
Abstract
Glucose-6-phosphate dehydrogenase (G6PD) deficiency and Southeast Asian ovalocytosis (SAO) caused by a 27-bp deletion in the band 3 gene (Band3Δ 27) are well-documented genetic traits resistant to malarial diseases; however, relationships between these traits and asymptomatic malaria infection hitherto had not been investigated. Filter-blotted blood samples were collected from a total of 210 healthy individuals, 100 males and 110 females, aged 6–17 years, in Sumba island, Indonesia, to survey for the presence of Plasmodium parasites, G6PD activity and the Band3Δ 27 mutation. Presence of P. falciparum and/or P. vivax was identified in 25 subjects (11.9%). In all, 24 subjects (11.4%) showed Band3Δ 27 heterozygously. In males and females, eight and nine subjects, respectively, showed G6PD deficiency. There was no significant difference in the prevalence of asymptomatic malaria infection between individuals with or without these traits (P>0.05). No alterations in the prevalence of asymptomatic malaria infection suggest that parasite invasion into erythrocytes is unlikely to be a target phase in which the two polymorphisms demonstrate possible protective effects against malaria.
Similar content being viewed by others

Introduction
Selection during longer periods of exposure to malaria may have played a role in modifying the genetic make-up of humans. This continual adaptation leads to enormous variation in the interaction between host and parasite. The molecular pathogenesis of Plasmodium infection, as well as host defense mechanisms against malaria infection, remain poorly understood. Well-known examples of host genetic factors related to erythrocyte abnormalities, glucose-6-phosphate dehydrogenase (G6PD) deficiency and hemoglobinopathies, including sickle cell disease and thalassaemia, have been studied in line with resistance to malaria. Another important malaria-related polymorphism in Asia and Oceania is genetically determined Southeast Asian ovalocytosis (SAO). One of the molecular bases for SAO is a 27-bp deletion in exon 11 of the band 3 anion exchanger 1 gene (Band3Δ 27) (Jarolim et al. 1991), which leads to the deletion of nine amino acids (codons 400–408) in the boundary between the cytoplasmic and the transmembrane domains of the gene product.
Several studies have confirmed the presence of G6PD deficiency in Southeast Asia, including several Indonesian islands (Iwai et al. 2001; Matsuoka et al. 2003). In vitro studies (Roth et al. 1983; Roth and Schulman 1988) and epidemiological evidence (Ruwende et al. 1995) have indicated that G6PD deficiency confers some resistance to P. falciparum, the primary human malaria parasite. The difference in the prevalence of G6PD deficiency in African and European populations may reflect selection by past P. falciparum infection (Saunders et al. 2002). As for SAO, epidemiological studies have also revealed a wide distribution of SAO in parts of Southeast Asia and Melanesia (Lie-Injo 1965; Amato and Booth 1977; Baer et al. 1976), which is particularly common in Austronesian populations (Kimura et al. 2003). The SAO is characterized by increased red-cell rigidity, which provides a barrier against malaria infection (Mohandas et al. 1984); however, a previous study resulted in no correlation between the presence of SAO and malaria in terms of parasite species and malaria development (Kimura et al. 2002). Although a feature of malaria infection in the endemic region is characterized by long periods of asymptomatic parasitemia punctuated by episodic clinical attacks (Marsh and Snow 1997; Rogier et al. 1996), little is known about effects of erythrocyte polymorphisms on the asymptomatic malaria. It was also found that erythrocytes of SAO are resistant to invasion by P. falciparum (Kidson et al. 1981). These studies strongly indicate protective effects of SAO against cerebral malaria (Genton et al. 1995).
For the laboratory diagnosis of malaria, polymerase chain reaction (PCR)-based molecular methods have been established to be more sensitive and specific than the conventional thin and thick blood-smear examinations or immunochromatographic techniques. As a result of the very high sensitivity and specificity in detecting the Plasmodium genome, more numbers of Plasmodium carriers have been identified and our notion of asymptomatic malaria carriers is changing. This enables us to detect tolerated parasitemia without symptoms.
To clarify relationships between these two erythrocyte polymorphisms and the presence of the malaria parasite among healthy individuals, we thus preliminary screened for the prevalence of G6PD deficiency and SAO caused by the Band3Δ 27 mutation, and the presence of malarial genomic DNA in healthy children in Sumba island, Indonesia, where P. falciparum and P. vivax are commonly distributed.
Materials and methods
Subjects
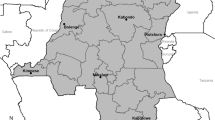
A total of 210 healthy individuals, 100 males and 110 females, aged between 6 and 17 years (mean age: 11.6±0.1 years), were recruited in East Sumba, East Nusa Tenggara Province, Indonesia (Fig. 1). Health conditions of the participants were monitored by two experienced pediatricians to exclude malaria patients. The exclusion criteria were having fever or diarrhea or malaria drug medication. After informed consent was obtained from each individual or their guardians, venous blood samples were collected and each blood specimen was blotted on a filter paper and dried.

Location of Sumba island, Indonesia
Detection of Plasmodium DNA
Genomic DNAs were extracted from blood-blotted filter papers with the standard phenol/chloroform method. Infection of P. falciparum and P. vivax was also screened with the use of nested PCR methods (Snounou et al. 1993). A pair of genus-specific primers constructed in the small subunit ribosomal RNA gene, rPLU5 (5′-CCTGTTGTTGCCTTAAACTTC-3′) and rPLU6 (5′-TTAAAATTGTTGCAGTTAAAACG-3′), were used for the first amplification; an initial denaturation at 95°C for 5 min, 45 cycles of denaturation at 94°C for 1 min and annealing at 58°C for 2 min and extension at 72°C for 2 min, and a final extension at 72°C for 5 min. PCR products of genus-specific amplification confirmed by gel electrophoresis were subjected to the second PCR for species-specific amplification. Species-specific primer sets for P. falciparum (rFAL1: 5′-TTAAACTGGTTTGGGAAAACCAAATATATT-3′ and rFAL2: 5′-ACACAATGAACTCAATCATGACTACCCGTC-3′) and P. vivax (rVIV1: 5′-CGCTTCTAGCTTAATCCACATAACTGATAC-3′ and rVIV2: 5′-ACTTCCAAGCCGAAGCAAAGAAAGTCCTTA-3′) were prepared. An initial denaturation at 95°C for 5 min was followed by 40 cycles of denaturation at 94°C for 1 min and annealing at 65°C for 30 s and extension at 72°C for 30 s, and a final extension at 72°C for 5 min. Size of PCR products positive for P. falciparum and P. vivax were 205 bp and 120 bp, respectively (Fig. 2).

Nested PCR assay for the detection of P. falciparum and P. vivax. The PCR products from three subjects were loaded after the first amplification by the primer sets of rPLU for Plasmodium species (P) and after the second amplification by the primer sets of rFAL for P. falciparum (F) and rVIV for P. vivax (V), corresponding to 1.2-kbp, 205-bp and 120-bp, respectively. A 100-bp marker (M) was also loaded. Applied products exhibited single infection with P. falciparum (Subject 1) and P. vivax (Subject 2), and both species (Subject 3)
Screening for the 27-bp deletion in the band 3 gene
Using isolated DNAs, PCR for the Band3Δ 27 mutation was performed with specific primers (5′-GGGCCCAGATGACCCTCTTGC-3′ and 5′-GCCGAAGGTGATGGCGGGTG-3′) that span the 27-bp deletion (Jarolim et al. 1991) and Ampli Taq gold (Applied Biosystems, Tokyo, Japan). An initial denaturation at 95°C for 5 min was followed by 40 cycles of denaturation at 94°C for 1 min and annealing and extension at 70°C for 1 min, with a final extension at 70°C for 5 min. The PCR products were separated on 2.5% agarose gel and visualized by ethidium bromide staining (Kimura et al. 1998). The wild-type and deletion alleles were identified by the size of the PCR products, corresponding to 175 bp and 148 bp, respectively (Fig. 3).

Identification of the Band3Δ 27 mutation. M 100-bp molecular size marker, Wild wild-type homozygous, Del deletion type heterozygous
Screening for G6PD deficiency
A mass screening for G6PD deficiency was conducted using a formazan ring method (Fujii et al. 1984). Plates of 1% agar gel in 0.1 M Tris-HCl (pH 6.5) and 10 mM MgCl2 containing 0.125% d-glucose-6-phosphate disodium salt (Wako Chem. Co., Tokyo, Japan), 0.025% β-nicotinamide-adenine dinucleotide phosphate oxidized form (Wako Chem. Co.), 0.025% phenazine methosulphate (Wako Chem. Co.) and 0.025% 3-(4,5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide (Wako Chem. Co) were prepared. A 3-mm diameter disk from each blood-blotted filter paper was placed on an agar plate and incubated at 37°C for at least 8 h. According to the size of the formazan ring, G6PD activity was interpreted as follows: formazan ring invisible or smaller than 5-mm diameter, bigger than 8-mm diameter and of a size in between was regarded as deficient, normal and intermediate, respectively (Fig. 4). All G6PD-deficient and intermediate samples were subjected to re-screening for confirmation.

G6PD activity visualized by the formazan ring method; normal (+), intermediate (±) and deficient (−)
Statistical analysis
Pearson’s chi-squared and Fisher’s exact tests were employed for the statistical analyses. Any P value less than 0.05 was considered as significantly different.
Results
Prevalence of Plasmodium infection
Prevalence of malaria infection was monitored by the presence of Plasmodium DNA (Fig. 2). As shown in Table 1, the presence of P. falciparum and/or P. vivax was identified in 25 subjects (11.9%). Co-infection with P. falciparum and P. vivax was observed only in males and not in females (P=0.0087).
Prevalence of the 27-bp deletion in the band 3 gene
Distribution of the Band3Δ 27 mutation is shown in Table 2. In all, 24 subjects (11.4%) showed the deletion heterozygously and none of the subjects showed homozygous deletion, as expected. Prevalence of Plasmodium was not different between deletion holders and non-holders (P=0.499).
Distribution of G6PD deficiency
Table 3 shows the prevalence of G6PD deficiency. Among 100 males, eight subjects showed G6PD deficiency, while eight subjects were intermediate. Nine and 16 out of 110 females were deficient and intermediate, respectively.
G6PD deficiency and Plasmodium distribution
Relationship between G6PD deficiency and Plasmodium infection is shown in Table 4. Among a total of 17 G6PD-deficient individuals, malaria infection was identified in only one female, who carried P. vivax; however, the parasite positivity was not statistically different between the G6PD-deficient and intermediate or normal group (P=0.382 or 0.698, respectively).
Discussion
Two erythrocyte abnormalities, G6PD deficiency and SAO, are considered to be important malaria-related polymorphisms. The information obtained in this study was based primarily on observation of naturally infected individuals living in an area where stable malaria transmission exists. Residents of such areas sometimes develop partial protection against severe malaria through genetic or epigenetic mechanisms. In this study, malaria infection detected in healthy child participants or asymptomatic Plasmodium carriers who did not develop fever and other symptoms of malaria indicates individuals living in this area harbor natural protective mechanism(s) against malaria. From the public health point of view, the presence of asymptomatic carriers should be examined because these carriers are able to be natural reservoirs of the Plasmodium parasites.
The presence of P. falciparum and P. vivax was molecularly confirmed in Sumba island; their presence had been reported by conventional microscopic observation. Although the number of samples was small, it is interesting that co-infection with P. falciparum and P. vivax was identified only in males. This sexual difference in the parasitemic status, if it occurs, will be reflected in the outcome of the malarial disease and eradication of the parasites from the carriers, since P. falciparum and P. vivax co-infected malaria patients have been observed both in males and females in Sumbanese (Soemantri, unpublished data).
In Sumba island, the prevalence of Band3Δ 27 was similar in past (12.6%) (Kimura et al. 2002) and present reports (11.4%). As for the relationships between the Band3Δ 27 mutation and malaria, it was reported that the deletion does not affect development of malaria disease and there was no preference for malaria species. The effects of the Band3Δ 27 mutation on the malaria parasitemic status had not been studied previously and the present study was the first to address this issue. As shown clearly in Table 2, the heterozygous presence of the Band3Δ 27 mutant gene does not alter the status of parasitemia. It is thus concluded that the protective effects of Band3Δ 27 against malaria is not exerted by acting against parasitic invasion, but through alleviating severe symptoms, as discussed previously (Kimura et al. 2002).
The frequency of G6PD deficiency in Sumbanese was 8.0% (based on the male subjects) and this figure is comparable to geographically close Flores islanders (6.2%) (Matsuoka et al. 2003). Our data showed that G6PD deficiency does not alter the prevalence of asymptomatic Plasmodium carriers. G6PD deficiency, therefore, has no apparent effect on malaria protection in this population; however, many studies have reported that G6PD deficiency does have protective effects against malaria. Mombo et al. (2003) showed that G6PD-deficient heterozygous females were significantly less infected with P. falciparum under asymptomatic conditions than wild-type homozygous females, but in males such a difference between deficient-allele and wild-type-allele holders was not observed. The mechanism of this gender-specific protection could be attributed to the failure of parasite adaptation to the heterogeneous cellular environment during recurrent erythrocyte infection. From the study in Flores island, two out of 16 subjects with severe G6PD deficiency had confirmed malaria parasites in their peripheral blood (Matsuoka et al. 2003). The prevalence of malaria parasitemia was low in G6PD-deficient subjects and none of the subjects with G6PD deficiency showed anemia. G6PD deficiency is the most common enzymopathy of humans. The geographical correlation of its distribution with past pandemics of malaria indicates that G6PD deficiency has risen in frequency through natural selection by malaria (Ruwende et al. 1995). Malaria appears to exert a selective pressure in favor of G6PD deficiency, since deficiency confers relative protection against P. falciparum.
As a preliminary study, we have simplified the frame of our study, employing the positivity of Plasmodium DNA as a marker for malaria infection. To evaluate protective effects of G6PD deficiency and SAO on malaria infection in detail, detection of quantitative differences in parasitemia between variants holders and non-holders should be performed. Malaria-resistant genetic traits are, of course, not only G6PD deficiency and ovalocytosis; we must take hemoglobinopathies such as thalassaemia into our consideration, which are expected to be highly prevalent in this region.
References
Amato D, Booth PB (1977) Hereditary ovalocytosis in Melanesians. Papua New Guinea Med J 20:26–32
Baer A, Lie-Injo LE, Welch QB, Lewis AN (1976) Genetic factors and malaria in the Temuans. Am J Hum Genet 28:179–188
Fujii H, Takahashi K, Miwa S (1984) A new simple screening method for glucose 6-phosphate dehydrogenase deficiency. Acta Haematol Jpn 47:185–188
Genton B, al-Yaman F, Mgone CS, Alexander N, Paniu MM, Alpers MP, Mokela D (1995) Ovalocytosis and cerebral malaria. Nature 378:564–565
Iwai K, Hirono A, Matsuoka H, Kawamoto F, Horie T, Lin K, Tantular IS, Dachlan YP, Notopuro H, Hidayah NI, Salim AM, Fujii H, Miwa S, Ishii A (2001) Distribution of glucose-6-phosphate dehydrogenase mutations in Southeast Asia. Hum Genet 108:445–449
Jarolim P, Palek J, Amato D, Hassan K, Sapak P, Nurse GT, Rubin HL, Zhai S, Sahr KE, Liu SC (1991) Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc Natl Acad Sci USA 88:11022–11026
Kidson C, Lamont G, Saul A, Nurse GT (1981) Ovalocytic erythrocytes from Melanesians are resistant to invasion by malaria parasites in culture. Proc Natl Acad Sci USA 78:5829–5832
Kimura M, Shimizu Y, Settheetham-Ishida W, Soemantri A, Tiwawech D, Romphruk A, Duangchan P, Ishida T (1998) Twenty-seven base pair deletion in erythrocyte band 3 protein gene responsible for Southeast Asian ovalocytosis is not common among Southeast Asians. Hum Biol 70:993–1000
Kimura M, Soemantri A, Ishida T (2002) Malaria species and Southeast Asian ovalocytosis defined by a 27-bp deletion in the erythrocyte band 3 gene. Southeast Asian J Trop Med Publ Health 33:4–6
Kimura M, Tamam M, Soemantri A, Nakazawa M, Ataka Y, Ohtsuka R, Ishida T (2003) Distribution of a 27-bp deletion in the band 3 gene in South Pacific islanders. J Hum Genet 48:642–645
Lie-Injo LE (1965) Hereditary ovalocytosis and haemoglobin E-ovalocytosis in Malayan Aborigines. Nature 208:1329
Marsh K, Snow RW (1997) Host-parasite interaction and morbidity in malaria endemic areas. Philos Trans R Soc Lond B Biol Sci 352:1385–1394
Matsuoka H, Arai M, Yoshida S, Tantular IS, Pusarawati S, Kerong H, Kawamoto F (2003) Five different glucose-6-phophate dehydrogenase (G6PD) variants found among 11 G6PD-deficient persons in Flores Island, Indonesia. J Hum Genet 48:541–544
Mohandas N, Lie-Injo LE, Friedman M, Mak JW (1984) Rigid membranes of Malayan ovalocytes: a likely genetic barrier against malaria. Blood 63:1385–1392
Mombo LE, Ntoumi F, Bisseye C, Ossari S, Lu CY, Nagel RL, Krishnamoorthy R (2003) Human genetic polymorphisms and asymptomatic Plasmodium falciparum malaria in Gabonese schoolchildren. Am J Trop Med Hyg 68:186–190
Rogier C, Commenges D, Trape JF (1996) Evidence for an age-dependent pyrogenic threshold of Plasmodium falciparum parasitemia in highly endemic populations. Am J Trop Med Hyg 54:613–619
Roth E Jr, Schulman S (1988) The adaptation of Plasmodium falciparum to oxidative stress in G6PD deficient human erythrocytes. Br J Haematol 70:363–367
Roth E Jr, Raventos-Suarez C, Rinaldi A, Nagel RL (1983) Glucose-6-phosphate dehydrogenase deficiency inhibits in vitro growth of Plasmodium falciparum. Proc Natl Acad Sci USA 80:298–299
Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, Warn P, Allsopp CE, Gilbert SC, Peschu N, Newbold CI, Greenwood BM, Marsh K, Hill AVS (1995) Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature 376:246–249
Saunders MA, Hammer MF, Nachman MW (2002) Nucleotide variability at G6PD and the signature of malarial selection in humans. Genetics 162:1849–1861
Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, Thaithong S, Brown KN (1993) High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol 61:315–320
Acknowledgements
We are grateful to Dr. Matius Kitu, Dinas Kesehatan, Sumba Timor, Nusa Tenngara Timor, Indonesia for his arrangements for this study and to Dr. Ryutaro Ohtsuka, Department of Human Ecology, School of International Health, Graduate School of Medicine, University of Tokyo (currently at National Institute for Environmental Studies) for providing us with the opportunity of undertaking this study. We also thank Dr. Masako Kimura for her helpful advice on Plasmodium PCR. This study was partly supported by a Grant-in-Aid for Scientific Research from JSPS and MEXT Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shimizu, H., Tamam, M., Soemantri, A. et al. Glucose-6-phosphate dehydrogenase deficiency and Southeast Asian ovalocytosis in asymptomatic Plasmodium carriers in Sumba island, Indonesia. J Hum Genet 50, 420–424 (2005). https://doi.org/10.1007/s10038-005-0271-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-005-0271-7
Keywords
This article is cited by
-
Prevalence and proportion estimate of asymptomatic Plasmodium infection in Asia: a systematic review and meta-analysis
Scientific Reports (2023)
-
Association between ovalocytosis and Plasmodium infection: a systematic review and meta-analysis
Scientific Reports (2023)
-
Association of glucose-6-phosphate dehydrogenase deficiency and malaria: a systematic review and meta-analysis
Scientific Reports (2017)
-
Review of key knowledge gaps in glucose-6-phosphate dehydrogenase deficiency detection with regard to the safe clinical deployment of 8-aminoquinoline treatment regimens: a workshop report
Malaria Journal (2013)
-
Solenostemon monostachyus, Ipomoea involucrata and Carica papaya seed oil versus Glutathione, or Vernonia amygdalina: Methanolic extracts of novel plants for the management of sickle cell anemia disease
BMC Complementary and Alternative Medicine (2012)
