
Abstract
The alkaline protease genes (cDNAALP2 gene and ALP2 gene) were amplified from complementary DNA (cDNA) and genomic DNA of the marine yeast Aureobasidium pullulans HN2-3, respectively. An open reading frame of 1,248 bp encoding a 415-amino acid protein with a calculated molecular weight of 42.9 kDa was characterized. The ALP2 gene contained two introns, which had 54 and 52 bp, respectively. When the cDNAALP2 gene was cloned into the multiple cloning sites of the surface display vector pINA1317-YlCWP110 and expressed in cells of Yarrowia lipolytica, the cells displaying protease could form a clear zone on the double plate containing milk protein and had protease activity. The cells displaying alkaline protease were also found to be able to produce bioactive peptides from different sources of proteins. The peptides produced from single-cell protein of marine yeast strain G7a had the highest angiotensin-converting enzyme inhibitory activity, while the peptides produced from spirulina protein had the highest antioxidant activity. This is the first report that the yeast cells displaying alkaline protease were used to produce bioactive peptides.
Similar content being viewed by others


Introduction
It has been well-known that bioactive peptides from different sources of proteins have opioid agonistic and antagonistic activity, angiotensin-converting enzyme (ACE) inhibitory activity, immunomodulatory effects, antimicrobial activity, and antioxidant activity (Silva and Malcata 2005). The protein sources include soy protein (Gibbs et al. 2004), fish protein (Kristinsson and Rasco 2000), milk protein (Gobbetti et al. 2000), and shrimp and spirulina protein (He et al. 2006; Ma et al. 2007; Ni et al. 2008a). The means for production of the bioactive peptides include enzymatic hydrolysis of proteins by using proteases, fermentation by using lactic bacteria and chemical synthesis (Gibbs et al. 2004; Gobbetti et al. 2000; Kristinsson and Rasco 2000). The most commonly used proteases are those from Bacillus sp, lactic bacteria, and marine yeasts (He et al. 2006; Ma et al. 2007; Minervini et al. 2003; Ni et al. 2008a; Okamoto et al. 1997). Sodium caseinates prepared from bovine, sheep, goat, pig, buffalo, or human milk were hydrolyzed by a partially purified proteinase of Lactobacillus helveticus PR4 and peptides in each hydrolysate have ACE-inhibitory and antibacterial activities (Minervini et al. 2003). ACE-inhibitory peptides were also produced in fermented milks started by Lactobacillus delbrueckii subsp. bulgaricus SS1 and L. lactis subsp. cremoris FT4 (Gobbetti et al. 2000). A glutamic-acid-specific endopeptidase from Bacillus subtilis ATCC 6051 was applied in the recovery of bioactive peptides from fusion proteins (Okamoto et al. 1997). The prawn of Penaeus japonicus was hydrolyzed by various proteases, and antioxidant activity of the hydrolysates was examined. Among the digests, pepsin digest showed the most potent antioxidant activity (Suetsuna et al. 2000). In our previous studies (Ma et al. 2007; Ni et al. 2008a), ACE-inhibitory activity of the peptides in the hydrolysate of shrimp protein produced by the purified protease from Aureobasidium pullulans HN2-3 was the highest, while antioxidant activity in the hydrolysate of spirulina protein produced by the purified protease from A. pullulans N13d was the highest.
In recent years, yeast surface display techniques have received increasing attention as they have many applications in biotechnological and industrial fields, such as cell adhesion, molecular recognition, immobilized biocatalysis, bioconversion, bioremediation, change of cell function, signal transduction, biosensor, live vaccine development, and ultra-high-throughput screening for the identification of novel biocatalysts (Becker et al. 2004; Ueda and Tanaka 2000; Won et al. 2006; Yue et al. 2008; Zhu et al. 2006). Amylases, cellulases, xylanases, hemolysin, and other proteins have been successfully immobilized on yeast cells and their potential applications were evaluated (Yue et al. 2008; Zhu et al. 2006; Ueda and Tanaka 2000). However, alkaline protease has not been displayed on yeast cells for the production of bioactive peptides so far. A surface display vector for protein display on the yeast Yarrowia lipolytica was constructed in our laboratory (Yue et al. 2008). The vector takes advantage of the following features: it uses a strong recombinant growth-phase-dependent promoter, hp4d (Madzak et al. 2004), and belongs to a series of auto-cloning vectors, able to integrate in the genome of any Y. lipolytica strain. These vectors carry zeta elements (LTRs from Ylt1 retrotransposon), which allow them to integrate either by homolog in Y. lipolytica strains carrying Ylt1, or by non-homologous recombination in strains devoid of this retrotransposon. Integration of auto-cloning vectors in Ylt1-free strains occurs at random loci, and can be combined with an amplification of the number of copies when a defective selective marker is used. The multiple copies are dispersed in the genome, increasing the stability of transformants compared to tandem-repeated integration (Nicaud et al. 2002). Furthermore, the bacterial moiety of these auto-cloning vectors can be removed, being allowed to use only a “yeast expression cassette” for the transformation of the recipient strain. The resulting strain is devoid of bacterial DNA, retaining GRAS (generally regarded as safe) status and avoiding the spread of antibiotic resistance genes in the environment. We found that enhanced green fluorescent protein and hemolysin can be displayed on all of the yeast cells using the surface display vector (Yue et al. 2008). In this study, in order to use the alkaline protease displayed on cells of Y. lipolytica to produce bioactive peptides, alkaline protease genes were cloned from the marine yeast A. pullulans HN2-3 and ligated into the multiple cloning sites of the surface display vector pINA1317-YlCWP110 and expressed in cells of Y. lipolytica. Then bioactive peptide production by the yeast cells displaying the alkaline protease was carried out.
Materials and Methods
Strains and Media
Marine yeast strain HN2-3, which could produce a large amount of extracellular protease, was isolated from sediment of sea saltern in Qingdao, China and was identified as A. pullulans (Ni et al. 2008a). The Y. lipolytica yeast strain used for cell surface display was Po1h (genotype: MatA, ura3-302, xpr2-322, axp1-2; phenotype: Ura −, AEP, AXP, Suc + (Madzak et al. 2004). Yeast strains were grown in yeast peptone dextrose (YPD) (1.0% yeast extract, 2.0% bacto peptone, 2.0% glucose). The yeast transformants were selected on YNB-N5000 (0.17% yeast nitrogen base without amino acids and ammonium sulfate, 1.0% glucose, 0.5% ammonium sulfate). In order to produce displayed protein by yeast transformants, the PPB medium was used (Jolivalt et al. 2005). The Escherichia coli strain used in this study for plasmid recovery and cloning experiments was DH5α [F − endA1 hsdR17 (rK − /mK +) supE44 thi-1λ − recA1 gyr96ΔlacU169 (φ80lacZΔM15)] and was grown in Luria–Bertani broth (LB). The E. coli transformants were grown in LB medium with 100 μg/ml of ampicillin or 30 μg/ml of kanamycin. The marine yeast Cryptococcus aureus G7a, which contained 53.0% (w/w) of crude protein, was grown in liquid YPD medium (Gao et al. 2007).
Plasmids
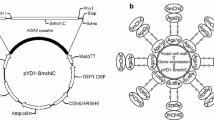
The surface display vector pINA1317-YlCWP110, which contains the C-terminal end of YlCWP1 from Y. lipolytica, was constructed in this laboratory (Yue et al. 2008). pMD19-T and pMD-19T simple vector were purchased from TaKaRa (Japan).
Isolation of DNA and RNA, Restriction Digestions, and Transformation
DNA manipulations were carried out using standard methods (Sambrook et al. 1989). Bacterial plasmid DNA was purified using Perfectprep plasmid minikit (Eppendorf). Yeast genomic DNA for amplification of the extracellular alkaline protease gene was isolated as described by Chi et al. (2007). Total RNA was extracted from A. pullulans HN2-3 using RNA simple Total RNA Kit (TianGen) according to the manufacturer’s protocol. The concentration and quality of the total RNA were estimated by measuring the absorbance ratio of 260/280 nm and agarose-gel electrophoresis, respectively. The RNA was reversely transcribed using RevertAidTM First Strand cDNA Synthesis Kit (Fermentas Life Sciences). Restriction endonuclease digestions and DNA ligations were performed according to the manufacture’s recommendations. E. coli was transformed with plasmid DNA according to Sambrook et al. (1989). Transformants were plated out onto LB medium containing 100 μg/ml of ampicillin or 30 μg/ml of kanamycin. Y. lipolytica was transformed according to the methods described by Xuan et al. (1988).
DNA Sequence and Computer Analysis
BLAST and ORF Finder programs at the National Center for Biotechnology Information (NCBI) were used for the nucleotide sequence analysis, deduction of the amino acid sequence, and database searches. Protein sequences were aligned using CLUSTAL W program (Thompson et al. 1994).
Cloning of Alkaline Protease Gene from A. pullulans HN2-3
The primers for cloning of ORF encoding extracellular alkaline protease were designed according to ALP1 gene (accession number EF198023). The forward primer and the reverse primer were Pu ATGTGGAAGAAGAGTGTTGC and Pd TAACGACCGCTGTTGTTGTAAAC, respectively. The genes encoding extracellular alkaline protease were amplified by polymerase chain reaction (PCR) using the first-strand cDNA and genomic DNA obtained above as templates. The reaction system (50 μl) was composed of 5.0 μl of 10 × buffer, 4.0 μl (2.5 mM) of deoxyribonucleotide triphosphates (dNTPs), 1.0 μl (50 mM) of Pu,1.0 μl (50 mM) of Pd, 1.0 μl of ExTaq DNA polymerase, 2.0 μl (10.0 ng/ml) of template DNA, and 36.0 μl of H2O. The conditions for the PCR amplification were as follows: initial denaturation at 94°C for 8 min, denaturation at 94°C for 1 min, annealing temperature at 50°C for 1 min, extension at 72°C for 1 min, final extension at 72°C for 8 min. PCR was run for 30 cycles and the PCR cycler was GeneAmp PCR System 2400 (PerkinElmer, Waltham, MA). PCR products were separated by agarose gel electrophoresis and recovered by using UNIQ-column DNA gel recovery kits (BIOASIA, Shanghai). The recovered PCR products were ligated into pMD19-T and transformed into competent cells of E. coli DH5α. The transformants were selected on plates with ampicillin. The plasmids in the transformant cells were extracted by using the methods described by Sambrook et al. (1989). The cloned DNA fragments inserted on the vector were sequenced by Shanghai Sangon Company. The gene, which was amplified from cDNA, was named cDNAALP2 gene, while the gene, which was amplified from genomic DNA, was named ALP2 gene.
Alkaline Protease Display on Cells of Y. lipolytica
In order to amplify the cDNAALP2 gene encoding alkaline protease by PCR, the forward primer was: P1 SfiI5′-ATATGGCCGTTCTGGCCGCTCCTGTTCCTCAGGAT-3′ (underlined bases encode SfiI restriction site) and the reverse primer was P2 HindIII 5’-AAGCTT GTGATGGTGATGGTGATGACGACCGCTGTTGTTGTAAAC-3’ (underlined bases encode HindIII restriction site and bold bases encode 6× His tag). The gene amplification by PCR, the gene expression, and alkaline protease display on cells of Y. lipolytica were performed as described by Yue et al. (2008). The resulting plasmid carrying cDNAALP2 gene was designated as pINA1317-YlCWP110-ALP2 (Fig. 2). Integration of cDNAALP2 gene into genomic DNA in the positive transformants was confirmed as described below. The positive transformants carrying cDNAALP2 gene were grown in PPB liquid medium for 96 h. The alkaline protease activity of different positive transformants was determined as described below. The cells of Y. lipolytica Po1h only carrying yeast cassette without cDNAALP2 gene were used as controls.
Determination of Alkaline Protease Activity
The cells of the positive transformants carrying the cDNAALP2 gene and Y. lipolytica Po1h only carrying yeast cassette without the cDNAALP2 gene were grown in PPB medium for 96 h, respectively. The cultures were washed three times with sterile saline water by centrifugation at 6,000×g and 4°C for 5 min. The recombinant alkaline protease activity displayed on the yeast cells was determined according to Inamura et al. (1985) with minor modifications. One unit of protease activity was defined as the increase of 0.001 absorbance unit at 440 nm. Cell dry weight of the cultures was determined as described by Gao et al. (2007). The specific alkaline protease activity was defined as units per gram of cell dry weight. Protease activity was also estimated using the double plate in which upper medium was PPB medium (pH 7.4) and bottom medium contained 2.0% milk. Different colonies of the transformants were transferred to the double plates and incubated at 28°C for 3 days and the clear zones around the colonies were observed and photographed.
Immunofluorescence Microscopy
In order to test if the alkaline protease is displayed on Y. lipolytica cell wall, immunofluorescence microscopy was conducted according to the methods described by Adams et al. (1998) and in the manual of pYD1 Yeast Display 223 Vector Kit (Invitrogen, USA), with minor modifications, using the 6× His monoclonal antibody produced by Clontech (USA) as primary antibody, and IgG/FITC produced by ZSGB-BIO (China) as secondary antibody. Y. lipolytica Po1h cells carrying YlCWP110-cDNAALP2 were grown in PPB liquid medium for 96 h. The cells were collected and washed three times by centrifugation with phosphate-buffered saline (PBS). The pellets were suspended in 3.7% formaldehyde and incubated overnight at 28°C with shaking. Then, the yeast cells were labeled using the primary and secondary antibodies mentioned above.
Confirmation of Integration of the Target Gene into Y. lipolytica Genome
To get the evidence that DNA fragments carrying YlCWP110-cDNAALP2 have been integrated into Y. lipolytica genome, genomic DNAs from the corresponding transformants were extracted and used as templates for PCR. The forward primer and the reverse primer used for this checking were P1 SfiI and P2 HindIII as described above, respectively. PCR amplification was performed as described above. The sizes of the PCR products were estimated using the Automated Gel Documentation & Analysis System (Gene-Genius, USA).
Preparation of Protein Sample
-
1.
Preparation of the marine yeast cells. The cells of the marine yeast C. aureus G7a with high protein content (Gao et al. 2007) were cultivated in YPD medium at 28°C by shaking for 48 h and collected by centrifugation (6,225×g, 5 min, 4°C) and washed three times with ice-cold distilled water. The pellet was suspended in 2.0 ml of ice-cold buffer containing (0.05 M glycine–NaOH, pH 9.0) to make a thick paste.
-
2.
Preparation of the spirulina suspension. 1.0 g of the spirulina (Arthospira platensis) powder was suspended in 2.0 ml of the ice-cold buffer to make a thick suspension.
-
3.
Preparation of the sliced prawn. 2.0 g of the fresh prawn muscle (Penaeus vannamei) was sliced into small pieces and the small pieces were suspended in 2.0 ml of the ice-cold buffer.
The thick yeast paste, spirulina suspension, and sliced prawn muscle were homogenized in a DY89-I Type Electric Glass Homogenizer (Xinzhi, Zhejiang, China) and homogenization proceeded for 1 h on the ice. The cell debris was removed by centrifugation (10,000×g, 30 min, 4°C) and about 2.0 ml of the supernatants was ultrafiltrated by using 10 kDa cutoff with a Labscales™ TFF System (Millipore, USA), and proteins in the ultrafiltrate with more than 10 kDa were used as the protein samples. Protein concentration in all the samples mentioned above was measured by the method of Bradford, and bovine serum albumin served as standard (Bradford 1976). Finally, the protein concentration in all the samples was adjusted to 0.5 mg/ml.
Protein Digestion
Protein digestion and preparation of the proteins with less than 3 kDa were performed according the methods described by Ma et al. (2007).
Assay of Angiotensin-converting Enzyme Inhibitory Activity and Antioxidant Activity
Angiotensin-converting enzyme (ACE) was prepared following the procedure of Yang and Li (2003). Rabbit lungs were supplied by Medical College of Qingdao. ACE inhibitory activities of the peptide samples were determined by direct spectrophotometric measurement (Li et al. 2005; Ma et al. 2007). ACE inhibitory activity of the peptide sample was calculated as follows:
where B is the absorbance of control (buffer added instead of test sample), C is the absorbance of the reaction blank (HCl was added before ACE), and A is the absorbance in the presence of sample.
The antioxidant activity in the peptide sample was determined by the improved 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS) radical cation decolorization assay (Re et al. 1999). The absorbance of 1.0 ml of the diluted ABTS+ [2, 2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid)] solution in 10 ml of distilled water at 734 nm was regarded as no inhibition (control) and the percentage inhibition of absorbance at 734 nm for the diluted ABTS+ solution with the peptide sample was calculated. The absorbance of 1.0 ml of the diluted ABTS+ solution in 10 ml of vitamin C solution (10 μg/ml) at 734 nm was regarded as 100% inhibition.
Results and Discussion
Cloning and Analysis of Alkaline Protease Gene
In our previous study (Ni et al. 2008a), we found that ACE-inhibitory activity of the peptides in the hydrolysate of shrimp protein produced by the purified protease from A. pullulans HN2-3 was the highest. Therefore, the gene-encoding alkaline protease in this marine yeast strain was cloned in this study. The results in Fig. 1 revealed that after sequencing of cDNAALP2 gene, ORF was found to have 1,248 bp (accession number EU224431), while after sequencing of ALP2 gene, ORF was found to contain 1,354 bp in length (accession number EU331441). After the sequence of cDNAALP2 gene was aligned with that of ALP2 gene, we found that there were two introns, which had 54 and 52 bp, respectively (in shade in Fig. 1). However, ALP1 gene from A. pullulans 10 contains two introns with 54 and 50 bp, respectively (Ni et al. 2008b). After the sequence of ALP2 gene cloned in this study was aligned with that of ALP1 gene from A. pullulans 10 by using nucleotide-translated nucleotide BLAST (blastn) at http://www.ncbi.nlm.nih.gov/Blast, it was found that the sequence of ALP2 gene had very high similarity (85.01%) to that of ALP1 gene isolated from A. pullulans 10, confirming that extracellular alkaline protease gene of A. pullulans HN2-3 was isolated (Ni et al. 2008b). The alignment and comparison of the protein (ALP2) sequence deduced from cDNAALP2 gene with sequences in the protein databases using the BLAST program show that the deduced protein had very high similarity (92.53%) to that from cDNAALP1 (Ni et al. 2008b). The calculated molecular mass of the protein deduced from cDNAALP2 gene was 42.9 kDa, and the protein contained 415 amino acids. Signal peptide analysis of the protein deduced from cDNAALP2 gene at http://cbs.dtu.dk/services/SignalP/ showed that the signal peptide had 18 amino acids and the peptide bond between 18th and 19th amino acid would be cleaved by signal peptidase (Fig. 1). N-glycosylation sites of the protein was also analyzed at http://cbs.dtu.dk/services/NetNGlyc and the results indicated that there was only potential N-linked glycosylation site of the protein, that was -N-G-T from 68 amino acid to 70 amino acid.

Nucleotide sequence of the alkaline protease gene of A. pullulans HN2-3 and its amino acid sequence deduced from the gene. The DNA sequence of introns is shown in shade. The signal peptide is also strongly shaded. The N-linked glycosylation site is boxed in gray. Serine active and histidine active sites are underlined
Analysis of the protein deduced from cDNAALP2 gene at http://motif.genome.jp/ showed that the protein had the conserved serine active site and histidine active site of serine proteases in the subtilase family from 356 to 366 amino acids and from 188 to 198 amino acids, respectively (Fig. 1). Therefore, the results demonstrated that like ALP1, ALP2 obtained in this study belonged to one member of serine proteases in the subtilisin family (Ni et al. 2008b).
Alkaline Protease Immobilization on Cells of Y. lipolytica
The cloned cDNAALP2 gene was ligated into the multiple cloning sites of the surface display vector pINA1317-YlCWP110 and expressed in cells of Y. lipolytica Po1h (Yue et al. 2008). The positive transformants carrying YlCWP110-cDNAALP2 and the control cells carrying only YlCWP-110 without cDNAALP2 gene were both grown in PPB medium for 96 h. Protease activity in supernatant of the culture and protease activity of the washed cells were determined. It was found that the washed transformants carrying YlCWP110-cDNAALP2 had alkaline protease activity and the activity was 691.26 U/g of cell dry weight (the cell dry weight per 1,000 ml of the culture was 12.0 g, therefore, 8,295.12 U per 1,000 ml of the culture) and protease activity in the supernatant of the culture was 2,000.0 U/l, while no protease activity of the control cells was detected (data not shown). The positive transformants carrying YlCWP110-cDNAALP2 also form the clear zone on the double plate containing milk protein (Fig. 2), while the control cells only carrying YlCWP-110 without cDNAALP2 gene only form dim zone because of citric acid produced by the host cells (Madzak et al. 2004).

The clear zones formed on the double plates with milk protein by the transformants carrying cDNA ALP2 gene and the dim zone formed by the control cells only carrying YlCWP-110 without cDNAALP2 gene
In order to confirm the presence of the 6× His-alkaline protease-YlCWP110 fusion protein on the cell-surface, immunofluorescence labeling of the cells was performed with 6× His monoclonal antibody as primary antibody and IgG/FITC as secondary antibody. YlCWP110-cDNAALP2-carrying Y. lipolytica cells were grown in PPB medium for 96 h. The Y. lipolytica cells displaying 6× His-alkaline protease-YlCWP110 fusion protein were labeled by IgG/FITC, and green fluorescence was observed on these cells, while no fluorescence was observed on control YlCWP110-carrying Y. lipolytica cells (Fig. 4). These results clearly demonstrate that the 6× His-alkaline protease-YlCWP110 fusion protein was displayed on the cell surface, allowing its recognition by the antibodies. It can also be noticed from these results in Fig. 3 that 100% of the observed cells displayed alkaline protease. In our previous study (Yue et al. 2008), it was also found that 100% of the Y. lipolytica cells displays enhanced green fluorescent protein or hemolysin using a GPI-anchor-fusion expression system and the Y. lipolytica cells displaying hemolysin exhibit hemolytic activity toward erythrocytes from flounder.

Immunofluorescent labeling of transformed Y. lipolytica cells using 6× His monoclonal antibody as primary antibody and IgG/FITC as secondary antibody. Microphotographs were taken under visible light (B and D), and immunofluorescence microphotographs were taken under emission at 550 nm (A and C). A and B Y. lipolytica control cells harboring YlCWP110; C and D Y. lipolytica cells harboring YlCWP110-cDNAALP2. Magnification 40 × 10
After the fragments without the signal sequence of cDNAALP2 gene were amplified from genomic DNA in Y. lipolytica carrying the cDNAALP2 gene by PCR as described in “Materials and Methods,” we found that the expected sizes of PCR products were 1,210 bp (data not shown), which was shorter than cDNAALP2 gene (1,248 bp) because of lack of signal sequence and the restriction site sequences of SfiI and HindIII added to the primer. This means that cDNAALP2 gene indeed has been integrated into the genomic DNAs in Y. lipolytica.
Bioactive Peptide Production using the Yeast Cells Displaying Alkaline Protease
The proteins were extracted from shrimp, spirulina, and single cells of marine yeast strain G7a. The cell-free extracts, milk, and casein solution were filtrated. The filtrates with proteins more than 10 kDa were digested by the yeast cells displaying alkaline protease. The supernatants obtained were filtrated again and the filtrates with short peptides less than 3 kDa were collected. After determination of ACE inhibitory activity and antioxidant activity of the filtrates, it was found that although all the filtrates had ACE inhibitory activity and antioxidant activity, ACE inhibitory activity of the filtrate from digest of single-cell protein of the marine yeast strain G7a was the highest (80.82%), while antioxidant activity of the filtrate from digest of spirulina (A. platensis) powder was the highest (73.97%) (Fig. 4). It was also found that all the filtrates had no antimicrobial activity (data not shown). Therefore, the results in Fig. 4 demonstrate that the yeast cells displaying alkaline protease can be used for bioactive peptide production. This is the first report that the yeast cells displaying alkaline protease were used for bioactive peptide production. As discussed in “Introduction” section, the surface display vector used in this study has many advantages. Therefore, the recombinant vector and the yeast cells displaying alkaline protease had the promising uses in biotechnology, food industry, and pharmaceutical industry. In our previous studies (Ma et al. 2007), it was indicated that the purified alkaline protease from the marine yeast A. pullulans 10 had potential uses in the production of bioactive peptides from shrimp (Trachypenaeus curvirostris) and spirulina (A. platensis) powder and ACE inhibitory activity of the filtrate from digest of shrimp (T. curvirostris) was the highest (85.3%), while antioxidant activity of the filtrate from digest of spirulina (A. platensis) powder was the highest (54.6%). The marine yeast C. aureus G7a used in this study contains a high level of protein (53.0 g of crude protein per 100 g of cell dry weight), a large amount of C16:0 (19.0%), C18:0 (46.3%), and C18:1 (33.3%) fatty acids and had a large amount of essential amino acids, especially lysine (12.6%) and leucine (9.1%), and vitamin C (2.2 mg per 100 g of cell dry weight) when it grows on Jerusalem artichoke extract (Gao et al. 2007). Spirulina (A. platensis) powder produced by many biotech companies in China is widely available in markets. So, the protein resources for bioactive peptide production are very rich in China.

ACE inhibitory activity and antioxidant activity of peptides from different proteins. Data are given as means ± SD, n = 3. Calculation of ACE inhibitory activity and antioxidant activity was described in “Materials and Methods”
Summary
It has been well documented that bioactive peptides have many functions and potential applications. So far, the means for production of the bioactive peptides include enzymatic hydrolysis of proteins by using proteases, fermentation by using lactic bacteria, and chemical synthesis. However, bioactive peptide production using the yeast cells displaying alkaline protease has not been tried yet. In the present study, the alkaline protease gene (cDNAALP2) was amplified from the cDNA of the marine yeast A. pullulans HN2-3 and characterized (Fig. 1). When the cDNAALP2 gene was cloned into the multiple cloning sites of the surface display vector pINA1317-YlCWP110 and expressed in the cells of Y. lipolytica, the cells displaying protease could form a clear zone on the double plate containing milk protein, and had protease activity (Fig. 2). The cells displaying alkaline protease were also found to be able to produce bioactive peptides from different sources of proteins. The peptides produced from single-cell protein of marine yeast strain G7a had the highest ACE inhibitory activity, while the peptides produced from spirulina protein had the highest antioxidant activity (Fig. 4). This is the first report that yeast cells displaying alkaline protease were used to produce bioactive peptides. As the protein resources for bioactive peptide production are very rich in China, this technique has many promising applications.
References
Adams A, Gottschling DE, Kaiser CA, Stearms T (1998) Yeast immunofluorescence. In: Burke D, Dawson D (eds) Methods in yeast genetics: a Cold Spring Harbor Laboratory course manual. Cold Spring Harbor Laboratory, New York, p. 100
Becker S, Schmoldt HU, Adams TM, Wilhelm S, Kolmar H (2004) Ultra-high-throughput screening based on cell-surface display and fluorescence-activated cell sorting for the identification of novel biocatalysts. Curr Opin Biotechnol 15:323–329
Bradford MM (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–253
Chi ZM, Ma CL, Wang P, Li HF (2007) Optimization of medium and cultivation conditions for alkaline protease production by the marine yeast Aureobasidium pullulans. Bioresour Technol 98:534–538
Gao LM, Chi ZM, Jun S, Ni XM, Wang L (2007) Single-cell protein production from Jerusalem artichoke extract by a recently isolated marine yeast Cryptococcus aureus G7a and its nutritive analysis. Appl Microbiol Biotechnol 77:825–832
Gibbs BF, Zougman A, Masse R, Mulligan C (2004) Production and characterization of bioactive peptides from soy hydrolysate and soy-fermented food. Food Res Int 37:123–131
Gobbetti M, Ferranti P, Smacchi E, Goffredi F, Addeo F (2000) Production of angiotensin-I-converting-enzyme-inhibitory peptides in fermented milks started by Lactobacillus delbrueckii subsp. bulgaricus SS1 and Lactococcus lactis subsp. cremoris FT4. Appl Environ Microbiol 66:3898–3904
He H, Chen X, Sun C, Zhang Y, Gao P (2006) Preparation and functional evaluation of oligopeptide-enriched hydrolysate from shrimp (Acetes chinensis) treated with crude protease from Bacillus sp. SM98011. Bioresour Technol 97:385–390
Inamura H, Nakai T, Muroga K (1985) An extracellular protease produced byVibrio anguillarum. Bull Jpn Soc Sci Fish 51:1915–1920
Jolivalt C, Madzak C, Brault A, Caminade E, Malosse C, Mougin C (2005) Expression of laccase lllb from the white-rot fungus Trametes versicolor in the yeast Yarrowia lipolytica for environmental applications. Appl Microbiol Biotechnol 66:450–456
Kristinsson HG, Rasco BA (2000) Biochemical and functional various alkaline proteases. J Agric Food Chem 48:657–666
Li GH, Liu H, Shi YH, Le GM (2005) Direct spectrophotometric measurement of angiotensin I-converting enzyme inhibitory activity for screening bioactive peptides. J Pharm Biomed Anal 37:219–224
Ma CL, Ni XM, Chi ZM, Ma LY, Gao LM (2007) Purification and characterization of an alkaline protease from the marine yeast Aureobasidium pullulans for bioactive peptide production from different sources. Mar Biotechnol 9:343–351
Madzak C, Gaillardin C, Beckerich JM (2004) Heterologous protein expression and secretion in the non-conventional yeast Yarrowia lipolytica: a review. J Biotechnol 109:63–81
Minervini F, Algaron F, Rizzello CG, Fox PF, Monnet V, Gobbetti M (2003) Angiotensin I-converting-enzyme-inhibitory and antibacterial peptides from Lactobacillus helveticus PR4 proteinase-hydrolyzed caseins of milk from six species. Appl Environ Microbiol 69:5297–5305
Ni XM, Chi ZM, Liu ZQ, Yue LX (2008a) Screening of protease-producing marine yeasts for production of the bioactive peptides. Acta Oceanol Sin 27:116–125
Ni XM, Chi ZM, Ma CL, Madzak C (2008b) Cloning, characterization and expression of the gene encoding alkaline protease in the marine yeast Aureobasidium pullulans 10. Mar Biotechnol 10:319–327
Nicaud JM, Madzak C, van den Broek P, Gysler C, Duboc P, Niederberger P, Gaillardin C (2002) Protein expression and secretion in the yeast Yarrowia lipolytica. FEMS Yeast Research 2:371–379
Okamoto H, Fujiwara T, Nakamura E, Katoh T, Iwamoto H, Tsuzuki H (1997) Purification and characterization of a glutamic-acid-specific endopeptidase from Bacillus subtilis ATCC 6051; application to the recovery of bioactive peptides from fusion proteins by sequence-specific digestion. Appl Microbiol Biotechnol 48:27–33
Re R, Pellergini N, Proteggente A, Pann A, Yang M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26:1231–1237
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, New York, pp 367–370 (Chinese translated edn.)
Silva SV, Malcata FX (2005) Caseins as source of bioactive peptides. Int Dairy J 15:1–15
Suetsuna K (2000) Antioxidant peptides from the protease digest of prawn (Penaeus japonicus) muscle. Mar Biotechnol 2:5–10
Thompson JD, Higgins DD, Gibson JJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4688
Ueda M, Tanaka A (2000) Genetic immobilization of proteins on the yeast cell surface. Biotechnol Adv 18:121–140
Won H, Lee SH, Lee KJ, Park J, Kim S, Kwon MH, Kim YS (2006) Construction and characterization of a pseudo-immune human antibody library using yeast surface display. Biochem Biophy Res Commun 346:896–903
Xuan JM, Fournier P, Gaillardin C (1988) Cloning of the LYS5 gene encoding saccharopine dehydrogenase. Curr Genet 14:5–21
Yang Y, Li J (2003) Isolation and properties of angiotensin converting enzyme from rabbit lung. Food Ferment Ind 8:53–56
Yue LX, Chi ZM, Wang L, Liu J, Madzak C, Li J, Wang XH (2008) Construction of a new plasmid for surface display on cells of Yarrowia lipolytica. J Microbiol Methods 72:116–123
Zhu KL, Chi ZM, Li J, Zhang FL, Li MJ, Yasoda HN, Wu LF (2006) The surface display of haemolysin from Vibrio harveyi on yeast cells and their potential applications as live vaccine in marine fish. Vaccine 24:6046–6052
Acknowledgments
This work was supported by Hi-Tech Research and Development Program of China (863). The grant no. is 2006AA09Z403.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ni, X., Yue, L., Chi, Z. et al. Alkaline Protease Gene Cloning from the Marine Yeast Aureobasidium pullulans HN2-3 and the Protease Surface Display on Yarrowia lipolytica for Bioactive Peptide Production. Mar Biotechnol 11, 81–89 (2009). https://doi.org/10.1007/s10126-008-9122-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-008-9122-9