
Abstract
This Perspective, arising from a workshop held in July 2008 in Buffalo NY, provides an overview of the role NMR has played in the United States Protein Structure Initiative (PSI), and a vision of how NMR will contribute to the forthcoming PSI-Biology program. NMR has contributed in key ways to structure production by the PSI, and new methods have been developed which are impacting the broader protein NMR community.
Similar content being viewed by others


The mission of structural coverage of most protein domain families, pioneered in PSI phases 1 and 2, is well on its way to completion [6]. NMR has played an integral role in this endeavor [35, 43]. The goal of structural coverage at a sequence identity level of ~30% for most protein domains in nature will represent a monumental achievement for humankind, contributing in many ways toward our understanding of the relationships between protein sequence, structure, and function. As we ponder the future contributions of structural genomics (SG) for biomedical research, we envision many future opportunities beyond structure production that have been created by these high throughput structural biology platforms.
In the coming years, target selection strategies likely will go beyond the current sparse sampling of representative members of protein families to strategies aimed at providing extensive structural coverage of functional biological systems at high resolution. These systems could include (i) signaling networks and metabolic pathways, (ii) proteomes of medically important species, particularly humans, (iii) human disease-related proteins including infectious diseases, (iv) the human and environmental microbiomes (‘metagenomics’), and (v) comparative analysis of structure, dynamics, and biochemical function across protein families. The application of SG platforms to one or more of these biological systems would leverage NIH’s investment in SG pipelines to further our understanding of fundamental mechanisms of protein function, molecular evolution, biological processes, and human disease at a reduced cost. Alternatively, SG centers could be redefined to focus on increasing the range and types of structures that presently cannot be routinely determined or modeled; for example, membrane proteins, higher order protein complexes, and eukaryotic proteins with extensive natively disordered regions and/or posttranslational modifications.
In considering future efforts, we note that the purified proteins themselves are among the most valuable products of SG efforts. The largest expense in SG is the preparation of pure, soluble protein. Much more could be done with these proteins, particularly the large fraction that does not readily yield structures. Given that all proteins carry out their biochemical function through their interactions with other molecules, we propose that the full realization of the potential of SG platforms must integrate studies of functionally relevant interacting molecules for each protein target. Therefore, we envision that a key element of future SG projects or platforms would include a systematic attempt to integrate experimental protein binding, and/or biochemical information with structural data. Examples of such strategies, which would include HTP biochemical characterization of proteins, are (i) screening of ligand binding coupled with 3D structure analysis of functional protein-ligand complexes (see, for example, [23, 37], (ii) screening or characterization of enzymatic activity coupled with 3D structures of relevant protein substrate/cofactor/inhibitor complexes (see, for example, [28], and (iii) identification of protein-protein interaction partners coupled with 3D structures of relevant multiprotein complexes. A particularly powerful application of such integrated SG/functional studies would be the systematic and comprehensive characterization of the structural basis of ligand (or substrate) binding specificity of proteins with related, but distinct, binding profiles, so as to understand the structural basis of their specificity. Here we define “ligand” as any small molecule or macromolecule that interacts functionally with a protein. By adopting this approach, SG would have stronger synergy with functional genomics activities, and better integration with systems biology. These studies would also identify complexes that stabilize protein structures, and enable structures to be determined for otherwise refractory proteins.
NMR spectroscopy has a unique and valuable role in SG
During the course of PSI phases 1 and 2, we have shown that NMR is a highly complementary approach to X-ray crystallography for protein structure determination [32, 44]. Many proteins that provide good NMR spectra have not been successfully crystallized. In particular, in contrast to X-ray crystallography, NMR is about equally successful for prokaryotic and eukaryotic proteins. Therefore, comprehensive structural coverage of any protein system involving small to medium sized proteins would benefit from an NMR component.
NMR data provide the basis for extending the static structural view of proteins, through the rapid identification of natively unfolded proteins and residue-specific characterization of disordered protein segments, including functionally important flexible surface loops. NMR is also an essential tool for characterizing alternative conformations and allosteric states. In some cases, the minor conformational states that can only be characterized by NMR studies are critically important for biological function. NMR can also be used to measure the rates of transitions between these conformational states. As such, future SG efforts seeking to understand the evolution of structural, functional, and dynamic diversity across a protein family will require NMR studies to provide dynamic information.
NMR is also a powerful method for screening of functional protein-ligand, protein-protein, and protein-nucleic acid interactions. While other biophysical techniques are also capable of identifying such interactions, NMR is uniquely able to identify even transient, but functionally important, interactions. The protein samples, and most of the instrumentation and techniques required for rapid NMR screening studies, are the same as those already used in PSI NMR structure determination pipelines, allowing easy integration of functional screening techniques. NMR methods are also valuable for validating initial ‘hits’ identified in HTP screening. It is important to recognize that the use of NMR as a HTP screening tool is not limited by protein size, since one may monitor either the protein or the ligand to detect the interaction.
Finally, NMR data are used to generate new functional hypotheses, and to confirm functional annotations, interactions, or biochemical reaction rates revealed in other “omics” projects (e.g., functional genomics, transcriptomics, or metabonomics). Hence, we envision that NMR will play a key role to connect SG with these ‘omics’ approaches, thereby better integrating SG into systems biology.
Accomplishments of NMR SG groups during PSI
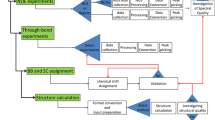
One Large Scale Center, The Northeast Structural Genomics Consortium (NESG), and one Specialized Center, the Center for Eukaryotic Structural Genomics (CESG), have made major commitments to protein NMR sample and structure production. The two centers have deposited into the PDB some 300 protein NMR structures (>90% of the PSI NMR structures) over the first 8 years of the PSI program. Thus, with ~12% of PSI resources dedicated to NMR pipelines, ~10% of PSI structures have been determined by NMR. Given similar levels of support and priority in these two centers, NMR makes contributions to structure production that are comparable to X-ray crystallography (Fig. 1, left panel). The Joint Center for Structural Genomics (JCSG), Center for Structure of Membrane Proteins (CSMP), and New York Center on Membrane Protein Structure (NYCOMPS) have also used NMR effectively, though with a smaller percentage effort. Many of these structures would not have been solved without the participation of NMR. Indeed, ~15% of small proteins provided by other Large Scale Centers to NESG NMR groups, because they could not be crystallized successfully, subsequently provided 3D structures by NMR. Many other potential opportunities to solve PSI target structures may have been missed by the other PSI centers, where NMR-tractable proteins have been produced, but not pursued by NMR analysis.

(left panel) In the two PSI centers with major commitments to NMR sample and structure production some 37% of structures were determined by NMR (42% and 22% in NESG and CESG, respectively). (right panel) MW distributions for protein NMR structures (>50 residues) determined by PSI groups and non SG groups in the same time period are similar. Inset—histogram plot of MW distribution of PSI NMR structures. Statistics were compiled in October 2008
Comparison of PSI and non-SG protein NMR structures deposited in the PDB during the same time period reveals that (i) the average molecular weight (MW) of PSI NMR structures, ~13 kDa, is similar to that of non-SG structures (Fig. 1, right panel), (ii) the fraction of homo-oligomeric protein structures (~15%) is also about the same, but (iii) the quality of PSI NMR structures is significantly better, when considering PROCHECK dihedral angle distribution and MOLPROBITY atomic clash scores (Fig. 2). As a consequence, PSI NMR structures are generally of sufficiently high accuracy to be used in crystallographic molecular replacement studies [30], and as useful as medium-resolution (1.8–2.5 Å) X-ray crystal structures for high-quality homology modeling (e.g., [22, 24]). The PSI NMR structure pipelines have also demonstrated that they can address challenging protein targets, including proteins with MW 20–35 kDa (Fig 1, right panel), dimeric and tetrameric proteins, and membrane proteins.

NMR structure quality assessed by MolProbity [7], using Z scores defined in Protein Structure Validation Suite (PSVS) software [5], are significantly better for structures generated in the PSI Centers (median Z score = −4.4 over 8 years) compared with NMR structures deposited in the PDB by groups not involved in structural genomics (median Z score = −8.6), during the same time periods. The analysis included statistics for all NMR structures deposited in the PDB by PSI centers (left panel) compared with statistics for 50 NMR structures chosen randomly from those deposited in the PDB by nonSG research groups (right panel) during the same time period. In these box plots, the central horizontal line is the median value, the bottom and top of the box denote the first and third quantiles, and the vertical “whiskers” denote ±1.5 times the interquartile range (approximately two standard deviations). Outlier values are indicated by open circles
NESG, CESG, and JCSG have also developed new methodology for lowering the costs per NMR structure, including (i) protocols for HTP preparation of 13C/15N- and 13C/15N/2H- enriched samples using novel eukaryotic wheat-germ based cell-free expression systems [39, 40] and bacterial single protein production (SPP) systems [29, 33, 34], (ii) HTP NMR screening platforms using microprobe robotics for buffer and construct optimization [1], (iii) GFT NMR [2, 3, 19, 20, 36], and related HIFI [8] and APSY [13–15] NMR experiments for reducing NMR measurement times by more than an order of magnitude, (iv) software for semi-automated data analysis and structure calculations [4, 9–18, 21, 25, 26, 41, 46], (v) software and protocols for structure validation and refinement based on residual dipolar couplings (RDCs) and chemical shifts [31, 38, 42], and (vi) software and servers for comprehensive structure quality assessment [5, 17] and refinement [30]. These methods have reduced the average time required per structure to 2–3 weeks for small to medium sized proteins; in favorable cases, NMR structures are determined in only a few days. Although not in the original charge to the PSI NMR groups, recent efforts in technology development have focused on addressing larger proteins, oligomeric structures, and protein-protein complexes. For example, the NYCOMPS and CSMP have made significant advances in developing new methods for sample preparation and NMR analysis of membrane protein structures [45, 27].
A promising future for NMR contributions to SG and the larger biomedical community
NMR’s role in structural biology is still rapidly evolving. Unlike x-ray crystallography, which has matured to a state in which almost all aspects can be highly automated, NMR is still approaching this goal. We are very optimistic that over the next decade NMR will continue to make gains analogous to those seen for crystallography over the past few decades. For example, recent advances demonstrate that sparse constraints, such as chemical shift, residual dipolar coupling data, and/or small numbers of long-range distance constraints, can be combined with conformational energy calculations to provide good quality protein structures. These emerging technologies will expand the range of proteins that can be addressed at high resolution by NMR, as well as the speed with which this can be done. The new avenues of biological research opened by SG platforms will be tremendously enhanced by these NMR technologies. Clearly, NMR approaches offer tremendous opportunities for SG projects, and will be required in order to extract the greatest knowledge and understanding of whichever biological systems are targeted in the next phase of SG research.
References
Aramini JM, Rossi P, Anklin C, Xiao R, Montelione GT (2007) Microgram-scale protein structure determination by NMR. Nat Methods 4:491–493. doi:10.1038/nmeth1051
Atreya HS, Szyperski T (2004) G-matrix Fourier transform NMR spectroscopy for complete protein resonance assignment. Proc Natl Acad Sci USA 101:9642–9647. doi:10.1073/pnas.0403529101
Atreya HS, Szyperski T (2005) Rapid NMR data collection. Methods Enzymol 394:78–108. doi:10.1016/S0076-6879(05)94004-4
Baran MC, Huang YJ, Moseley HN, Montelione GT (2004) Automated analysis of protein NMR assignments and structures. Chem Rev 104:3541–3556. doi:10.1021/cr030408p
Bhattacharya A, Tejero R, Montelione GT (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66:778–795. doi:10.1002/prot.21165
Burley SK, Joachimiak A, Montelione GT, Wilson IA (2008) Contributions to the NIH–NIGMS protein structure initiative from the PSI production centers. Structure 16:5–11. doi:10.1016/j.str.2007.12.002
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. doi:10.1093/nar/gkm216
Eghbalnia HR, Bahrami A, Tonelli M, Hallenga K, Markley JL (2005) High-resolution iterative frequency identification for NMR as a general strategy for multidimensional data collection. J Am Chem Soc 127:12528–12536. doi:10.1021/ja052120i
Eghbalnia HR, Bahrami A, Wang L, Assadi A, Markley JL (2005) Probabilistic Identification of Spin Systems and their Assignments including Coil-Helix Inference as Output (PISTACHIO). J Biomol NMR 32:219–233. doi:10.1007/s10858-005-7944-6
Eghbalnia HR, Wang L, Bahrami A, Assadi A, Markley JL (2005) Protein Energetic Conformational Analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements. J Biomol NMR 32:71–81. doi:10.1007/s10858-005-5705-1
Fiorito F, Hiller S, Wider G, Wuthrich K (2006) Automated resonance assignment of proteins: 6D APSY-NMR. J Biomol NMR 35:27–37. doi:10.1007/s10858-006-0030-x
Grishaev A, Steren CA, Wu B, Pineda-Lucena A, Arrowsmith C, Llinas M (2005) ABACUS, a direct method for protein NMR structure computation via assembly of fragments. Proteins 61:36–43. doi:10.1002/prot.20457
Hiller S, Fiorito F, Wuthrich K, Wider G (2005) Automated projection spectroscopy (APSY). Proc Natl Acad Sci USA 102:10876–10881. doi:10.1073/pnas.0504818102
Hiller S, Wasmer C, Wider G, Wuthrich K (2007) Sequence-specific resonance assignment of soluble nonglobular proteins by 7D APSY-NMR spectroscopy. J Am Chem Soc 129:10823–10828. doi:10.1021/ja072564+
Hiller S, Wider G, Wuthrich K (2008) APSY-NMR with proteins: practical aspects and backbone assignment. J Biomol NMR 42:179–195. doi:10.1007/s10858-008-9266-y
Huang YJ, Moseley HN, Baran MC, Arrowsmith C, Powers R, Tejero R, Szyperski T, Montelione GT (2005) An integrated platform for automated analysis of protein NMR structures. Methods Enzymol 394:111–141. doi:10.1016/S0076-6879(05)94005-6
Huang YJ, Powers R, Montelione GT (2005) Protein NMR recall, precision, and F-measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics. J Am Chem Soc 127:1665–1674. doi:10.1021/ja047109h
Huang YJ, Tejero R, Powers R, Montelione GT (2006) A topology-constrained distance network algorithm for protein structure determination from NOESY data. Proteins 62:587–603. doi:10.1002/prot.20820
Kim S, Szyperski T (2003) GFT NMR, a new approach to rapidly obtain precise high-dimensional NMR spectral information. J Am Chem Soc 125:1385–1393. doi:10.1021/ja028197d
Kim S, Szyperski T (2004) GFT NMR experiments for polypeptide backbone and 13Cbeta chemical shift assignment. J Biomol NMR 28:117–130. doi:10.1023/B:JNMR.0000013827.20574.46
Lemak A, Steren CA, Arrowsmith CH, Llinas M (2008) Sequence specific resonance assignment via multicanonical Monte Carlo search using an ABACUS approach. J Biomol NMR 41:29–41. doi:10.1007/s10858-008-9238-2
Liu G, Li Z, Chiang Y, Acton T, Montelione GT, Murray D, Szyperski T (2005) High-quality homology models derived from NMR and X-ray structures of E. coli proteins YgdK and Suf E suggest that all members of the YgdK/Suf E protein family are enhancers of cysteine desulfurases. Protein Sci 14:1597–1608. doi:10.1110/ps.041322705
Mercier KA, Baran M, Ramanathan V, Revesz P, Xiao R, Montelione GT, Powers R (2006) FAST-NMR: functional annotation screening technology using NMR spectroscopy. J Am Chem Soc 128:15292–15299. doi:10.1021/ja0651759
Mirkovic N, Li Z, Parnassa A, Murray D (2007) Strategies for high-throughput comparative modeling: applications to leverage analysis in structural genomics and protein family organization. Proteins 66:766–777. doi:10.1002/prot.21191
Moseley HN, Monleon D, Montelione GT (2001) Automatic determination of protein backbone resonance assignments from triple resonance nuclear magnetic resonance data. Methods Enzymol 339:91–108. doi:10.1016/S0076-6879(01)39311-4
Moseley HN, Riaz N, Aramini JM, Szyperski T, Montelione GT (2004) A generalized approach to automated NMR peak list editing: application to reduced dimensionality triple resonance spectra. J Magn Reson 170:263–277. doi:10.1016/j.jmr.2004.06.015
Poget SF, Girvin ME (2007) Solution NMR of membrane proteins in bilayer mimics: small is beautiful, but sometimes bigger is better. Biochem Biophys Acta 1768:3098–3106
Proudfoot M, Kuznetsova E, Sanders SA, Gonzalez CF, Brown G, Edwards AM, Arrowsmith CH, Yakunin AF (2008) High throughput screening of purified proteins for enzymatic activity. Methods Mol Biol 426:331–341. doi:10.1007/978-1-60327-058-8_21
Qing G, Ma LC, Khorchid A, Swapna GV, Mal TK, Takayama MM, Xia B, Phadtare S, Ke H, Acton T et al (2004) Cold-shock induced high-yield protein production in Escherichia coli. Nat Biotechnol 22:877–882. doi:10.1038/nbt984
Ramelot TA, Raman S, Kuzin AP, Xiao R, Ma LC, Acton TB, Hunt JF, Montelione GT, Baker D, Kennedy MA (2009) Improving NMR protein structure quality by Rosetta refinement: a molecular replacement study. Proteins 75:147–167
Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A et al (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA 105:4685–4690. doi:10.1073/pnas.0800256105
Snyder DA, Chen Y, Denissova NG, Acton T, Aramini JM, Ciano M, Karlin R, Liu J, Manor P, Rajan PA et al (2005) Comparisons of NMR spectral quality and success in crystallization demonstrate that NMR and X-ray crystallography are complementary methods for small protein structure determination. J Am Chem Soc 127:16505–16511. doi:10.1021/ja053564h
Suzuki M, Roy R, Zheng H, Woychik N, Inouye M (2006) Bacterial bioreactors for high yield production of recombinant protein. J Biol Chem 281:37559–37565. doi:10.1074/jbc.M608806200
Suzuki M, Zhang J, Liu M, Woychik NA, Inouye M (2005) Single protein production in living cells facilitated by an mRNA interferase. Mol Cell 18:253–261. doi:10.1016/j.molcel.2005.03.011
Szyperski T (2008) On NMR-based structural proteomics. In: Sussman JL, Silman I (eds) Structural proteomics. World Scientific Publishing, Hackensack, NJ
Szyperski T, Yeh DC, Sukumaran DK, Moseley HN, Montelione GT (2002) Reduced-dimensionality NMR spectroscopy for high-throughput protein resonance assignment. Proc Natl Acad Sci USA 99:8009–8014. doi:10.1073/pnas.122224599
Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H et al (2006) Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA 103:15835–15840. doi:10.1073/pnas.0605224103
Vila JA, Aramini JM, Rossi P, Kuzin A, Su M, Seetharaman J, Xiao R, Tong L, Montelione GT, Scheraga HA (2008) Quantum chemical 13C(alpha) chemical shift calculations for protein NMR structure determination, refinement, and validation. Proc Natl Acad Sci USA 105:14389–14394. doi:10.1073/pnas.0807105105
Vinarov DA, Lytle BL, Peterson FC, Tyler EM, Volkman BF, Markley JL (2004) Cell-free protein production and labeling protocol for NMR-based structural proteomics. Nat Methods 1:149–153. doi:10.1038/nmeth716
Vinarov DA, Newman CL, Markley JL (2006) Wheat germ cell-free platform for eukaryotic protein production. FEBS J 273:4160–4169
Volk J, Herrmann T, Wuthrich K (2008) Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J Biomol NMR 41:127–138. doi:10.1007/s10858-008-9243-5
Wang X, Bansal S, Jiang M, Prestegard JH (2008) RDC-assisted modeling of symmetric protein homo-oligomers. Protein Sci 17:899–907. doi:10.1110/ps.073395108
Yee A, Gutmanas A, Arrowsmith CH (2006) Solution NMR in structural genomics. Curr Opin Struct Biol 16:611–617. doi:10.1016/j.sbi.2006.08.002
Yee AA, Savchenko A, Ignachenko A, Lukin J, Xu X, Skarina T, Evdokimova E, Liu CS, Semesi A, Guido V et al (2005) NMR and X-ray crystallography, complementary tools in structural proteomics of small proteins. J Am Chem Soc 127:16512–16517. doi:10.1021/ja053565+
Zhang Q, Atreya HS, Kamen DE, Girvin ME, Szyperski T (2008) GFT projection NMR based resonance assignment of membrane proteins: application to subunit C of E. coli F(1)F (0) ATP synthase in LPPG micelles. J Biomol NMR 40:157–163. doi:10.1007/s10858-008-9224-8
Zheng D, Huang YJ, Moseley HN, Xiao R, Aramini J, Swapna GV, Montelione GT (2003) Automated protein fold determination using a minimal NMR constraint strategy. Protein Sci 12:1232–1246. doi:10.1110/ps.0300203
Acknowledgments
We thank B. Mao and J. Everett for their assistance in statistical analyses. This work was supported by National Institutes of Health Grants U54-GM074958 (Northeast Structure Genomics Consortium), U54-GM75026 (New York Consortium on Membrane Protein Structure), U54-GM074901 (Center for Eukaryotic Structural Genomics), and P41-RR02301 (National Magnetic Resonance Facility at Madison).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Montelione, G.T., Arrowsmith, C., Girvin, M.E. et al. Unique opportunities for NMR methods in structural genomics. J Struct Funct Genomics 10, 101–106 (2009). https://doi.org/10.1007/s10969-009-9064-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10969-009-9064-0