
Abstract
Genistein is a small, biologically active flavonoid that is found in high amounts in soy. This important compound possesses a wide variety of biological activities, but it is best known for its ability to inhibit cancer progression. In particular, genistein has emerged as an important inhibitor of cancer metastasis. Consumption of genistein in the diet has been linked to decreased rates of metastatic cancer in a number of population-based studies. Extensive investigations have been performed to determine the molecular mechanisms underlying genistein’s antimetastatic activity, with results indicating that this small molecule has significant inhibitory activity at nearly every step of the metastatic cascade. Reports have demonstrated that, at high concentrations, genistein can inhibit several proteins involved with primary tumor growth and apoptosis, including the cyclin class of cell cycle regulators and the Akt family of proteins. At lower concentrations that are similar to those achieved through dietary consumption, genistein can inhibit the prometastatic processes of cancer cell detachment, migration, and invasion through a variety of mechanisms, including the transforming growth factor (TGF)-β signaling pathway. Several in vitro findings have been corroborated in both in vivo animal studies and in early-phase human clinical trials, demonstrating that genistein can both inhibit human cancer metastasis and also modulate markers of metastatic potential in humans, respectively. Herein, we discuss the variety of mechanisms by which genistein regulates individual steps of the metastatic cascade and highlight the potential of this natural product as a promising therapeutic inhibitor of metastasis.
Similar content being viewed by others


1 Introduction
Genistein, or 4′,5,7-trihydroxyisoflavone, is a small molecule found in high natural abundance in soy products [1]. Individuals who consume soy as a dietary staple have elevated blood concentrations of genistein when compared to those who consume a Western-style red-meat-based diet [2]. This difference in genistein concentrations has been associated with differential outcomes in a variety of clinical contexts including cholesterol regulation, osteoporosis, and cancer. Interest in genistein as a potential therapeutic agent has recently risen in the field of oncology as population-based studies have linked genistein consumption with a decreased risk of mortality from several types of cancer, most notably prostate and breast cancer [3–9]. This important small molecule possesses a relatively wide array of biological activities that may underlie its ability to reduce the mortality associated with various cancer types. In particular, several studies have indicated that inhibition of metastasis by genistein represents an important mechanism by which it is able to reduce mortality associated with solid organ cancer. This is highly relevant, as solid organ cancer has an extremely poor prognosis once it has progressed to the metastatic stage. Genistein has therefore become one of the most widely studied small-molecule inhibitors of both cancer cell growth and metastasis and has the potential to be of substantial clinical benefit to patients with various types of cancer.
This article will review the structural, pharmacologic, and biological characteristics of genistein that are relevant to its ability to inhibit metastasis of various cancer types. We will describe the structural characteristics of genistein and its related isoflavone family members and will demonstrate how these structural features confer biological activity onto this small molecule. In addition, we will discuss genistein’s pharmacologic characteristics in relation to epidemiological studies, which provide one measure of genistein’s antimetastatic activity. The main focus of this article will be on genistein’s ability to regulate individual steps of the metastatic cascade, including cell proliferation, cell detachment, cell migration, and cell invasion. Finally, preclinical models of metastasis, as well as prospective human studies of pharmacology, toxicity, and efficacy, will be discussed.
2 Genistein: structure and epidemiology
2.1 Structural characteristic and synthesis of genistein
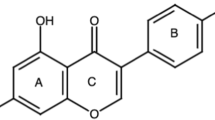
Genistein was originally isolated by Perkin and Newbury in 1899 from Dyer’s Broom (Genista tinctoria) [10]. This naturally derived compound is a member of the isoflavone branch of the flavonoid family of small molecules, which includes over 5,000 compounds [11, 12]. The isoflavones are structurally characterized by their 3-phenylchromen-4-one backbone, which consists of two benzene rings linked by a heterocyclic pyran ring. In addition to this heterocyclic core, genistein and its related isoflavone family members are polyphenols, in that they contain several hydroxyl groups attached to core phenyl rings. These phenols lend significant antioxidant activity to this class of compounds, with genistein and other related flavonoids, such as epigallocatechin 3-gallate possessing significant activity against free radicals in tissue [11]. Importantly, genistein is a recognized protein-tyrosine kinase inhibitor [13]. Although this has not been proven, this activity is presumed to stem from genistein’s C4′ phenolic group (Fig. 1), which structurally resembles the phosphoacceptor moiety of tyrosine.

Structures of genistein (left) and 17-β-estradiol (right). Genistein’s structure is characterized by a 3-phenylchromen-4-one core (highlighted in red) and phenolic substitution at the C4′, C5, and C7 positions. The C4′ and C7 phenols of genistein are in very similar positions to key hydroxyl groups on estradiol (highlighted in green), allowing genistein to bind to the ER
Genistein’s structural characteristics also impart this compound with the ability to act as a weak estrogen mimic, leading to its classification as a phytoestrogen (i.e., an estrogen-like compound derived from a plant source) [14]. In particular, compared to 17-β-estradiol, the predominant sex hormone present in females, genistein shares both a near identical molecular weight as well as a similar hydroxylation pattern, with two key phenolic groups at C7 and C4′ (Fig. 1) [15]. Importantly, the C7 hydroxyl group is needed for genistein to bind to the estrogen receptor (ER), as it mimics the A ring of the steroidal estrogen core. Furthermore, the distance (∼11.5 Å) between the C7 and C4′ phenolic groups (highlighted in green, see Fig. 1) allows for optimal binding of genistein to the ER, as they are in very similar positions to the key hydroxyl groups on the estradiol core [11]. Both the C4′ and C7 phenolic groups have been shown in a crystal structure to form key contacts with ERβ, with the C4′ phenol binding to Glu305 and Arg306 and the C7 phenol to His475 [16]. Because of these structural characteristics, genistein can bind to both α and β isoforms of the ER [17, 18], although it binds to ERβ with 20-fold higher affinity than ERα [19]. Genistein’s ability to both bind and stimulate the ER has led to several studies on its effects in postmenopausal women, with results suggesting that genistein could restore bone mass and relieve menopausal symptoms such as hot flashes and vaginitis [11]. Therefore, the structural similarities between genistein and estradiol have important functional consequences, which could lead to improvements in overall health or potentially detrimental side effects.
Due to the various biological activities that are associated with genistein, material for further clinical studies is in high demand. Although genistein and its related isoflavone family member, daidzein, are the most prominent isoflavones found in soy products, concentrations of these small molecules are usually quite low and can vary depending upon the type of soy preparation, making extraction from the natural source inefficient. Efforts have been made to increase the concentrations of genistein and other isoflavones in other plant products, such as corn and tobacco [20, 21], but this approach currently remains a challenge. Therefore, in addition to obtaining genistein from natural sources, several synthetic methods have also been developed in an effort to generate more material for biological analysis.
The first synthesis of genistein was in 1928 by Baker and Robinson, in which they employed a one-carbon homologation of a deoxybenzoin substrate to obtain the 2-methyl precursor of genistein [22]. This deoxybenzoin (2-hydroxyphenyl benzyl ketone) route still remains one of the most prominent routes that is used to generate isoflavones, along with the closely related chalcone route (Fig. 2a, b) [15]. In the deoxybenzoin route, the deoxybenzoin substrate is constructed under Lewis acidic conditions by condensation of a free phenol with a reagent containing an electrophilic carboxylic acid (Friedel-Crafts reaction). Treatment of this deoxybenzoin substrate with an activated one-carbon donor (e.g., N,N-DMF dimethyl acetal or DMF/PCl5) then leads to formation of the pyran ring to generate the desired isoflavone [23–25]. In the chalcone route, a chalcone substrate is generated by an aldol reaction between an appropriate acetophenone and an aromatic aldehyde. Oxidative rearrangement of the chalcone substrate by utilizing either a thallium(III) or hypervalent iodide reagent in methanol, followed by base-mediated ring closure, yields the final desired products [26]. Although these two routes are used to construct bulk quantities of synthetic genistein, other methods such as the Suzuki-Miyaura cross-coupling of arylboronic acids and halochromones are often used to generate smaller amounts of material for biological analysis [27, 28].

a The deoxybenzoin route for the synthesis of genistein and other isoflavones. The deoxybenzoin substrate is constructed with a Friedel-Crafts reaction between a substituted phenol and a phenylacetic acid derivative. A one-carbon homologation then completes the synthesis of the isoflavone core. b The chalcone route for isoflavone synthesis. The chalcone substrate is obtained from an aldol reaction between an acetophenone and an aromatic aldehyde. Oxidative rearrangement with diacetoxyiodo benzene or thallium(III) nitrate in MeOH then provides the intermediate acetal, which cyclizes to the isoflavone under base-catalyzed conditions
2.2 Dietary consumption and epidemiology of genistein
Although genistein can be synthetically produced to increase the supply of this natural product, humans most often ingest genistein through dietary consumption of soy products. Estimates of average daily genistein consumption by individuals who consume a soy-based diet range from 0.3 to 1.0 mg genistein per kilogram body weight per day [29–41]. The primary chemical form present in soybeans and nonfermented soy products is the C7-glycosylated form, also known as genistin [42]. After ingestion of genistin, hydrolysis within the intestine yields the aglycone, genistein, and the free sugar [43]. Rodent-based pharmacologic studies demonstrate that, after absorption from the intestine, genistein undergoes first-pass metabolism in the liver, as well as enterohepatic circulation [44]. These findings are corroborated with preliminary human dietary studies that demonstrate that ∼90% of the total amount of genistein in the blood has undergone extensive liver metabolism and exists in either the glucuronidated or sulfated form [2], while only 10% exists in the free (i.e., nonconjugated) form. Though studies investigating the biological activity of free genistein compared to its conjugated metabolic products are limited, it appears that the metabolic products do not have the same biologic activity [45, 46].
Therefore, it is important to consider that the concentration of free genistein in the blood of soy consumers is in the low nanomolar range. Effects found to be induced by genistein in preclinical model systems at low nanomolar concentrations therefore represent potentially important mechanisms that could be operative in humans. However, when considering effects induced by genistein in preclinical models that require concentrations in the low-to-mid micromolar range, their potential clinical relevance becomes less clear. It is clear that micromolar concentrations of total genistein (i.e., conjugated plus free forms) can readily be attained in the blood after high-dose genistein administration (see Section 5). However, because the majority of total genistein has undergone metabolic inactivation, its ability to induce a pharmacologic effect is questionable. On the other hand, some animal studies (see Section 4) appear to suggest that effects requiring high concentrations in vitro may be operative in vivo at lower concentrations.
Dietary studies of soy consumption provide a measure of the steady-state blood concentrations of genistein in humans after chronic exposure. The blood concentrations of the free and conjugated forms of genistein are 100 to 1,000 times lower in low-soy consumers (i.e., those who subsist on a Western red-meat-based diet) than those found in high-soy consumers [2]. This increased blood concentration of genistein in soy consumers has been linked to the differential clinical outcomes observed between high- and low-soy-consuming populations. However, it is important to consider that the differential dietary consumption of genistein occurs alongside of a much broader spectrum of dietary and lifestyle differences across populations. These other factors likely contribute to the different and sometimes conflicting results of population-based studies. In this review, we will highlight several investigations that identify positive links between dietary genistein consumption and cancer reduction. However, care should be taken to consider additional factors that may have an impact in these studies.
Population-based studies have linked decreased cancer incidence to increased genistein consumption for several cancer types, with most studies focused on prostate or breast cancer [3–9]. A recent meta-analysis of five cohort studies and eight case–control studies indicates that there is an inverse relationship between dietary genistein consumption and prostate cancer incidence and mortality [6]. Age-adjusted rates of metastatic prostate cancer are approximately tenfold lower among high-soy-consuming Southeast Asians than among low-soy consumers in the West (i.e., USA and Western Europe) [3–5]. However, after migration of Southeast Asians to the West, immigrant risk of prostate cancer approaches that of Western populations within one generation, indicating that differences in prostate cancer incidence are not entirely genetic and that they can be pharmacologically altered. Interestingly, the rates of localized or early-stage prostate cancer appear to be similar across different populations. The prevalence of primary prostate cancer in Chinese men that were born in China is only slightly lower than of American-born Chinese men (i.e., twofold lower of equal) [3, 47]. Similarly, while the rates of early-stage prostate cancer are similar between Japanese and American men, death from metastatic prostate cancer is much higher in men living in the USA [48]. The increased prevalence of clinical cancer may occur within a single generation after migration from the East to West [5]. These data support the notion that dietary constituents, such as genistein, and/or lifestyle factors may modulate the metastatic behavior of prostate cancer and reduce the incidence of more aggressive disease.
Although most of the epidemiological data regarding genistein’s anticancer activity are focused on prostate cancer, the effects of this natural product have also been studied in the context of other cancer types. In breast cancer, genistein consumption has been associated with a decrease in disease incidence [7, 8], potentially as a result of genistein’s ability to mimic natural estrogens (see Section 2.1), which are known to play a role in breast cancer progression. These studies have not specifically looked at factors related to the metastatic incidence of breast cancer. Several studies have also reported an inverse association between genistein consumption and colon cancer risk [9, 49], but these studies were also focused on the initial development of the disease as opposed to the progression to metastatic disease. Taken together, population-based studies have definitively demonstrated that those who subsist on a soy-based diet have blood concentrations of genistein much higher than those who do not. Further, they suggest that genistein may play a role in initial cancer prevention and/or in cancer progression. Confounding dietary and lifestyle factors cannot be excluded and likely contribute to differences between studies.
3 Genistein’s effects on the metastatic cascade
As detailed in the previous sections, several population-based epidemiological studies suggest that genistein can inhibit the development of metastatic disease in a variety of cancer types. Metastasis is a multistep process wherein a cancer cell leaves the site of the primary lesion, passes through the circulatory system, and establishes a secondary tumor at a new distant organ site. The individual cell-based steps that together constitute the metastatic cascade are depicted in Fig. 3. The metastatic process begins with the growth of cancer cells at the primary site of development, followed by decreased cell adhesion and local invasion through the basement membrane. The diagnosis of clinical cancer typically requires the presence of cancer cells that have invaded through the basement membrane of the primary organ. In contrast, when cancer cells are present in a clinical tissue specimen but have not invaded through the basement membrane, it is generally not termed cancer. Various terms have been applied, including preclinical cancer, intraepithelial cancer, or in situ cancer. After cells have invaded the basement membrane, their continued growth and associated induction of angiogenesis lead to the formation of a primary tumor. Cells then invade through the stroma of the primary organ, followed by intravasation into the vessels of the circulatory and lymphatic systems. Once cells have gained access to the circulatory system, they are transported to a distant site where they must attach to the vessel wall and extravasate, thereby entering a distinct organ. After invasion and implantation into the distant organ, cells resume the process of increased proliferation and angiogenesis, and begin to form a metastatic tumor. In the following sections, we will discuss genistein’s ability to inhibit these various steps of the metastatic cascade in greater detail. We will begin by describing the various mechanisms by which genistein can inhibit primary tumor growth, including its effects on cell proliferation and apoptosis. We will then describe genistein’s ability to regulate the later stages of the metastatic process, including cell adhesion, migration, and invasion.

The metastatic cascade
3.1 Genistein can alter cell proliferation and cell death
In this section, we will discuss genistein’s ability to inhibit growth of the primary tumor, which is the critical first step in the progression of metastatic disease. The larger the primary tumor grows, the more likely that the tumor will be able to acquire traits that lead to a metastatic phenotype [50]. Increased tumor growth can arise from an increase in the rate of cell division (i.e., proliferation) and/or a decrease in the rate of cell death (i.e., apoptosis or programmed cell death). Genistein can affect both of these processes, as well as modulate key regulatory protein such as Akt and nuclear factor κB (NF-κB). In general, low-to-mid micromolar concentrations of genistein are required for these effects in cell-culture-based models, though interestingly, effects in animal models have been observed at lower concentrations.
3.1.1 Effects of genistein on cell cycle progression
The cell cycle is a tightly regulated process, with each step carefully monitored by a specific group of proteins that act as checkpoints for proper cell division. The balance between these key checkpoint proteins is critical for progression through each step of the cell cycle to occur. The cyclin-dependent kinase (Cdk) family of proteins and their associated activating cyclins are particularly important regulators of the cell cycle (reviewed in [51]). For the G1/S-phase transition, cyclin D-Cdk4/Cdk6, cyclin E-Cdk2, and cyclin A-Cdk2 complexes are necessary for progression through the checkpoint, while the cyclin B-Cdk1 complex is necessary for the G2/M-phase transition. Perturbations in the cell cycle, particularly those that disrupt this Cdk family of proteins, have a significant impact on the rate of cell division and are often implicated in the development of primary cancerous tumors. Therefore, therapeutic modulation of these proteins has been extensively studied in an attempt to inhibit the development of a variety of cancers.
Several reports have demonstrated that genistein can induce cell cycle arrest and that it can therapeutically modulate key regulator cell cycle proteins at concentrations ranging from 5 to 200 μM [52]. It is important to note that these concentrations are greater than the blood levels that are observed with dietary consumption, indicating that this is likely not the primary mechanism by which genistein inhibits metastasis. However, it is theoretically possible to achieve these levels in humans, and various animal studies have also demonstrated that genistein can reduce the primary tumor size in certain contexts (see Section 4). Therefore, genistein’s effects on the cell cycle are potentially relevant to its ability to modulate the ultimate development of metastasis in humans.
Although several studies have described genistein’s ability to induce cell cycle inhibition, an exact mechanism of action has not been identified. Most studies indicate that genistein potentially targets proteins involved in the G2/M checkpoint, as it has been shown to induce arrest in this checkpoint in breast cancer [53–55], prostate cancer [54], and other cancers [55–57]. The main regulator of the G2/M checkpoint is Cdc2, also known as Cdk1. In ovarian and neuroblastoma cancer cells, genistein activates Chk1 and Chk2, which in turn activate, or dephosphorylate, Cdc25C and Cdc25A [56, 58]. The Cdc25 proteins are phosphatases that activate Cdc2; therefore, their inactivation by genistein thereby inactivates Cdc2 and induces G2/M-phase arrest. An additional regulator of the G2/M checkpoint is cyclin B1, which spikes at the G2/M transition, resulting in progression through this checkpoint. Genistein decreases cyclin B1 in breast cancer cells, which prevents cells from passing through this checkpoint and entering mitosis [59]. These combined effects of genistein on Cdc2, cyclin B1, as well as other cell cycle proteins, all serve to decrease cancer cell proliferation and therefore represent potential therapeutic targets.
3.1.2 The effect of genistein on apoptosis
In addition to inhibiting cellular proliferation, genistein also increases the rate of cancer cell death. Programmed cell death, or apoptosis, is a carefully regulated process that can be induced by a variety of stimuli, including programmed tissue remodeling, cell detachment (when it is then termed anoikis), genomic damage, hypoxia, signaling pathway derangement, growth factor or cytokine limitations, and infection [60–62]. Dysregulation of apoptosis contributes to the development of cancer, as decreased rates of cell death facilitate an overall increase in cell number. For this reason, the apoptotic pathway, similar to the cell cycle proteins described above, has been an attractive target for therapeutic development [63].
Genistein can induce apoptosis in a variety of epithelial cancers [52, 64–66]. These proapoptotic effects occur at genistein concentrations of 10–200 μM, similar to those that alter cellular proliferation. Again, as these levels are higher than the measured blood concentrations of high-soy consumers, genistein’s ability to inhibit apoptosis may not be the primary mechanism by which it regulates metastasis. These mechanisms might still be clinically relevant with regard to the development of the primary tumor, as these effects may be of primary importance in the context of high-dose pharmacologic therapy, in which elevated blood concentrations of genistein can be obtained.
A major regulator of apoptosis is the Bcl-2 family of proteins, such as Bcl-2, Bcl-xl, and Bcl-w. These proteins promote cell survival by sequestering the proapoptotic proteins Bax and Bak [67, 68]. Antagonizing this reaction are the BH3-only proteins, which promote apoptosis by displacing Bax and Bak bound to the Bcl-2 proteins. When Bax and Bak are released, they oligomerize to the outer membrane of the mitochondria, providing a channel for cytochrome c to move into the cytoplasm [69]. Upon release, cytochrome c interacts with Apaf-1 to bind to procaspase 9, cleaving this protein to activate caspase 9 [70]. Caspase 9 activation promotes the activation of caspases 3, 6, and 7, leading to rapid cell death.
Although the exact molecular mechanism of genistein’s effect on apoptosis has not been identified, most studies indicate that genistein acts by altering the expression of the various proapoptotic and antiapoptotic proteins described above. Specifically, Bcl-xl, an antiapoptotic protein, has been shown to be decreased in breast cancer [71–73], prostate cancer [74], and other cancers [75–77] upon treatment with genistein. Conversely, proapoptotic proteins Bax, caspase 3, and caspase 9 have all been shown to increase in vitro with genistein treatment. Increased Bax was observed in breast cancer [71, 73, 78], prostate cancer [74, 78], and other cancers [75, 76, 79]. Increased caspase 3 was seen in breast cancer [72, 80], prostate cancer [81, 82], colon cancer [83], and other cancers [79, 84–87], and increased caspase 9 was observed in pancreatic cancer [87], glioblastoma [85], and several other cancer types [84, 86].
3.1.3 Genistein treatment can decrease activation of Akt and NF-κB
Akt proteins are serine/threonine kinases that act as master regulators of cellular proliferation, protein synthesis, and apoptosis. As these kinases are upstream of a wide variety of other pathways that are involved in cell survival and proliferation, derangements of these pathways are often implicated in the development of a variety of cancer types. Akt is able to inhibit Bad activity, which decreases the chances of apoptosis [88, 89]. Additionally, Akt can activate mTOR, which results in stimulation of protein synthesis and cell growth [90]. Akt can also inhibit GSK-3β, which is an inhibitor of cyclin D1, a protein critical for early cell cycle progression [91]. Thus, activation of Akt in this context results in increased stimulation of cell proliferation. Finally, Akt activates Rho signaling, which results in increased cell motility. Based on these combined results, Akt and its associated signaling pathways have been attractive therapeutic targets for inhibiting cancer growth and metastasis [92].
At concentrations similar to those used in studies on cellular proliferation and apoptosis, genistein has been shown to decrease phosphorylated-Akt in prostate cancer [82, 93], breast cancer (both in vitro and in vivo) [94–96], colon cancer [97], and several other cancer types [77, 98–100]. It has been hypothesized that this inhibition of Akt is partially responsible for the effects on cell proliferation that were described in the previous two sections (reviewed in [65]).
Another major target of Akt is the NF-κB pathway. Upon activation by an IκB kinase, NF-κB translocates into the nucleus where it regulates gene expression. Many of its target genes have antiapoptotic effects, such as Bcl-2, or regulate the cell cycle, such as myc and cyclin D1 [50]. Inhibition of NF-κB by genistein has been observed in prostate cancer [93, 101, 102], breast cancer [94], and other cancers [98, 103] and may also partially explain the effects of genistein on cell cycle progression and apoptosis.
3.2 Genistein can alter cell adhesion
Concurrent with the process of primary tumor growth, cancer cells also acquire additional molecular aberrations that allow them advance further in the metastatic process. Decreased cell adhesion, or cell detachment, is an initial step in this metastatic transformation. In order for cells to move from their primary site, they must first detach from both the neighboring cells as well as from the extracellular matrix (ECM). In the literature, cell adhesion generally refers to the attachment of the cells to the ECM, whereas the process of cells adhering to one another is termed cell–cell adhesion. In both instances, these processes are mediated by transmembrane cell adhesion molecules. Cell–cell adhesion is canonically mediated by the binding of the cadherin family of proteins to each other, which is known as homotypic binding. Cell adhesion to the ECM is mediated by heterotypic binding, which involves two different proteins binding to one another, in order to anchor the cell. In epithelial cells, the major proteins involved in heterotypic binding are the integrin family of proteins [104]. Transmembrane integrin receptors are heterodimeric complexes, composed of α and β integrin subunits. There are at least eighteen α and eight β integrin isoforms, and the combination of α/β integrin subtypes determines the specific ECM protein to which each integrin binds. The expression of specific integrin subtypes, such as α3β1 and α6β1, has been associated with invasive cancer [105–107] and has also been implicated in genistein-mediated effects on adhesion. Integrins anchor the cell by the binding of the extracellular domain to the ECM proteins, while the integrin cytoplasmic domain binds to the actin cytoskeleton [104, 108]. This cytoplasmic domain connects to actin filaments via the structural adapter proteins, talin and vinculin [109, 110]. The associated macromolecular complex formed from these protein interactions is termed a focal adhesion complex. This complex provides a central location for cell signaling proteins that are involved in cellular adhesion and cellular migration, such as focal adhesion kinase (FAK), paxillin, and Src. FAK is a downstream target of several growth factor receptors, including epidermal growth factor receptor and platelet-derived growth factor receptor, and upon activation FAK recruits a variety of signaling factors that can regulate cell adhesion and cell motility [111]. FAK therefore plays a central role in integrin signaling, and many groups have studied its ability to regulate cell adhesion, migration, and invasion [112–114]. Increased FAK expression has been reported in several different cancers and can lead to decreased apoptosis and increased cell motility [115–119].
Our group has demonstrated that genistein increases human prostate cancer cell adhesion both in vitro [120] and in prostate cancer cells orthotopically implanted into mice [121]. This indicates that genistein can act to inhibit cell detachment, which is an early step in the metastatic cascade. Genistein-mediated increases in cell adhesion are time and concentration dependent and can be observed at low nanomolar concentrations of genistein. Though the exact pharmacologic mechanism by which genistein induces changes in cell adhesion is not known, we have shown that genistein increases FAK translocation to the focal adhesion complexes [120]. FAK then forms a complex with β1 integrin, and this interaction restores polar β1 integrin expression in cells that have lost integrin polarity [115, 120].
In other cancer types, genistein’s effects upon cell adhesion have been less extensively investigated. Genistein increases the adhesion of breast cancer [120, 122] and melanoma cells [122] in vitro at submicromolar concentrations, similar to the findings for prostate cancer. In contrast, genistein can inhibit the adhesion of hepatocarcinoma cells and pancreatic cells in vitro [123, 124]. These contrasting effects on cellular adhesion could stem from differences in cell type or assay methodology. In particular, changes in the type of ECM proteins and/or changes in the timing of drug treatment can have profound effects on the outcome of adhesion assays. Interestingly, studies that have measured adhesion along with other motility-associated parameters, such as migration and invasion, have shown that genistein inhibits cell motility regardless of its effects upon cell adhesion. Since adhesion is a kinetic process, involving cyclical detachment and attachment of integrins, it is possible that genistein is affecting the rate of focal adhesion complex formations. This would also explain the inconsistencies in genistein’s effect on cellular adhesion in different cancer cell types. Genistein consistently inhibits FAK activation in all of these studies, irrespective of its effect upon adhesion [120, 123, 125]. Given FAK’s role in regulating focal adhesion complex formation and turnover, it is likely an important pharmacological target for genistein’s effects upon adhesion.
3.3 Genistein can decrease the rate of cell migration
Once cells have detached from the ECM, they then acquire the ability to increase their motility. Cell migration is an important part of the metastatic cascade; it is necessary for cells to move through the primary organ. However, migration is a complex process that is poorly understood. A wide variety of proteins that can alter cell migration have been identified, but it is unclear how these are regulated in the cell [126, 127]. Changes in focal adhesion complex have been implicated to play a role in cell migration, as FAK has been shown to increase this phenotype in Chinese hamster ovary cells [112]. Despite preliminary studies, however, migration still remains one of the least extensively studied aspects of cancer metastasis.
Due to the limited study of this phenotype, the effects of genistein on cellular migration have only been studied on a limited basis. Studies have shown that genistein treatment can decrease cell migration in rat prostate carcinoma cells in a dose-dependent manner, over the noncytotoxic concentration range of 1 to 10 μM [124]. Over a similar concentration range, genistein can inhibit the migration of murine breast cancer and melanoma cells [122]. Importantly, studies have demonstrated that genistein inhibits phosphorylation of FAK on Y387, which is necessary for increases in cell migration [64]. These results imply that genistein’s effects on both cell adhesion (see Section 3.2) and cell migration are in part regulated by its ability to inhibit activation of FAK. However, the exact mechanism by which genistein can alter these processes is not known.
3.4 Genistein can decrease matrix metalloproteinase expression
The process of cell invasion is a combination of in cell migration with concurrent degradation of the surrounding ECM. This degradation of ECM proteins is mediated largely by matrix metalloproteinases (MMPs) [128]. MMPs constitute a family of zinc endopeptidases that cleave ECM proteins, thereby contributing to a variety of functions involved with normal homeostatic cell movement and tissue remodeling. MMPs have been categorized based upon the proteins that they degrade. These classifications include: collagenases, gelatinases, stromelysins, matrilysins, and membrane-type MMPs [129]. MMPs play a role in normal cell growth and division, as cells are continually cleaving and rebuilding the surrounding extracellular matrix as they proliferate. However, MMPs also play a role in aberrant cell growth and tumor formation, since they provide space for the tumor to grow and release various growth factors that drive tumor proliferation. This dual function makes targeting these molecules difficult, as these proteins are promoted in cancer development but are also critical in normal homeostatic processes. Therefore, global inhibition of MMPs also impacts basic and critical cellular process. This has created a difficulty in targeting MMPs therapeutically, as small-molecule inhibitors designed to bind the MMP active site are not able to effectively discriminate between individual MMP subtypes. For this reason, when tested in clinical trials, MMP inhibitors have induced general toxicity, such as muscular and skeletal joint pain, thereby preventing their further clinical development [130].
MMP-2, along with MMP-9, makes up the gelatinase family of MMPs. These proteins degrade a variety of ECM proteins, including type IV collagen [128]. This is significant as the basement membrane is made up primarily of type IV collagen [131], although it also contains laminins, osteonectin, heparin proteoglycans, and enactin [126, 132]. These extracellular membrane proteins are the first to be cleaved during the invasive stage of cancer progression. Increased expression of MMP-2 has been shown to predict a poor prognosis in a variety of cancers, including ovarian, pancreatic, prostate, and colorectal cancer (reviewed in [128]). In addition, MMP-2-deficient mice show decreased rates of cancer progression and angiogenesis in melanoma cells [133].
Our group has shown that genistein is able to decrease MMP-2 expression in a panel of human prostate cancer cell lines ranging from primary noncancerous cells to established metastatic variant cells, although little to no effect on MMP-9 was observed in this study at [134]. Effects were observed with genistein concentrations down to 10 nM. Reduction of MMP-2 by genistein treatment has also been confirmed by other groups in prostate cancer [135], breast cancer [136, 137], and glioblastoma cell lines [138]. Decreased expression of MMP-7 and MMP-9 has been in observed in breast cancer lines treated with genistein [136, 137], and decreased expression MMP-9 has also been observed in prostate cancer [139], glioblastoma [138], and pancreatic cancer [140] in vitro. In melanoma, breast, and prostate cancer cells, treatment with genistein decreases general zymographic activity, which is a measure of MMP activity [122, 141]. These combined studies demonstrate that genistein can selectively inhibit the production of individual MMPs, which could have significant impact on a cancer cell’s ability to invade deeper into the surrounding tissue.
3.5 Mechanisms of genistein-mediated decrease in cell invasion
Cell invasion represents a critical characteristic of the metastatic phenotype. In order for invasion to occur physiologically, cells must invade through the basement membrane, through the primary organ, and through the distant organ to which they eventually metastasize. Cells that are more phenotypically invasive, as well as those that show molecular alterations characteristic of invasive potential, show a greater propensity to metastasize [50].
Our group has shown that genistein is able to inhibit MMP-2 production in multiple studies, as described in Section 3.4. Through an additional series of studies, we have elucidated a key signaling pathway in human prostate cancer cells that plays a role in regulating cell invasion and metastasis and is also inhibited at genistein concentrations as low as 10 nM [134, 142–144]. This pathway is as follows: TGF-β→MEK4→p38 MAPK→MAPKAPK2→HSP27→MMP-2→invasion, where TGF-β is transforming growth factor β, MEK4 is mitogen-activated protein kinase kinase 4, p38 MAPK is p38 mitogen-activated protein kinase, MAPKAPK2 is mitogen-activated protein kinase-activated protein kinase 2, and HSP27 is heat shock protein 27 (see Fig. 4). Many of the proteins in this pathway have been linked to cancer progression and metastasis. Studies have shown that MEK4 is a key tyrosine/threonine kinase that is upregulated in advanced human and mouse prostate cancer progression [145] and can increase invasion of breast and pancreatic cells [146]. We have demonstrated that genistein directly binds to and inhibits the kinase activity of MEK4, thereby inhibiting the production of MMP-2 and decreasing cellular invasion [143]. p38 MAPK mediates a variety of cellular processes, ranging from proliferation and cell survival to invasion and metastasis (reviewed in [147]). HSP27 is upregulated in human prostate cancer progression [148] and regulates the structure and function of the actin cytoskeleton, which has important consequences for cell motility [149]. When dephosphorylated, HSP27 acts as an actin capping protein and thus inhibits actin polymerization [150, 151]. However, when phosphorylated, HSP27 induces increased F-actin formation [152, 153], increased cell migration [154, 155], and increased cell invasion [144].

Genistein inhibits critical pathways in cancer invasion. Our group has demonstrated that genistein can specifically target MEK4. This inhibition results in inactivation of the MEK4 pathway, decreased MMP-2 production, and decreased cell invasion. Genistein also activates Smad1, which is activated by the endoglin signaling pathway, and causes decreased cell invasion. Additionally, genistein inhibits FAK activation, resulting in increased cell adhesion. At this time, it is unclear whether the activation of Smad1 and FAK are due to genistein’s inhibition of MEK4 or via a different signaling mechanism
The MEK4 pathway has also been shown to crosstalk with the endoglin signaling pathway via the Smad family of transcription factors [156]. We have also shown that the endoglin pathway suppresses human prostate cancer cell invasion and that genistein activates endoglin pathway signaling [157–159]. Endoglin is an accessory TGF-β superfamily receptor that is lost during prostate cancer progression [157]. Endoglin signaling in prostate cancer was shown by our group to be dependent upon the type I TGF-β superfamily receptor, ALK-2. Endoglin’s suppression of prostate cancer cell invasion is mediated by the activation of the transcription factor Smad1 [158] (see Fig. 4). Ultimately, the state of cellular invasion is regulated by the ratio of Smad1 to Smad3. Smad1 is activated by the endoglin/ALK-2 signaling pathway and acts to suppress invasion. In contrast, Smad3 is activated by the TGF-β/ALK-5 signaling pathway, also known as the canonical TGF-β signaling pathway, and acts to increase invasion. Genistein is able to act on both branches of this pathway. Genistein acts to inhibit the canonical TGF-β pathway at the levels of Smad3 by virtue of its ability to inhibit MEK4. This is due to the fact that p38 MAPK, which is downstream of MEK4, activates Smad3 through signaling crosstalk [156]. Conversely, genistein can activate Smad1 in a manner that is dependent on ALK-2 kinase activity, but that is independent of endoglin [159]. This is important, as genistein can therapeutically compensate for endoglin loss, which occurs in prostate cancer progression. Therefore, these two linked signaling pathways have been identified as important regulators of human prostate cancer cell invasion, and genistein induces anti-invasive effects on both pathways.
Other groups have demonstrated that genistein suppresses invasion of a variety of prostate cancer cell lines through mechanisms other than inhibition of TGF-β signaling [139, 160, 161]. In androgen-responsive human prostate cancer cells, low micromolar concentrations of genistein significantly inhibited invasion [161]. This study proposed that genistein acted by causing a reversion of the epithelial to mesenchymal transition, or EMT. The EMT is a process by which cells revert from a more differentiated, epithelial state to a more undifferentiated, or mesenchymal, state, which involves the perturbation of a variety of proteins involved in cellular differentiation. This process commonly occurs as cancer cells progress to a more advanced stage. In this study, genistein caused a reversion of this EMT phenotype by increasing levels of E-cadherin, a membrane protein that is associated with increased cell–cell adhesion and reduced invasive potential. Genistein also decreased the levels of vimentin, a protein that is typically associated with a mesenchymal, or invasive, state (see Section 3.2).
Multiple groups have also demonstrated that genistein can inhibit invasion of breast cancer cells [137, 162–164]. In one study, the authors proposed that decreased invasion with genistein treatment was a result of decreased production of MMPs (see Section 3.4), although an exact molecular target was not discussed [137]. A second study demonstrated that genistein is able to downregulate the levels of the chemokine receptors CXCR4 and CXCL12 when the breast cancer cells were treated with low micromolar concentrations of genistein [162]. These receptors direct the homing of primary cancer cells to a metastatic site and can also alter cellular adhesion.
Genistein can also decrease cell invasion in other cancers such as pancreatic cancer [140] and colon cancer [165]. In pancreatic cancer, the protein Forkhead box protein M1 (FoxM1), which is normally associated with cell cycle progression through the G2/M-phase checkpoint, was implicated as a target of genistein action at 50 to 100 μM [140]. This inhibition of FoxM1 resulted in a decrease in MMP-9, as well as decreases in proteins that regulate the cell cycle and angiogenesis. Similarly, genistein was shown to inhibit invasion of colon cancer at similar concentrations to those used in the studies on pancreatic cancer, although the effects were thought to be a result of genistein’s antioxidant properties (see Section 2.1), rather than inhibition of any specific proteins [165].
Taken together, the above studies demonstrate that genistein is capable of inhibiting invasion through a variety of mechanisms in multiple cell types. In human prostate cancer, MEK4 is a key pharmacologic target of genistein’s anti-invasion action. However, for other cancer types, it is not yet known whether genistein’s effects stem from regulation of the same or a different pharmacologic target. Further, it is not clear whether genistein is acting upon multiple pharmacologic targets or whether it is acting upon a single target which then regulates a variety of other pathways. This situation could be clinically relevant, as cancer cells contain multiple molecular abnormalities that require therapeutic correction in order to restore the desired phenotype. Therefore, while these studies demonstrate that genistein has a multitude of effects on cancer invasion, they also suggest that genistein represents a promising multitarget therapeutic agent.
4 Genistein’s effects upon metastasis in preclinical animal models
As an extension of the in vitro studies discussed in Section 4, our group has demonstrated that genistein can inhibit prostate cancer metastasis in mice [121]. In this study, human prostate cancer cells were implanted into the prostate glands of mice fed with 0, 100, or 250 mg genistein per kilogram of soy-free chow. The human prostate cancer cells in this study were metastatic variant PC3-M cells, which are androgen and estrogen receptor negative and hormonally unresponsive. The genistein treatment regime yielded plasma concentrations of total genistein of 0, 290, and 1,307 nM, respectively. As only 10% of circulating genistein is in the free, unconjugated form, free plasma concentrations of genistein were estimated to be 0, 29, and 131 nM, respectively. These concentrations correspond to those measured in humans who subsist on a soy-based diet (see Section 2.2). Genistein treatment significantly decreased the formation of distant soft tissue metastasis in a dose-dependent fashion. There was no effect upon primary tumor growth, which is consistent with genistein’s primary role as an inhibitor of metastasis, rather than a cytotoxic agent. Analysis of primary tumor tissue indicated that genistein was inhibiting the detachment of prostate cancer cells, as assessed by quantitative image analysis. Importantly, genistein decreased phosphorylation of both p38 MAPK and HSP27 in tumor tissue, consistent with inhibition of MEK4. Further, genistein treatment decreased phosphorylation of FAK, but this trend was not statistically significant.
Other groups have also demonstrated that consumption of soy or genistein can inhibit human prostate cancer progression or metastasis in vivo [166]. In one report, the authors used hormonally responsive LNCaP cells and investigated three different soy-based dietary preparations. Each preparation decreased tumor weight in a statistically significant manner. However, since each of the preparations also decreased serum androgen levels, effects on tumor growth were likely mediated through primary effects on androgen concentrations. Furthermore, mice that were fed genistin, or the glycosylated form of genistein, did not have decreased metastasis, while mice fed soy phytochemical concentrate, which is high in genistein, did have decreased metastasis. While it would be expected that genistein would readily be converted to genistein, these findings call this assumption into question. Therefore, the type of preparation appears to have a significant impact on outcome. Similarly, genistein has also been shown to decrease cancer proliferation in a rat model of spontaneous prostate cancer, which was constructed using a Simian Virus-40 T-antigen targeted probasin promoter [167]. In this study, a dose of 250 mg genistein per kilogram chow (which gave to a blood concentration of 2,160 nM of total genistein) suppressed the development of prostate cancer, although a dose of 83 mg genistein per kilogram was not effective. Treatment with genistein caused decreased cell proliferation and increased apoptosis in the prostate, as well as decreased expression of insulin-like growth factor 1 and steroid receptor coactivator 3 proteins, which have been implicated in cell invasion and metastasis. Finally, a third group has reported that genistein decreased cancer progression in a murine model of prostate cancer that involved crosses between TRAMP and C57BL/6 nontransgenic mice [160, 168, 169]. Interestingly, genistein increased cancer progression when TRAMP and FVB crosses were used [139]. FVB and C57BL/6 mice have established genetic background differences that can affect a wide variety of biological and therapeutic response profiles [170], and these differences are likely responsible for the differential outcomes observed with genistein in these and other model systems.
Genistein has also been extensively studied in mouse models of breast cancer. However, genistein’s weak estrogenic activity (see Section 2.1) has been the central focus of these studies, as breast cancer is known to be hormonally responsive. These studies indicate that the effect of genistein upon breast cancer is dependent upon the nature of the estrogenic environment in which the study is conducted. If endogenous estrogen is low, genistein can bind the ER receptor and exert progrowth effects upon responsive systems. If endogenous estrogen is high and potent, genistein can act as a competitor to estrogen and thus antagonize this hormone’s progrowth effects (reviewed in [171]). Therefore, while genistein has shown promise as an anticancer agent for breast cancer, the results are much more complex and dependent on hormone levels than those for prostate cancer.
In non-estrogen-dependent cancer types, genistein has shown strong promise as an inhibitor of metastasis in mice. Soy concentrate and genistin both inhibited the growth of human bladder cancer cells in mice, and soy concentrate inhibited formation of metastasis [79]. In an orthotopic model of pancreatic cancer in mice, genistein induced apoptosis of the primary tumor and inhibited metastasis [87]. Additionally, another group has shown that genistein reduced metastasis of murine colon cancer cells to the lungs in a murine model [165]. In an azoxymethane-induced intestinal adenocarcinoma model in Wistar rats, genistein had no effect upon primary tumors but inhibited metastasis [172]. Similarly, genistein also decreased primary tumor growth as well as the formation of metastasis in an orthotopic murine model of human hepatocellular carcinoma cells [123]. Therefore, these examples demonstrate that genistein consistently reduces the formation of metastasis in a variety of tumor types, while it has varied effects on the primary tumor.
5 Prospective studies of genistein in humans
Most clinical trials of genistein have examined dose ranges that are similar to those consumed in the diet. To date, both phase I and phase II clinical trials have been performed with doses of genistein that were chosen to approximate the dietary consumption level of regular soy consumers, which ranges from 0.3 to 1 mg per kg body weight per day [29–41]. Phase I trials were designed to characterize the toxicity and pharmacology of genistein in order to better understand its metabolism and safety in the body, while phase II trials were designed to test genistein’s efficacy in humans.
In order to better understand the pharmacokinetic parameters of genistein in humans, we have performed an initial phase I single-dose study of genistein in a cohort of men with prostate cancer [173]. In this study, we examined doses of 2–8 mg genistein per kilogram body weight. The 2 mg per kg dose was chosen to correspond with an amount equivalent to two to seven times the average daily genistein consumption, while the 8 mg per kg dose corresponded to eight to 27 times the average daily consumption. Genistein was well tolerated, with little to no side effects observed. The peak blood concentrations of total genistein ranged from 4.3 to 16.3 μM, and the peak concentrations of free genistein ranged from 66 to 170 nM, indicating that 90% of blood genistein was conjugated (consistent with the population-based studies, see Section 2.2). The half-life of genistein was 15–22 h, and its clearance was not altered by body mass, indicating that dosing does not need to be calculated based upon weight (i.e., fixed amounts of drug can be administered to humans). A similar study focused upon a cohort of younger healthy men and reported similar findings, with the exception that genistein showed a half-life of only 9 h [174]. Another study examined doses of 4–8 mg genistein per kilogram given daily for 3 months to a cohort of men with prostate cancer [175]. This study showed a similar pharmacokinetic profile for genistein and, importantly, demonstrated that pharmacokinetic parameters were not affected after chronic dosing. Interestingly, this study also reported estrogen-related side effects, such as breast tenderness and flushing. However, the men in the study cohort were also on antiandrogen hormone therapy for prostate cancer, and these side effects are commonly associated with this therapy. Therefore, it is unclear whether these side effects were due to genistein or to the hormone therapy. These side effects were not seen in any of the other studies that were performed at similar dose levels, suggesting that genistein does not induce estrogenic side effects in men, at least with the doses and treatment times examined to date.
Phase I trials have also been conducted in a cohort of healthy postmenopausal women, where doses of 2–16 mg genistein per kilogram were examined [176]. Breast tenderness, an indicator of estrogen action, was reported in this study, though it was unclear whether it was due to genistein treatment. Pharmacokinetics for genistein were similar to those reported by the same group in a cohort of healthy men, suggesting that administration of genistein to varied populations results in a consistent pharmacokinetic profile [174].
In addition to these phase I studies, several phase II studies of genistein in subjects with prostate cancer have also been reported. In a recent study by our group, subjects were randomized to treatment with 2 mg genistein per kilogram body weight versus no treatment prior to undergoing radical prostatectomy for localized prostate cancer [143]. After treatment, histologically normal prostate epithelial cells were then selectively removed from intact prostate tissue by laser capture microdissection. These cells, present within a prostate gland that also harbors prostate cancer, represent an “at-risk” target cell type and are therefore a viable target for a compound that inhibits conversion to an invasive phenotype. MMP-2 transcript levels were evaluated in these cells by qRT/PCR, and it was shown that genistein decreased MMP-2 gene expression to 24% of the level seen in control subjects. Blood concentrations of free genistein were approximately 140 nM in the genistein-treated cohorts, while the levels in the control group were below detection. This study demonstrated for the first time that it is possible to inhibit prometastatic processes in humans through a targeted therapeutic intervention [143]. Findings from this prospective study are consistent with epidemiological studies that link genistein consumption to decreased prostate cancer metastasis and decreased mortality from prostate cancer. It will be important for future studies to evaluate the ability of genistein to inhibit prostate cancer metastasis in the context of a prospective randomized study.
In another phase II trial, subjects with progressive prostate cancer were treated with soy milk three times daily for 12 months, which roughly equated to a genistein concentration of 1 mg genistein per kilogram per day [177]. This treatment regimen gave a median blood concentration of free genistein of 44 nM. Soy milk treatment decreased the rate of increase of serum prostate-specific antigen (PSA) when compared to the rate of increase of PSA that was seen in subjects prior to entering the study. PSA is used clinically to track prostate cancer growth in humans, and in normal cancer progression, the rate of PSA increase tends to accelerate with time. Therefore, the fact that this treatment regime slowed this process even as time progressed represents a significant accomplishment. Finally, a third phase II study of genistein in men with various stages of prostate cancer was performed by administering soy extract at an approximate dose of 6 mg genistein per kilogram per day for 6 months [178]. This therapy was similarly well tolerated, as less than 10% of participants experienced mild diarrhea, and 17% of participants experienced a decrease in their PSA levels.
Clinical trials are now increasingly testing genistein in combination with other agents. The primary focus of these studies is typically on advanced cancer, with the primary goal of identifying combination therapies that are able to effectively kill cancer cells. One such study examined genistein in combination with gemcitabine in subjects with advanced pancreatic adenocarcinoma [179], while another has examined the combination of genistein and lycopene in subjects with recurring prostate cancer [180]. While no definitive activity was observed in either study, these investigations are still at the early stages. In addition, several phase II trials of genistein in breast, prostate, and pancreatic cancer are ongoing, as listed in the NIH Clinical Trials database. With the early promise of genistein as a single-agent therapeutic in prostate cancer, it will be important to determine if this effect is translatable to other cancers.
6 Conclusion
Genistein is a biologically important isoflavone that is found in high amounts in soy products. This small compound has garnered a great deal of attention in the field of oncology research, as it exerts a wide range of biological effects of direct relevance to cancer. In particular, genistein has proven to be a valuable tool for the inhibition of cancer metastasis, exerting effects on both the initial steps of primary tumor growth as well as the later steps of the metastatic cascade. The concentration at which genistein is used is a critical determinant of the range of targets that it affects and the cellular phenotypes that result from treatment. At concentrations of genistein near the micromolar range, which have been achieved in prospective phase I studies, genistein exhibits significant inhibition of tumor growth. This inhibition of proliferation has been linked to genistein’s interaction with a variety of different protein classes, including cell cycle regulators, proapoptotic and antiapoptotic proteins, and the Akt/NF-κB family of proteins. Additional preclinical studies have indicated that inhibition of the later steps of the metastatic cascade may be an important biological effect of genistein at low nanomolar concentrations, which are achieved through dietary consumption. Genistein has been shown to exert inhibitory effects on targets that are involved in almost every step of the metastatic cascade, including FAK, integrins, TGF-β signaling molecules, and MMPs, which together are critical to the processes of cell adhesion and invasion. Early positive clinical trial studies have corroborated these preclinical findings, as subjects administered genistein show decreased markers of advanced metastatic disease, such as MMP-2 and PSA. These preliminary evaluations will need to be pursued by larger confirmatory prospective clinical studies, but the findings from ongoing trials have already provided critical information about the clinical importance of these mechanisms. Overall, genistein represents a powerful tool for understanding cancer development and holds great promise as a therapeutic inhibitor of metastasis in a variety of cancer types. Furthermore, findings to date suggest that genistein has the potential to have a high clinical impact, and it is clear that expanding our knowledge of this important natural product will lead to novel future developments in the field of cancer treatment.
References
Messina, M., Nagata, C., & Wu, A. H. (2006). Estimated Asian adult soy protein and isoflavone intakes. Nutrition and Cancer, 55(1), 1–12.
Adlercreutz, H., Markkanen, H., & Watanabe, S. (1993). Plasma concentrations of phyto-oestrogens in Japanese men. Lancet, 342(8881), 1209–1210.
Adlercreutz, H. (1990). Western diet and western diseases: Some hormonal and biochemical mechanisms and associations. Scandinavian Journal of Clinical and Laboratory Investigation. Supplementum, 201, 3–23.
Severson, R. K., Nomura, A. M., Grove, J. S., & Stemmermann, G. N. (1989). A prospective study of demographics, diet, and prostate cancer among men of Japanese ancestry in Hawaii. Cancer Research, 49(7), 1857–1860.
Shimizu, H., Ross, R. K., Bernstein, L., Yatani, R., Henderson, B. E., & Mack, T. M. (1991). Cancers of the prostate and breast among Japanese and white immigrants in Los Angeles county. British Journal of Cancer, 63(6), 963–966.
Hwang, Y. W., Kim, S. Y., Jee, S. H., Kim, Y. N., & Nam, C. M. (2009). Soy food consumption and risk of prostate cancer: A meta-analysis of observational studies. Nutrition and Cancer, 61(5), 598–606.
Lee, H. P., Gourley, L., Duffy, S. W., Esteve, J., Lee, J., & Day, N. E. (1991). Dietary effects on breast-cancer risk in Singapore. Lancet, 337(8751), 1197–1200.
Wu, A. H., Ziegler, R. G., Horn-Ross, P. L., Nomura, A. M., West, D. W., Kolonel, L. N., et al. (1996). Tofu and risk of breast cancer in Asian-Americans. Cancer Epidemiology, Biomarkers & Prevention, 5(11), 901–906.
Bobe, G., Sansbury, L. B., Albert, P. S., Cross, A. J., Kahle, L., Ashby, J., et al. (2008). Dietary flavonoids and colorectal adenoma recurrence in the polyp prevention trial. Cancer Epidemiology, Biomarkers & Prevention, 17(6), 1344–1353.
Perkin, A. G., & Newbury, F. G. (1899). The colouring matters contained in dyer’s broom (Genista tinctoria) and heather (Calluna vulgaris). Journal of the Chemical Society, 75, 830–839.
Andersen, Ø. M., & Markham, K. R. (2006). Flavonoids: Chemistry, biochemistry, and applications. Boca Raton: CRC.
Veitch, N. C., & Grayer, R. J. (2008). Flavonoids and their glycosides, including anthocyanins. Natural Product Reports, 25(3), 555–611.
Akiyama, T., Ishida, J., Nakagawa, S., Ogawara, H., Watanabe, S., Itoh, N., et al. (1987). Genistein, a specific inhibitor of tyrosine-specific protein kinases. The Journal of Biological Chemistry, 262(12), 5592–5595.
Matsumura, A., Ghosh, A., Pope, G. S., & Darbre, P. D. (2005). Comparative study of oestrogenic properties of eight phytoestrogens in mcf7 human breast cancer cells. The Journal of Steroid Biochemistry and Molecular Biology, 94(5), 431–443.
Dixon, R. A., & Ferreira, D. (2002). Genistein. Phytochemistry, 60(3), 205–211.
Pike, A. C., Brzozowski, A. M., Hubbard, R. E., Bonn, T., Thorsell, A. G., Engstrom, O., et al. (1999). Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. The EMBO Journal, 18(17), 4608–4618.
Kuiper, G. G., Lemmen, J. G., Carlsson, B., Corton, J. C., Safe, S. H., van der Saag, P. T., et al. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology, 139(10), 4252–4263.
Mueller, S. O., Simon, S., Chae, K., Metzler, M., & Korach, K. S. (2004). Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor alpha (ER alpha) and ER beta in human cells. Toxicological Sciences, 80(1), 14–25.
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Haggblad, J., Nilsson, S., et al. (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology, 138(3), 863–870.
Jung, W., Yu, O., Lau, S. M., O’Keefe, D. P., Odell, J., Fader, G., et al. (2000). Identification and expression of isoflavone synthase, the key enzyme for biosynthesis of isoflavones in legumes. Nature Biotechnology, 18(2), 208–212.
Yu, O., Jung, W., Shi, J., Croes, R. A., Fader, G. M., McGonigle, B., et al. (2000). Production of the isoflavones genistein and daidzein in non-legume dicot and monocot tissues. Plant Physiology, 124(2), 781–794.
Walter, E. (1941). Genistin (an isoflavone glucoside) and its aglucone, genistein, from soybeans. Journal of the American Chemical Society, 63(12), 3273–3276.
Balasubramanian, S., & Nair, M. G. (2000). An efficient “one-pot” synthesis of isoflavones. Synthetic Communications, 30, 469–484.
Wahala, K., & Hase, T. A. (1991). Expedient synthesis of polyhydroxyisoflavones. Journal of the Chemical Society. Perkin Transactions, 1, 3005–3008.
Chang, Y. C., Nair, M. G., Santell, R. C., & Helferich, W. G. (1994). Microwave-mediated synthesis of anticarcinogenic isoflavones from soybeans. Journal of Agricultural and Food Chemistry, 42, 1869–1871.
Kawamura, Y., Maruyama, M., Tokuoka, T., & Tsukayama, M. (2002). Synthesis of isoflavones from 2′-hydroxychalcones using poly[4-(diacetoxy)iodo]styrene or related hypervalent iodine reagent. Synthesis-Stuttgart, 17, 2490–2496.
Gammill, R. B. (1979). A new and efficient synthesis of 3-halogenated 4h-1-benzopyran-4-ones. Synthesis-Stuttgart, 11, 901–903.
Hoshino, Y., Miyaura, N., & Suzuki, A. (1988). Novel synthesis of isoflavones by the palladium-catalyzed cross-coupling reaction of 3-bromochromone with arylboronic acids or its esters. Bulletin. Chemical Society of Japan, 61, 3008–3010.
Adlercreutz, H., Honjo, H., Higashi, A., Fotsis, T., Hamalainen, E., Hasegawa, T., et al. (1991). Urinary excretion of lignans and isoflavonoid phytoestrogens in Japanese men and women consuming a traditional Japanese diet. The American Journal of Clinical Nutrition, 54(6), 1093–1100.
Arai, Y., Watanabe, S., Kimira, M., Shimoi, K., Mochizuki, R., & Kinae, N. (2000). Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. The Journal of Nutrition, 130(9), 2243–2250.
Horiuchi, T., Onouchi, T., Takahashi, M., Ito, H., & Orimo, H. (2000). Effect of soy protein on bone metabolism in postmenopausal Japanese women. Osteoporosis International, 11(8), 721–724.
Kimira, M., Arai, Y., Shimoi, K., & Watanabe, S. (1998). Japanese intake of flavonoids and isoflavonoids from foods. Journal of Epidemiology, 8(3), 168–175.
Nagata, C., Inaba, S., Kawakami, N., Kakizoe, T., & Shimizu, H. (2000). Inverse association of soy product intake with serum androgen and estrogen concentrations in Japanese men. Nutrition and Cancer, 36(1), 14–18.
Nagata, C., Kabuto, M., Kurisu, Y., & Shimizu, H. (1997). Decreased serum estradiol concentration associated with high dietary intake of soy products in premenopausal Japanese women. Nutrition and Cancer, 29(3), 228–233.
Nagata, C., Shimizu, H., Takami, R., Hayashi, M., Takeda, N., & Yasuda, K. (2000). Relations of insulin resistance and serum concentrations of estradiol and sex hormone-binding globulin to potential breast cancer risk factors. Japanese Journal of Cancer Research, 91(9), 948–953.
Nagata, C., Takatsuka, N., Kawakami, N., & Shimizu, H. (2000). Association of diet with the onset of menopause in Japanese women. American Journal of Epidemiology, 152(9), 863–867.
Nagata, C., Takatsuka, N., Kurisu, Y., & Shimizu, H. (1998). Decreased serum total cholesterol concentration is associated with high intake of soy products in Japanese men and women. The Journal of Nutrition, 128(2), 209–213.
Takatsuka, N., Nagata, C., Kurisu, Y., Inaba, S., Kawakami, N., & Shimizu, H. (2000). Hypocholesterolemic effect of soymilk supplementation with usual diet in premenopausal normolipidemic Japanese women. Preventive Medicine, 31(4), 308–314.
Wakai, K., Egami, I., Kato, K., Kawamura, T., Tamakoshi, A., Lin, Y., et al. (1999). Dietary intake and sources of isoflavones among Japanese. Nutrition and Cancer, 33(2), 139–145.
Arai, Y., Uehara, M., Sato, Y., Kimira, M., Eboshida, A., Adlercreutz, H., et al. (2000). Comparison of isoflavones among dietary intake, plasma concentration and urinary excretion for accurate estimation of phytoestrogen intake. Journal of Epidemiology, 10(2), 127–135.
Yamamoto, S., Sobue, T., Sasaki, S., Kobayashi, M., Arai, Y., Uehara, M., et al. (2001). Validity and reproducibility of a self-administered food-frequency questionnaire to assess isoflavone intake in a Japanese population in comparison with dietary records and blood and urine isoflavones. The Journal of Nutrition, 131(10), 2741–2747.
Fukutake, M., Takahashi, M., Ishida, K., Kawamura, H., Sugimura, T., & Wakabayashi, K. (1996). Quantification of genistein and genistin in soybeans and soybean products. Food and Chemical Toxicology, 34(5), 457–461.
Rowland, I., Faughnan, M., Hoey, L., Wahala, K., Williamson, G., & Cassidy, A. (2003). Bioavailability of phyto-oestrogens. The British Journal of Nutrition, 89(Suppl 1), S45–S58.
Sfakianos, J., Coward, L., Kirk, M., & Barnes, S. (1997). Intestinal uptake and biliary excretion of the isoflavone genistein in rats. The Journal of Nutrition, 127(7), 1260–1268.
Murata, M., Midorikawa, K., Koh, M., Umezawa, K., & Kawanishi, S. (2004). Genistein and daidzein induce cell proliferation and their metabolites cause oxidative DNA damage in relation to isoflavone-induced cancer of estrogen-sensitive organs. Biochemistry, 43(9), 2569–2577.
Pugazhendhi, D., Watson, K. A., Mills, S., Botting, N., Pope, G. S., & Darbre, P. D. (2008). Effect of sulphation on the oestrogen agonist activity of the phytoestrogens genistein and daidzein in mcf-7 human breast cancer cells. The Journal of Endocrinology, 197(3), 503–515.
Cook, L. S., Goldoft, M., Schwartz, S. M., & Weiss, N. S. (1999). Incidence of adenocarcinoma of the prostate in Asian immigrants to the United States and their descendants. The Journal of Urology, 161(1), 152–155.
Yatani, R., Shiraishi, T., Nakakuki, K., Kusano, I., Takanari, H., Hayashi, T., et al. (1988). Trends in frequency of latent prostate carcinoma in Japan from 1965–1979 to 1982–1986. Journal of the National Cancer Institute, 80(9), 683–687.
Toyomura, K., & Kono, S. (2002). Soybeans, soy foods, isoflavones and risk of colorectal cancer: A review of experimental and epidemiological data. Asian Pacific Journal of Cancer Prevention: APJCP, 3(2), 125–132.
Weinberg, R. (2007). The biology of cancer. New York: Garland Science.
Malumbres, M., & Barbacid, M. (2009). Cell cycle, cdks and cancer: A changing paradigm. Nature Reviews. Cancer, 9(3), 153–166.
Ramos, S. (2007). Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. The Journal of Nutritional Biochemistry, 18(7), 427–442.
Cappelletti, V., Fioravanti, L., Miodini, P., & Di Fronzo, G. (2000). Genistein blocks breast cancer cells in the g(2)m phase of the cell cycle. Journal of Cellular Biochemistry, 79(4), 594–600.
Choi, Y. H., Lee, W. H., Park, K. Y., & Zhang, L. (2000). P53-independent induction of p21 (waf1/cip1), reduction of cyclin b1 and g2/m arrest by the isoflavone genistein in human prostate carcinoma cells. Japanese Journal of Cancer Research, 91(2), 164–173.
Yan, G. R., Xiao, C. L., He, G. W., Yin, X. F., Chen, N. P., Cao, Y., et al. (2010). Global phosphoproteomic effects of natural tyrosine kinase inhibitor, genistein, on signaling pathways. Proteomics, 10(5), 976–986.
Ouyang, G., Yao, L., Ruan, K., Song, G., Mao, Y., & Bao, S. (2009). Genistein induces g2/m cell cycle arrest and apoptosis of human ovarian cancer cells via activation of DNA damage checkpoint pathways. Cell Biology International, 33(12), 1237–1244.
Su, S. J., Chow, N. H., Kung, M. L., Hung, T. C., & Chang, K. L. (2003). Effects of soy isoflavones on apoptosis induction and g2-m arrest in human hepatoma cells involvement of caspase-3 activation, bcl-2 and bcl-xl downregulation, and cdc2 kinase activity. Nutrition and Cancer, 45(1), 113–123.
Ismail, I. A., Kang, K. S., Lee, H. A., Kim, J. W., & Sohn, Y. K. (2007). Genistein-induced neuronal apoptosis and g2/m cell cycle arrest is associated with mdc1 up-regulation and plk1 down-regulation. European Journal of Pharmacology, 575(1–3), 12–20.
Choi, Y. H., Zhang, L., Lee, W. H., & Park, K. Y. (1998). Genistein-induced g2/m arrest is associated with the inhibition of cyclin b1 and the induction of p21 in human breast carcinoma cells. International Journal of Oncology, 13(2), 391–396.
Adams, J. M., & Cory, S. (2007). The bcl-2 apoptotic switch in cancer development and therapy. Oncogene, 26(9), 1324–1337.
Adams, J. M. (2003). Ways of dying: Multiple pathways to apoptosis. Genes & Development, 17(20), 2481–2495.
Danial, N. N., & Korsmeyer, S. J. (2004). Cell death: Critical control points. Cell, 116(2), 205–219.
Fesik, S. W. (2005). Promoting apoptosis as a strategy for cancer drug discovery. Nature Reviews. Cancer, 5(11), 876–885.
Kyle, E., Neckers, L., Takimoto, C., Curt, G., & Bergan, R. (1997). Genistein-induced apoptosis of prostate cancer cells is preceded by a specific decrease in focal adhesion kinase activity. Molecular Pharmacology, 51(2), 193–200.
Banerjee, S., Li, Y., Wang, Z., & Sarkar, F. H. (2008). Multi-targeted therapy of cancer by genistein. Cancer Letters, 269(2), 226–242.
Sarkar, F. H., & Li, Y. (2004). The role of isoflavones in cancer chemoprevention. Frontiers in Bioscience, 9, 2714–2724.
Cuconati, A., Degenhardt, K., Sundararajan, R., Anschel, A., & White, E. (2002). Bak and bax function to limit adenovirus replication through apoptosis induction. Journal of Virology, 76(9), 4547–4558.
Willis, S. N., Fletcher, J. I., Kaufmann, T., van Delft, M. F., Chen, L., Czabotar, P. E., et al. (2007). Apoptosis initiated when bh3 ligands engage multiple bcl-2 homologs, not bax or bak. Science, 315(5813), 856–859.
Green, D. R., & Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science, 305(5684), 626–629.
Acehan, D., Jiang, X., Morgan, D. G., Heuser, J. E., Wang, X., & Akey, C. W. (2002). Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Molecular Cell, 9(2), 423–432.
Tophkhane, C., Yang, S., Bales, W., Archer, L., Osunkoya, A., Thor, A. D., et al. (2007). Bcl-2 overexpression sensitizes mcf-7 cells to genistein by multiple mechanisms. International Journal of Oncology, 31(4), 867–874.
Li, Y., Upadhyay, S., Bhuiyan, M., & Sarkar, F. H. (1999). Induction of apoptosis in breast cancer cells mda-mb-231 by genistein. Oncogene, 18(20), 3166–3172.
Upadhyay, S., Neburi, M., Chinni, S. R., Alhasan, S., Miller, F., & Sarkar, F. H. (2001). Differential sensitivity of normal and malignant breast epithelial cells to genistein is partly mediated by p21(waf1). Clinical Cancer Research, 7(6), 1782–1789.
Davis, J. N., Singh, B., Bhuiyan, M., & Sarkar, F. H. (1998). Genistein-induced upregulation of p21waf1, downregulation of cyclin b, and induction of apoptosis in prostate cancer cells. Nutrition and Cancer, 32(3), 123–131.
Lian, F., Li, Y., Bhuiyan, M., & Sarkar, F. H. (1999). P53-independent apoptosis induced by genistein in lung cancer cells. Nutrition and Cancer, 33(2), 125–131.
Alhasan, S. A., Pietrasczkiwicz, H., Alonso, M. D., Ensley, J., & Sarkar, F. H. (1999). Genistein-induced cell cycle arrest and apoptosis in a head and neck squamous cell carcinoma cell line. Nutrition and Cancer, 34(1), 12–19.
Zhang, B., Shi, Z. L., Liu, B., Yan, X. B., Feng, J., & Tao, H. M. (2010). Enhanced anticancer effect of gemcitabine by genistein in osteosarcoma: The role of akt and nuclear factor-kappab. Anti-Cancer Drugs, 21(3), 288–296.
Kazi, A., Daniel, K. G., Smith, D. M., Kumar, N. B., & Dou, Q. P. (2003). Inhibition of the proteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochemical Pharmacology, 66(6), 965–976.
Singh, A. V., Franke, A. A., Blackburn, G. L., & Zhou, J. R. (2006). Soy phytochemicals prevent orthotopic growth and metastasis of bladder cancer in mice by alterations of cancer cell proliferation and apoptosis and tumor angiogenesis. Cancer Research, 66(3), 1851–1858.
Po, L. S., Wang, T. T., Chen, Z. Y., & Leung, L. K. (2002). Genistein-induced apoptosis in mcf-7 cells involves changes in bak and bcl-x without evidence of anti-oestrogenic effects. The British Journal of Nutrition, 88(5), 463–469.
Kumi-Diaka, J., Sanderson, N. A., & Hall, A. (2000). The mediating role of caspase-3 protease in the intracellular mechanism of genistein-induced apoptosis in human prostatic carcinoma cell lines, du145 and lncap. Biology of the Cell, 92(8–9), 595–604.
Oh, H. Y., Leem, J., Yoon, S. J., Yoon, S., & Hong, S. J. (2010). Lipid raft cholesterol and genistein inhibit the cell viability of prostate cancer cells via the partial contribution of EGFR-Akt/p70s6k pathway and down-regulation of androgen receptor. Biochemical and Biophysical Research Communications, 393(2), 319–324.
Chen, A. C., & Donovan, S. M. (2004). Genistein at a concentration present in soy infant formula inhibits caco-2bbe cell proliferation by causing g2/m cell cycle arrest. The Journal of Nutrition, 134(6), 1303–1308.
Baxa, D. M., Luo, X., & Yoshimura, F. K. (2005). Genistein induces apoptosis in T lymphoma cells via mitochondrial damage. Nutrition and Cancer, 51(1), 93–101.
Das, A., Banik, N. L., & Ray, S. K. (2010). Flavonoids activated caspases for apoptosis in human glioblastoma t98g and u87mg cells but not in human normal astrocytes. Cancer, 116(1), 164–176.
Jin, C. Y., Park, C., Kim, G. Y., Lee, S. J., Kim, W. J., & Choi, Y. H. (2009). Genistein enhances trail-induced apoptosis through inhibition of p38 MAPK signaling in human hepatocellular carcinoma hep3b cells. Chemico-Biological Interactions, 180(2), 143–150.
Buchler, P., Gukovskaya, A. S., Mouria, M., Buchler, M. C., Buchler, M. W., Friess, H., et al. (2003). Prevention of metastatic pancreatic cancer growth in vivo by induction of apoptosis with genistein, a naturally occurring isoflavonoid. Pancreas, 26(3), 264–273.
Datta, S. R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y., et al. (1997). Akt phosphorylation of bad couples survival signals to the cell-intrinsic death machinery. Cell, 91(2), 231–241.
del Peso, L., Gonzalez-Garcia, M., Page, C., Herrera, R., & Nunez, G. (1997). Interleukin-3-induced phosphorylation of bad through the protein kinase akt. Science, 278(5338), 687–689.
Wullschleger, S., Loewith, R., & Hall, M. N. (2006). Tor signaling in growth and metabolism. Cell, 124(3), 471–484.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., & Hemmings, B. A. (1998). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase b. Nature, 378, 785–789.
Mitsiades, C. S., Mitsiades, N., & Koutsilieris, M. (2004). The Akt pathway: Molecular targets for anti-cancer drug development. Current Cancer Drug Targets, 4(3), 235–256.
Li, Y., & Sarkar, F. H. (2002). Inhibition of nuclear factor kappab activation in pc3 cells by genistein is mediated via akt signaling pathway. Clinical Cancer Research, 8(7), 2369–2377.
Gong, L., Li, Y., Nedeljkovic-Kurepa, A., & Sarkar, F. H. (2003). Inactivation of NF-kappab by genistein is mediated via Akt signaling pathway in breast cancer cells. Oncogene, 22(30), 4702–4709.
Peng, J. H., Zhu, J. D., Mi, M. T., Li, F. J., Cai, L., Dong, J. Z., et al. (2010). Prepubertal genistein exposure affects erbb2/Akt signal and reduces rat mammary tumorigenesis. European Journal of Cancer Prevention, 19(2), 110–119.
Ferenc, P., Solar, P., Kleban, J., Mikes, J., & Fedorocko, P. (2010). Down-regulation of bcl-2 and akt induced by combination of photoactivated hypericin and genistein in human breast cancer cells. Journal of Photochemistry and Photobiology. B: Biology, 98(1), 25–34.
Nakamura, Y., Yogosawa, S., Izutani, Y., Watanabe, H., Otsuji, E., & Sakai, T. (2009). A combination of indol-3-carbinol and genistein synergistically induces apoptosis in human colon cancer ht-29 cells by inhibiting akt phosphorylation and progression of autophagy. Molecular Cancer, 8, 100.
El-Rayes, B. F., Ali, S., Ali, I. F., Philip, P. A., Abbruzzese, J., & Sarkar, F. H. (2006). Potentiation of the effect of erlotinib by genistein in pancreatic cancer: The role of akt and nuclear factor-kappab. Cancer Research, 66(21), 10553–10559.
Kim, S. H., Kim, Y. B., Jeon, Y. T., Lee, S. C., & Song, Y. S. (2009). Genistein inhibits cell growth by modulating various mitogen-activated protein kinases and Akt in cervical cancer cells. Annals of the New York Academy of Sciences, 1171, 495–500.
Park, S. J., Kim, M. J., Kim, Y. K., Kim, S. M., Park, J. Y., & Myoung, H. (2010). Combined cetuximab and genistein treatment shows additive anti-cancer effect on oral squamous cell carcinoma. Cancer Letters, 292(1), 54–63.
Li, Y., & Sarkar, F. H. (2002). Gene expression profiles of genistein-treated pc3 prostate cancer cells. The Journal of Nutrition, 132(12), 3623–3631.
Davis, J. N., Kucuk, O., & Sarkar, F. H. (1999). Genistein inhibits NF-kappa b activation in prostate cancer cells. Nutrition and Cancer, 35(2), 167–174.
Gadgeel, S. M., Ali, S., Philip, P. A., Wozniak, A., & Sarkar, F. H. (2009). Genistein enhances the effect of epidermal growth factor receptor tyrosine kinase inhibitors and inhibits nuclear factor kappa b in non small cell lung cancer cell lines. Cancer, 115(10), 2165–2176.
Giancotti, F. G., & Ruoslahti, E. (1999). Integrin signaling. Science, 285(5430), 1028–1032.
Nagle, R. B., Knox, J. D., Wolf, C., Bowden, G. T., & Cress, A. E. (1994). Adhesion molecules, extracellular matrix, and proteases in prostate carcinoma. Journal of Cellular Biochemistry. Supplement, 19, 232–237.
Cress, A. E., Rabinovitz, I., Zhu, W., & Nagle, R. B. (1995). The alpha 6 beta 1 and alpha 6 beta 4 integrins in human prostate cancer progression. Cancer and Metastasis Reviews, 14(3), 219–228.
Fornaro, M., Tallini, G., Zheng, D. Q., Flanagan, W. M., Manzotti, M., & Languino, L. R. (1999). P27(kip1) acts as a downstream effector of and is coexpressed with the beta1c integrin in prostatic adenocarcinoma. The Journal of Clinical Investigation, 103(3), 321–329.
Yamada, K. M. (1997). Integrin signaling. Matrix Biology, 16(4), 137–141.
van Nimwegen, M. J., & van de Water, B. (2007). Focal adhesion kinase: A potential target in cancer therapy. Biochemical Pharmacology, 73(5), 597–609.
Chen, H. C., Appeddu, P. A., Parsons, J. T., Hildebrand, J. D., Schaller, M. D., & Guan, J. L. (1995). Interaction of focal adhesion kinase with cytoskeletal protein talin. The Journal of Biological Chemistry, 270(28), 16995–16999.
Sieg, D. J., Hauck, C. R., Ilic, D., Klingbeil, C. K., Schaefer, E., Damsky, C. H., et al. (2000). Fak integrates growth-factor and integrin signals to promote cell migration. Nature Cell Biology, 2(5), 249–256.
Cary, L. A., Chang, J. F., & Guan, J. L. (1996). Stimulation of cell migration by overexpression of focal adhesion kinase and its association with src and fyn. Journal of Cell Science, 109(Pt 7), 1787–1794.
Burridge, K., Turner, C. E., & Romer, L. H. (1992). Tyrosine phosphorylation of paxillin and pp 125fak accompanies cell adhesion to extracellular matrix: A role in cytoskeletal assembly. The Journal of Cell Biology, 119(4), 893–903.
Webb, D. J., Donais, K., Whitmore, L. A., Thomas, S. M., Turner, C. E., Parsons, J. T., et al. (2004). Fak-src signalling through paxillin, erk and mlck regulates adhesion disassembly. Nature Cell Biology, 6(2), 154–161.
Liu, Y., Kyle, E., Lieberman, R., Crowell, J., Kellof, G., & Bergan, R. C. (2000). Focal adhesion kinase (FAK) phosphorylation is not required for genistein-induced FAK-beta-1-integrin complex formation. Clinical & Experimental Metastasis, 18(3), 203–212.
Liu, Y. Q., Kyle, E., Patel, S., Housseau, F., Hakim, F., Lieberman, R., et al. (2001). Prostate cancer chemoprevention agents exhibit selective activity against early stage prostate cancer cells. Prostate Cancer and Prostatic Diseases, 4(2), 81–91.
Tremblay, L., Hauck, W., Aprikian, A. G., Begin, L. R., Chapdelaine, A., & Chevalier, S. (1996). Focal adhesion kinase (pp 125fak) expression, activation and association with paxillin and p50csk in human metastatic prostate carcinoma. International Journal of Cancer, 68(2), 164–171.
Hiscox, S., Jordan, N. J., Morgan, L., Green, T. P., & Nicholson, R. I. (2007). Src kinase promotes adhesion-independent activation of FAK and enhances cellular migration in tamoxifen-resistant breast cancer cells. Clinical & Experimental Metastasis, 24(3), 157–167.
Meng, X. N., Jin, Y., Yu, Y., Bai, J., Liu, G. Y., Zhu, J., et al. (2009). Characterisation of fibronectin-mediated FAK signalling pathways in lung cancer cell migration and invasion. British Journal of Cancer, 101(2), 327–334.
Bergan, R., Kyle, E., Nguyen, P., Trepel, J., Ingui, C., & Neckers, L. (1996). Genistein-stimulated adherence of prostate cancer cells is associated with the binding of focal adhesion kinase to beta-1-integrin. Clinical & Experimental Metastasis, 14(4), 389–398.
Lakshman, M., Xu, L., Ananthanarayanan, V., Cooper, J., Takimoto, C. H., Helenowski, I., et al. (2008). Dietary genistein inhibits metastasis of human prostate cancer in mice. Cancer Research, 68(6), 2024–2032.
Farina, H. G., Pomies, M., Alonso, D. F., & Gomez, D. E. (2006). Antitumor and antiangiogenic activity of soy isoflavone genistein in mouse models of melanoma and breast cancer. Oncology Reports, 16(4), 885–891.
Gu, Y., Zhu, C. F., Dai, Y. L., Zhong, Q., & Sun, B. (2009). Inhibitory effects of genistein on metastasis of human hepatocellular carcinoma. World Journal of Gastroenterology, 15(39), 4952–4957.
Miekus, K., & Madeja, Z. (2007). Genistein inhibits the contact-stimulated migration of prostate cancer cells. Cellular and Molecular Biology Letters, 12, 348–361.
Sawai, H., Okada, Y., Funahashi, H., Matsuo, Y., Takahashi, H., Takeyama, H., et al. (2005). Activation of focal adhesion kinase enhances the adhesion and invasion of pancreatic cancer cells via extracellular signal-regulated kinase-1/2 signaling pathway activation. Molecular Cancer, 4, 37.
Hood, J. D., & Cheresh, D. A. (2002). Role of integrins in cell invasion and migration. Nature Reviews. Cancer, 2(2), 91–100.
Friedl, P., & Wolf, K. (2003). Tumour-cell invasion and migration: Diversity and escape mechanisms. Nature Reviews. Cancer, 3(5), 362–374.
Vihinen, P., Ala-aho, R., & Kahari, V. M. (2005). Matrix metalloproteinases as therapeutic targets in cancer. Current Cancer Drug Targets, 5(3), 203–220.
Jezierska, A., & Motyl, T. (2009). Matrix metalloproteinase-2 involvement in breast cancer progression: A mini-review. Medical Science Monitor, 15(2), RA32–RA40.
Wojtowicz-Praga, S. M., Dickson, R. B., & Hawkins, M. J. (1997). Matrix metalloproteinase inhibitors. Investigational New Drugs, 15(1), 61–75.
Di Lullo, G. A., Sweeney, S. M., Korkko, J., Ala-Kokko, L., & San Antonio, J. D. (2002). Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. The Journal of Biological Chemistry, 277(6), 4223–4231.
Collier, I. E., Wilhelm, S. M., Eisen, A. Z., Marmer, B. L., Grant, G. A., Seltzer, J. L., et al. (1988). H-ras oncogene-transformed human bronchial epithelial cells (tbe-1) secrete a single metalloprotease capable of degrading basement membrane collagen. The Journal of Biological Chemistry, 263(14), 6579–6587.
Itoh, T., Tanioka, M., Yoshida, H., Yoshioka, T., Nishimoto, H., & Itohara, S. (1998). Reduced angiogenesis and tumor progression in gelatinase a-deficient mice. Cancer Research, 58(5), 1048–1051.
Huang, X., Chen, S., Xu, L., Liu, Y., Deb, D. K., Platanias, L. C., et al. (2005). Genistein inhibits p38 map kinase activation, matrix metalloproteinase type 2, and cell invasion in human prostate epithelial cells. Cancer Research, 65(8), 3470–3478.
Kumi-Diaka, J. K., Hassanhi, M., Merchant, K., & Horman, V. (2006). Influence of genistein isoflavone on matrix metalloproteinase-2 expression in prostate cancer cells. Journal of Medicinal Food, 9(4), 491–497.
Lee, W. Y., Huang, S. C., Tzeng, C. C., Chang, T. L., & Hsu, K. F. (2007). Alterations of metastasis-related genes identified using an oligonucleotide microarray of genistein-treated hcc1395 breast cancer cells. Nutrition and Cancer, 58(2), 239–246.
Kousidou, O. C., Mitropoulou, T. N., Roussidis, A. E., Kletsas, D., Theocharis, A. D., & Karamanos, N. K. (2005). Genistein suppresses the invasive potential of human breast cancer cells through transcriptional regulation of metalloproteinases and their tissue inhibitors. International Journal of Oncology, 26(4), 1101–1109.
Puli, S., Lai, J. C., & Bhushan, A. (2006). Inhibition of matrix degrading enzymes and invasion in human glioblastoma (u87mg) cells by isoflavones. Journal of Neurooncology, 79(2), 135–142.
El Touny, L. H., & Banerjee, P. P. (2009). Identification of a biphasic role for genistein in the regulation of prostate cancer growth and metastasis. Cancer Research, 69(8), 3695–3703.
Wang, Z., Ahmad, A., Banerjee, S., Azmi, A., Kong, D., Li, Y., et al. (2010). Foxm1 is a novel target of a natural agent in pancreatic cancer. Pharmaceutical Research, 27, 1159–1168.
Skogseth, H., Follestad, T., Larsson, E., & Halgunset, J. (2006). Transcription levels of invasion-related genes in prostate cancer cells are modified by inhibitors of tyrosine kinase. APMIS: Acta Pathologica, Microbiologica, et Immunologica Scandinavica, 114(5), 364–371.
Xu, L., & Bergan, R. C. (2006). Genistein inhibits matrix metalloproteinase type 2 activation and prostate cancer cell invasion by blocking the transforming growth factor beta-mediated activation of mitogen-activated protein kinase-activated protein kinase 2–27-kDa heat shock protein pathway. Molecular Pharmacology, 70(3), 869–877.
Xu, L., Ding, Y., Catalona, W. J., Yang, X. J., Anderson, W. F., Jovanovic, B., et al. (2009). Mek4 function, genistein treatment, and invasion of human prostate cancer cells. Journal of the National Cancer Institute, 101(16), 1141–1155.
Xu, L., Chen, S., & Bergan, R. C. (2006). Mapkapk2 and hsp27 are downstream effectors of p38 map kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene, 25(21), 2987–2998.
Lotan, T. L., Lyon, M., Huo, D., Taxy, J. B., Brendler, C., Foster, B. A., et al. (2007). Up-regulation of mkk4, mkk6 and mkk7 during prostate cancer progression: An important role for sapk signalling in prostatic neoplasia. The Journal of Pathology, 212(4), 386–394.
Wang, L., Pan, Y., & Dai, J. L. (2004). Evidence of mkk4 pro-oncogenic activity in breast and pancreatic tumors. Oncogene, 23(35), 5978–5985.
Wagner, E. F., & Nebreda, A. R. (2009). Signal integration by jnk and p38 mapk pathways in cancer development. Nature Reviews. Cancer, 9(8), 537–549.
Cornford, P. A., Dodson, A. R., Parsons, K. F., Desmond, A. D., Woolfenden, A., Fordham, M., et al. (2000). Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Research, 60(24), 7099–7105.
Mounier, N., & Arrigo, A. P. (2002). Actin cytoskeleton and small heat shock proteins: How do they interact? Cell Stress & Chaperones, 7(2), 167–176.
Benndorf, R., Hayess, K., Ryazantsev, S., Wieske, M., Behlke, J., & Lutsch, G. (1994). Phosphorylation and supramolecular organization of murine small heat shock protein hsp25 abolish its actin polymerization-inhibiting activity. The Journal of Biological Chemistry, 269(32), 20780–20784.
Miron, T., Vancompernolle, K., Vandekerckhove, J., Wilchek, M., & Geiger, B. (1991). A 25-kD inhibitor of actin polymerization is a low molecular mass heat shock protein. The Journal of Cell Biology, 114(2), 255–261.
Lavoie, J. N., Hickey, E., Weber, L. A., & Landry, J. (1993). Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. The Journal of Biological Chemistry, 268(32), 24210–24214.
Lavoie, J. N., Gingras-Breton, G., Tanguay, R. M., & Landry, J. (1993). Induction of Chinese hamster hsp27 gene expression in mouse cells confers resistance to heat shock. Hsp27 stabilization of the microfilament organization. The Journal of Biological Chemistry, 268(5), 3420–3429.
Hedges, J. C., Dechert, M. A., Yamboliev, I. A., Martin, J. L., Hickey, E., Weber, L. A., et al. (1999). A role for p38(mapk)/hsp27 pathway in smooth muscle cell migration. The Journal of Biological Chemistry, 274(34), 24211–24219.
Rousseau, S., Houle, F., Landry, J., & Huot, J. (1997). P38 map kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene, 15(18), 2169–2177.
Hayes, S. A., Huang, X., Kambhampati, S., Platanias, L. C., & Bergan, R. C. (2003). P38 map kinase modulates smad-dependent changes in human prostate cell adhesion. Oncogene, 22(31), 4841–4850.
Liu, Y., Jovanovic, B., Pins, M., Lee, C., & Bergan, R. C. (2002). Over expression of endoglin in human prostate cancer suppresses cell detachment, migration and invasion. Oncogene, 21(54), 8272–8281.
Craft, C. S., Romero, D., Vary, C. P., & Bergan, R. C. (2007). Endoglin inhibits prostate cancer motility via activation of the alk2-smad1 pathway. Oncogene, 26(51), 7240–7250.
Craft, C. S., Xu, L., Romero, D., Vary, C. P., & Bergan, R. C. (2008). Genistein induces phenotypic reversion of endoglin deficiency in human prostate cancer cells. Molecular Pharmacology, 73(1), 235–242.
El Touny, L. H., & Banerjee, P. P. (2007). Genistein induces the metastasis suppressor kangai-1 which mediates its anti-invasive effects in tramp cancer cells. Biochemical and Biophysical Research Communications, 361(1), 169–175.
Zhang, L. L., Li, L., Wu, D. P., Fan, J. H., Li, X., Wu, K. J., et al. (2008). A novel anti-cancer effect of genistein: Reversal of epithelial mesenchymal transition in prostate cancer cells. Acta Pharmacologica Sinica, 29(9), 1060–1068.
Hsu, E. L., Chen, N., Westbrook, A., Wang, F., Zhang, R., Taylor, R. T., et al. (2009). Modulation of cxcr4, cxcl12, and tumor cell invasion potential in vitro by phytochemicals. Journal of Oncology, 2009, 491985.
Magee, P. J., McGlynn, H., & Rowland, I. R. (2004). Differential effects of isoflavones and lignans on invasiveness of mda-mb-231 breast cancer cells in vitro. Cancer Letters, 208(1), 35–41.
Scholar, E. M., & Toews, M. L. (1994). Inhibition of invasion of murine mammary carcinoma cells by the tyrosine kinase inhibitor genistein. Cancer Letters, 87(2), 159–162.
Ogasawara, M., Matsunaga, T., & Suzuki, H. (2007). Differential effects of antioxidants on the in vitro invasion, growth and lung metastasis of murine colon cancer cells. Biological & Pharmaceutical Bulletin, 30(1), 200–204.
Zhou, J. R., Yu, L., Zhong, Y., Nassr, R. L., Franke, A. A., Gaston, S. M., et al. (2002). Inhibition of orthotopic growth and metastasis of androgen-sensitive human prostate tumors in mice by bioactive soybean components. The Prostate, 53(2), 143–153.
Harper, C. E., Cook, L. M., Patel, B. B., Wang, J., Eltoum, I. A., Arabshahi, A., et al. (2009). Genistein and resveratrol, alone and in combination, suppress prostate cancer in sv-40 tag rats. The Prostate, 69(15), 1668–1682.
Mentor-Marcel, R., Lamartiniere, C. A., Eltoum, I. E., Greenberg, N. M., & Elgavish, A. (2001). Genistein in the diet reduces the incidence of poorly differentiated prostatic adenocarcinoma in transgenic mice (tramp). Cancer Research, 61(18), 6777–6782.
El Touny, L. H., & Banerjee, P. P. (2007). Akt gsk-3 pathway as a target in genistein-induced inhibition of tramp prostate cancer progression toward a poorly differentiated phenotype. Carcinogenesis, 28(8), 1710–1717.
Sato, Y., Seo, N., & Kobayashi, E. (2006). Genetic background differences between fvb and c57bl/6 mice affect hypnotic susceptibility to pentobarbital, ketamine and nitrous oxide, but not isoflurane. Acta Anaesthesiologica Scandinavica, 50(5), 553–556.
Messina, M., McCaskill-Stevens, W., & Lampe, J. W. (2006). Addressing the soy and breast cancer relationship: Review, commentary, and workshop proceedings. Journal of the National Cancer Institute, 98(18), 1275–1284.
Iishi, H., Tatsuta, M., Baba, M., Yano, H., Sakai, N., & Akedo, H. (2000). Genistein attenuates peritoneal metastasis of azoxymethane-induced intestinal adenocarcinomas in Wistar rats. International Journal of Cancer, 86(3), 416–420.
Takimoto, C. H., Glover, K., Huang, X., Hayes, S. A., Gallot, L., Quinn, M., et al. (2003). Phase I pharmacokinetic and pharmacodynamic analysis of unconjugated soy isoflavones administered to individuals with cancer. Cancer Epidemiology, Biomarkers & Prevention, 12(11 Pt 1), 1213–1221.
Busby, M. G., Jeffcoat, A. R., Bloedon, L. T., Koch, M. A., Black, T., Dix, K. J., et al. (2002). Clinical characteristics and pharmacokinetics of purified soy isoflavones: Single-dose administration to healthy men. The American Journal of Clinical Nutrition, 75(1), 126–136.
Fischer, L., Mahoney, C., Jeffcoat, A. R., Koch, M. A., Thomas, B. E., Valentine, J. L., et al. (2004). Clinical characteristics and pharmacokinetics of purified soy isoflavones: Multiple-dose administration to men with prostate neoplasia. Nutrition and Cancer, 48(2), 160–170.
Bloedon, L. T., Jeffcoat, A. R., Lopaczynski, W., Schell, M. J., Black, T. M., Dix, K. J., et al. (2002). Safety and pharmacokinetics of purified soy isoflavones: Single-dose administration to postmenopausal women. The American Journal of Clinical Nutrition, 76(5), 1126–1137.
Pendleton, J. M., Tan, W. W., Anai, S., Chang, M., Hou, W., Shiverick, K. T., et al. (2008). Phase II trial of isoflavone in prostate-specific antigen recurrent prostate cancer after previous local therapy. BMC Cancer, 8, 132.
deVere White, R. W., Hackman, R. M., Soares, S. E., Beckett, L. A., Li, Y., & Sun, B. (2004). Effects of a genistein-rich extract on PSA levels in men with a history of prostate cancer. Urology, 63(2), 259–263.
El-Rayes, B. F., Philip, P. A., Sarkar, F. H., Shields, A. F., Ferris, A. M., Hess, K., et al. (2010). A phase II study of isoflavones, erlotinib, and gemcitabine in advanced pancreatic cancer. Investigational New Drugs. doi:10.1007/s10637-010-9386-6.
Vaishampayan, U., Hussain, M., Banerjee, M., Seren, S., Sarkar, F. H., Fontana, J., et al. (2007). Lycopene and soy isoflavones in the treatment of prostate cancer. Nutrition and Cancer, 59(1), 1–7.
Acknowledgements
This work was supported by grants from the National Institutes of Health to R. C. B., CA122985, Prostate SPORE CA90386, and N01-CN-35157. J. M. P. is the recipient of National Institutes of Health T32 AG000260, “Drug Discovery in Age-Related Disorders.” R. L. F. acknowledges grant support from the National Institutes of Health predoctoral fellowship F31CA138097 and from the Malkin Scholars Program.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pavese, J.M., Farmer, R.L. & Bergan, R.C. Inhibition of cancer cell invasion and metastasis by genistein. Cancer Metastasis Rev 29, 465–482 (2010). https://doi.org/10.1007/s10555-010-9238-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-010-9238-z