheria toxin entry into cells is facilitated by low pH. J Cell Biol 87(3pt1):828–832
Sarkar DP et al (1989) Initial stages of influenza hemagglutinin-induced cell fusion monitored simultaneously by two fluorescent events: cytoplasmic continuity and lipid mixing. J Cell Biol 109(1):113–122
Scarselli E et al (2002) The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025
Schaeffer E et al (2004) Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in human CD4 T lymphocytes. J Virol 78(3):1375–1383
Schelhaas M et al (2007) Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131(3):516–529
Sieczkarski SB, Whittaker GR (2003) Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4(5):333–343
Stehle T, Harrison SC (1996) Crystal structures of murine polyomavirus in complex with straight-chain and branched-chain sialyloligosaccharide receptor fragments. Structure 4(2):183–194
Stein BS et al (1987) pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 49(5):659–668
Svensson U (1985) Role of vesicles during adenovirus 2 internalization into HeLa cells. J Virol 55(2):442–449
Tan K et al (1997) Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308
Tscherne DM et al (2006) Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80(4):1734–1741
Varga MJ (1991) Infectious entry pathway of adenovirus type 2. J Virol 65(11):6061–6070
Voss T et al (2008) Peptide-based entry inhibitors for influenza. Antivir Res 78(2):A18
Wakita T et al (2005) Production of hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11(7):791–796
Wang K et al (1998) Adenovirus internalization and infection require dynamin. J Virol 72(4):3455–3458
Wei BL et al (2005) Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J Virol 79(9):5705–5712
Weiss W et al (1988) Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333:426–431
Weissenhorn W et al (1997) Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430
Wickham TJ et al (1993) Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73(2):309–319
Wild CT et al (1994) Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci U S A 91:9770–9774
Yamaya M et al (2010) Clarithromycin inhibits type A seasonal influenza virus infection in human airway epithelial cells. J Pharmacol Exp Ther 333(1):81–90
Yang W et al (2008) Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J Biol Chem 283:8643–8653
Yoshimura A et al (1982) Infectious cell entry mechanism of influenza virus. J Virol 43(1):284–293
Yu YG et al (1994) Insertion of a coiled-coil peptide from influenza virus hemagglutinin into membranes. Science 266(5183):274–276
Zheng A et al (2007) Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J Virol 81:12465–12471
Zhong J et al (2005) Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102(26):9294–9299
Zullo J et al (1987) Regulation of c-myc and c-fos mRNA levels by polyomavirus: distinct roles for the capsid protein VP1 and the viral early proteins. Proc Natl Acad Sci U S A 84(5):1210–1214
Acknowledgements
The authors would like to thank the Medical Research Council (MRC) for funding JAT’s doctoral studentship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: David Robinson
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Thorley, J.A., McKeating, J.A. & Rappoport, J.Z. Mechanisms of viral entry: sneaking in the front door. Protoplasma 244, 15–24 (2010). https://doi.org/10.1007/s00709-010-0152-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-010-0152-6