
Abstract
Large-scale development of expressed sequence tag simple sequence repeat (EST-SSR) markers was performed in peanut (Arachis hypogaea L.) to obtain more informative genetic markers. A total of 10,102 potential non-redundant EST sequences, including 3,445 contigs and 6,657 singletons, were generated from cDNA libraries of the gynophore, roots, leaves and seedlings. A total of 3,187 primer pairs were designed on flanking regions of SSRs, some of which allowed one and two base mismatches. Among the 3,187 markers generated, 2,540 (80%) were trinucleotide repeats, 302 (9%) were dinucleotide repeats, and 345 (11%) were tetranucleotide repeats. Pre-polymorphic analyses of 24 Arachis accessions were performed using 10% polyacrylamide gels. A total of 1,571 EST-SSR markers showing clear polymorphisms were selected for further polymorphic analysis with a Fluoro-fragment Analyzer. The 16 Arachis accessions examined included cultivated peanut varieties as well as diploid species with the A or B genome. Altogether 1,281 (81.5%) of the 1,571 markers were polymorphic among the 16 accessions, and 366 (23.3%) were polymorphic among the 12 cultivated varieties. Diversity analysis was performed and the genotypes of all 16 Arachis accessions showed similarity coefficients ranging from 0.37 to 0.97.
Similar content being viewed by others


Introduction
Peanut or groundnut (Arachis hypogaea L.) is a major oil seed crop grown extensively in Africa, Asia and America. Annually, it is grown over a global area of nearly 25.2 million ha, with production totaling 35.9 million metric tons (FAOSTAT 2006). In addition to its role in the confectionery industry, peanut represents a rich source of protein, minerals and vitamins. The cake that remains after oil extraction can be used in human food or incorporated into animal feed (Savage and Keenen 1994). The haulm serves as excellent forage for cattle and may also be used in pasture and field improvement, since peanut fixes nitrogen via its symbiosis with Rhizobium spp. (Kerridge and Hardy 1994).
Peanut is an an allotetraploid species (AABB genome; 2n = 4x = 40) that belongs to the tribe Aeschynomeneae, subtribe Stylosantinae and family Leguminosae (Stalker 1997). It is divided into two subspecies, hypogaea and fastigata, which can be divided into two (hypogaea and hirsuta) and four (fastigata, vulgaris, aequatoriana and peruviana) types, respectively. In addition, modern cultivars are generally classified into four botanical types, i.e., Spanish, Valencia, Virginia and Southeast runner, and these are used widely as representatives of the botanical types in genetic variability analyses (Krapovickas and Gregory 1994). Polyploidy is often observed in higher plant species and is caused by the duplication of chromosomes during cell division. Polyploidy plays a major role in speciation and adaptation (Ramsey and Schemske 1998; Soltis and Soltis 2000). Peanut probably originated via a single hybridization event between the wild species Arachis duranensis Krapovickas and W.C. Gregory (AA genome) and Arachis ipaënsis Krapovickas and W.C. Gregory (BB genome), followed by a rare but spontaneous duplication of the chromosomes (Halward et al. 1991; Krapovickas and Gregory 1994; Kochert et al. 1996; Seijo et al. 2004, 2007). Apart from the above-mentioned species, several other species have been suggested as putative donors of the A and B genomes (Halward et al. 1991; Raina and Mukai 1999; Raina et al. 2001). The resulting tetraploid plant would have been reproductively isolated from other diploid species and this isolation may have been responsible for the limited genetic diversity of A. hypogaea. In contrast, wild diploid species are more diverse, and serve as potential germplasm for genetic and genomic studies (Stalker and Simpson 1995; Kameswara Rao et al. 2003; Bravo et al. 2006).
During the past two decades, various types of molecular markers have been developed for diversity analysis of the genus Arachis, including restriction fragment length polymorphism (RFLP), random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), and inter simple sequence repeat (ISSR) markers (Halward et al. 1991; Kochert et al. 1991, 1996; Paik-Ro et al. 1992; Hilu and Stalker 1995; He and Prakash 1997; Galgaro et al. 1998; Subramanian et al. 2000; Raina et al. 2001; Dwivedi et al. 2001; Gimenes et al. 2002; Herselman 2003). In addition, microsatellite or simple sequence repeat (SSR) markers have been developed using sequences derived from SSR-enriched genomic libraries and expressed sequence tags (ESTs) (Hopkins et al. 1999; Palmieri et al. 2002, 2005; He et al. 2003, 2005; Ferguson et al. 2004; Moretzsohn et al. 2004, 2005; Mace et al. 2006; Gimenes et al. 2007; Proite et al. 2007; Cuc et al. 2008; Naito et al. 2008; Guo et al. 2009). SSR markers have the advantage over many other types of markers as they are reproducible, multi-allelic, co-dominant, analytically simple and readily transferable (Rafalski et al. 1996; He et al. 2003). To date, fewer than 1,500 SSR markers, including the markers generated for linkage map construction, have been published and used for genetic studies of the genus Arachis.
A major hurdle in the development of peanut molecular genetics has been narrow genetic diversity. SSR-based linkage maps of the genus Arachis were first established from crosses between diploid wild species and either the AA genome (A. duranensis and A. stenosperma) or the BB genome (A. ipaënsis and A. magna) (Moretzsohn et al. 2005, 2009). Subsequently, chromosome segment substitution lines between a cultivated peanut (used as a backcross line) and a synthetic amphidiploid between A. duranensis and A. ipaënsis (Foncéka et al. 2009) were used to develop a linkage map that placed 298 SSR loci into 21 linkage groups. More recently, an SSR-based genetic linkage map of cultivated peanut was constructed using 135 loci mapped into 22 linkage groups (Varshney et al. 2009a, b). This was followed by a composite genetic linkage map of three recombinant inbred line (RIL) populations containing 175 SSR loci mapped into 22 linkage groups (Hong et al. 2010). Although a number of studies have attempted to develop DNA markers and genetic linkage maps for peanut, the linkage groups in the latest maps have not yet converged with the number of chromosome pairs (20), which suggests that the current DNA marker resources remain insufficient for molecular genetics in peanut.
cDNA libraries are constructed using mRNA isolated from a particular developmental stage or tissue. ESTs are generated by large-scale sequencing of randomly-picked clones from these cDNA libraries. The main advantage of EST-derived SSR (EST-SSR) markers over genomic SSR markers is that they can be rapidly and cheaply developed using EST database mining. Since these sequences represent transcribed regions of the genome, they can lead to the development of gene-based maps, which help in identifying candidate functional genes and increase the efficiency of marker assisted selection (MAS) (Gupta and Rustgi 2004). In addition, it is likely that these markers would show greater conservation and have a higher rate of transferability to closely related species than genomic SSR markers (Scott et al. 2000; Saha et al. 2004). EST-SSR markers can also serve as anchor markers for comparative and evolutionary studies (Kantety et al. 2002; Varshney et al. 2005a). However, the conserved nature of EST-SSR markers may limit their polymorphism. Although earlier investigations have developed EST-SSR markers and performed diversity analyses on cultivated and wild species of peanut, they found low levels of informative markers and very limited polymorphism (Luo et al. 2005; Proite et al. 2007; Liang et al. 2009; Jinyan et al. 2009; Song et al. 2010).
A large-scale development of peanut EST-SSR markers was undertaken to increase the number of informative markers. Marker polymorphisms were investigated in wild and cultivated peanut accessions belonging to different botanical types. The resultant SSR markers include markers that show polymorphism across different peanut accessions. These markers will provide a valuable resource for future genetic mapping, quantitative trait loci (QTL) analysis, comparative genetic studies and MAS.
Materials and methods
Plant materials
A cDNA library was constructed from Arachis hypogaea L. cv. Chibahandachi, a Japanese peanut variety. Polymorphic SSR markers were used to perform analyses on 30 Arachis accessions comprising different botanical varieties and landraces of cultivated peanut, as well as several diploid species. The information on botanical types, geographical origin and biological status is provided in Table 1. Three pairs of lines, ICGV86031 and TAG 24, K7988 and V10309, and K30076 and K30097, have been reported as the parental lines for linkage maps of the cultivated peanut (AABB), the AA genome of Arachis, and the BB genome of Arachis, respectively (Varshney et al. 2009a, b; Moretzsohn et al. 2005, 2009). DNA for these parental accessions was kindly provided by Dr. R. K. Varshney from the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT) and Dr. D. J. Bertioli from the Universidade Católica de Brasília. Seeds of other accessions were obtained from a seed collection at the Chiba Prefectural Agriculture and Forestry Research Center in Japan.
Development of EST-SSR markers
Plant RNA Reagent (Invitrogen, USA) was used to extract total RNA from 5 g samples of gynophores, roots, leaves and seedlings of the peanut variety Chibahandachi. Purification of polyadenylated RNA and conversion to cDNA were performed as described previously (Asamizu et al. 1999). Synthesized cDNA was resolved by 1% agarose gel electrophoresis and the fraction ranging from 1 to 3 kb was recovered. The recovered fragments were cloned into the EcoRI-XhoI site of a pBluescript II SK- plasmid vector (Stratagene, California, USA), which was then electroporated into an E. coli ElectroTen-Blue strain (Stratagene). For the generation of ESTs, plasmid DNAs were amplified from colonies using TempliPhi (GE Healthcare, Little Chalfont, UK) and then subjected to sequencing using the BigDye Terminator® Cycle Sequencing Ready Reaction Kit (Applied Biosystems, California, USA). Reaction mixtures were run on an ABI PRISM 3730 automated DNA sequencer (Applied Biosystems).
Sequencing chromatograms were evaluated with phred (Ewing et al. 1998; Ewing and Green 1998) and vector-derived sequences were trimmed with Cross match (Ewing and Green 1998). The EST reads were quality-trimmed with the phred quality score at a position where five ambiguous bases (phred score under 16) were found within 15 contiguous bases. Reads that comprised >50 bp of contiguous sequence of satisfying quality were submitted to the DDBJ/EMBL/GenBank databases with the accession numbers FS960760 to FS988327. To identify non-redundant peanut ESTs, the MIRA 3.0.5 program was used for clustering at the normal setting for EST assembly (Chevreux et al. 2004). Similarity searches were performed for non-redundant peanut ESTs using the BLASTX program against deduced protein-encoding genes in the genomes of Arabidopsis thaliana (Arabidopsis Genome Initiative 2000), Lotus japonicus (Sato et al. 2008), Glycine max (Schmutz et al. 2010), and Medicago truncatula (release 3.0: http://www.medicago.org/genome/). EST contigs were classified into eukaryotic clusters of orthologous groups (KOGs) categories according to the results of BLASTX searches against amino acid sequences in the KOG data set (http://www.ncbi.nlm.nih.gov/COG/) (Tatusov et al. 2003). These sequence similarities were judged to be significant when the E-value was less than 1E−10.
Microsatellite or SSRs ≥ 15 nucleotides in length, which contained all possible combinations of the dinucleotide (NN), trinucleotide (NNN) and tetranucleotide (NNNN) repeat, were identified from non-redundant peanut ESTs using the SSRIT (Simple Sequence Repeat Identification Tool) program (Temnykh et al. 2001) for perfect SSRs and the fuzznuc tool from EMBOSS version 6.1.0 (Rice et al. 2000) for SSRs with mismatches. For amplification of SSR-containing regions, the Primer 3 program (Rozen and Skaletsky 2000) was used to design primer pairs against the sequences flanking each SSR, so that amplified fragments were between 90 and 300 bp in length. Redundancy of the newly designed markers to those previously reported was checked by exact match searches using the BLASTX program of the primer sequences of 1,478 published SSR markers in peanut with the EST sequences obtained in this study (Hopkins et al. 1999; He et al. 2003, 2005; Luo et al. 2005; Ferguson et al. 2004; Moretzsohn et al. 2004, 2005 and 2009; Gimenes et al. 2007; Proite et al. 2007; Cuc et al. 2008; Naito et al. 2008; Liang et al. 2009; Song et al. 2010). Design details for the peanut EST-SSR primers, along with the corresponding SSR motifs, product sizes and primer sequences, are available on the web at http://marker.kazusa.or.jp/peanut.
Amplification of SSR-containing regions and detection of polymorphisms
DNA was extracted from single seeds (100 mg) using the DNeasy Plant Mini Kit (Qiagen Inc., Valencia, CA, USA). DNA quantification and quality checks were performed using a Nanodrop spectrophotometer ND1000 (NanoDrop Technologies, Wilmington, DE, USA) and 0.8% agarose gel electrophoresis, respectively.
A total of 3,187 EST-SSR markers were prescreened on 24 accessions of Arachis spp. (Table 1). PCR reactions were performed in 5 μl reaction volumes using 0.6 ng of genomic DNA in 1 × PCR buffer (Bioline, London, UK), 3 mM MgCl2, 0.08 U of BIOTAQ DNA polymerase (Bioline), 0.8 mM dNTPs, and 0.4 μM of each primer. A modified touchdown PCR protocol was followed, as described previously by Sato et al. (2005). The PCR products were separated by electrophoresis using 10% polyacrylamide gels to check for amplification. Data were analyzed for markers with locus specificity and 1,571 EST-SSR markers that could amplify the single and double polymorphic loci were selected. Markers that showed multiple loci or faint amplification were excluded from further analysis.
The Fluoro-fragment Analyzer (ABI 3730, Applied Biosystems) was used for a second polymorphic analysis of the 16 Arachis accessions with the selected 1,571 EST-SSR markers (Table 1). Of the 16 accessions, 10 overlapped during prescreening, while the other six were used only for the second polymorphic analysis (Table 1). PCR reactions were performed in 5 μl reaction volumes, as described above, with the exception that 1.0 ng of genomic DNA was used as the template. PCR products were diluted 1:1 in dH2O and 3 μl of diluted PCR product were used in labeling reactions in which the fluorescent dye R6G was included in the labeling mixture. After ethanol precipitation, 7 μl of HiDi formamide (Applied Biosystems) and 0.02 μl of Genescan 500 LIZ Size Standard (Applied Biosystems) were added to the dried samples. After denaturation at 95°C for 5 min, the samples were fractionated in the Fluoro-fragment Analyzer using a 36-cm capillary array. After electrophoresis, allele sizing was performed using GeneMapper version 3.5 software (Applied Biosystems).
Determination of allele frequency and diversity analysis
Allelic data derived from the Fluoro-fragment Analyzer was used to calculate the polymorphism information content (PIC), a measure of allelic diversity at a given locus. The following equation was used to determine PIC (Botstein et al. 1980; Anderson et al. 1993):
where P ij is the frequency of the jth allele for the ith marker. Since the Arachis accessions were from a self-pollinated species, heterozygous alleles should not be commonly observed. Therefore, in cases where more than two peaks were observed in an accession, each peak was regarded as an individual locus. Loci for which no amplification was observed were considered null alleles. The overall PIC values and number of alleles are considered to be measures of genetic diversity. These estimates were performed separately among all the tested accessions and among cultivated peanut accessions.
The allelic data were converted into a binary matrix using the scores 1, 0 for presence and absence of the allele. The binary data were analyzed using NTSYSpc version 2.0 software (Rohlf 1998). Dice coefficients were used to derive genetic similarity (GS) among the genotypes followed by phenetic clustering using the SAHN computational module via the average linkage between groups fusion method i.e., UPGMA (unweighted pair group method with arithmetic average).
Results
Features of peanut ESTs
A total of 27,568 cDNA clones, including 5,724 clones from the gynophore library, 6,623 clones from the root library, 6,740 clones from the leaf library and 8,481 clones from the seedling library, were sequenced from their 5′ ends. A total of 17,046,017 qualified bases were obtained and the average GC content was 47.0%.
To identify the number of independent EST species, clustering of the EST sequences was performed using the MIRA 3.0.5 program. This resulted in the generation of 10,102 potential non-redundant EST sequences, including 3,445 contigs and 6,657 singletons. When these non-redundant EST sequences were searched for similarity against the proteome databases of three legume genomes (G. max, L. japonicus and M. truncatula) and the A. thaliana genome, 8,918 non-redundant ESTs showed significant similarity (E-value < 1E−10) to registered sequences, and the remaining 1,184 non-redundant ESTs were unique to peanut (Electronic Supplementary Material Table 1).
To investigate the functional classification of peanut ESTs, non-redundant EST sequences were compared with KOGs by BLASTX, and classified into KOG categories (Tatusov et al. 2003). Among the 10,102 non-redundant peanut EST sequences, 4,380 showed similarity to KOG sequences with functional classifications, and their distribution into KOG functional categories is shown in Fig. 1. To analyze the expressed gene features at the organ level, KOG classification was carried out against non-redundant EST sequences obtained from the reads of each cDNA library. As a result, the proportion of non-redundant ESTs classified into the metabolism category was slightly higher in the gynophore and root libraries (MOESM6).

Functional classification of non-redundant peanut EST sequences into eukaryotic clusters of orthologous group (KOG) categories. BLASTX was used to compare non-redundant EST sequences with the KOG sequence set. EST sequences were then classified into the KOG categories according to sequence similarity
To identify differentially-expressed genes, the EST abundance in each contig of non-redundant peanut ESTs was analyzed. Of the 3,445 contigs, 1,090 (32%) candidates for differentially expressed genes were derived from a single cDNA library (MOESM1 and MOESM2). Analysis of EST abundance within contigs also showed genes with high levels of expression in each organ (Supplemental Table 1). The most abundant EST was identified in the gynophore cDNA library (779 ESTs; 13.6%), within the cluster for contig0003, which shows similarity to a senescence-associated protein (Glyma13g12070.1). The most common EST in the root cDNA library was identified for contig0007, in which 112 ESTs (1.7%) showed similarity to a lectin protein kinase (Glyma02g01590.1). The most abundant EST in both the leaf (318 ESTs; 4.7%) and seedling (571 ESTs; 6.7%) cDNA libraries was identified in the cluster for contig0002, and was similar to a chlorophyll A-B binding protein (Glyma16g28070.1).
Microsatellite features and marker development
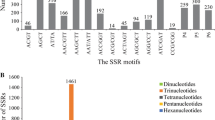
A total of 1,254 di-, tri- and tetranucleotide SSRs of 15 bp or longer were identified in the non-redundant EST sequences. Given that the total size of the non-redundant peanut EST sequences is 6.9 Mbp, the average frequency of SSRs in transcribed regions of the peanut genome can be estimated to be one SSR in every 5.5 kb. Di-, tri- and tetranucleotide SSRs accounted for 24.2, 66.8 and 8.9% of identified SSRs, respectively (Table 2). Among the 1,254 SSR regions assigned, qualified primer pairs could be designed against 382 SSR regions. These primer pairs were used for the validation of the EST-SSR markers. To increase the number of candidate EST-SSR markers, additional primer pairs were designed that allowed either single-base mismatches (662 primer pairs) or two-base mismatches (2,143 primer pairs) in the SSR regions (Table 2). A total of 3,187 EST-SSR primer pairs was therefore designed. In terms of the repeat motifs among the 3,187 markers, 2,540 (80%) were trinucleotide repeats (TNRs), whereas 302 (9%) and 345 (11%) were di- and tetranucleotide repeats, respectively. Four types of dinucleotide repeats were found. Poly(AG)n and poly(AT)n were the most frequently observed motifs, constituting 90% of all dinucleotide repeats (Table 2). Among the trinucleotide repeats, the poly(AAG)n motif was most abundant (626, 19.6%), followed by poly(ATC)n (310, 9.7%), poly(GGA)n (306, 9.6%), and poly(GGT)n (262, 8.2%). Poly(AAAG)n, poly(AAAT)n, and poly(AAAC)n were the most commonly observed tetranucleotide repeats and comprised 62% of these repeats.
Exact match searches of primer sequences of a total of 1,478 peanut SSR markers published previously with the EST sequences obtained here indicated that primer sequences of 18 reported markers showed exact matches to EST sequences for either one or both primers. Twelve of these were used for marker development in this study, as shown in MOESM3. The peanut EST-SSR primer sequences, along with the corresponding SSR motifs, product sizes, primer sequences and pictures of 10% polyacrylamide gel electrophoresis, are provided on the web at http://marker.kazusa.or.jp/peanut and in Supplemental Table 3.
Polymorphic analysis of the EST-SSR markers
The amplification status of 3,187 EST-SSR primer pairs was assessed for 24 Arachis accessions using 10% polyacrylamide gel electrophoresis. Amplified DNA fragments were observed with 2,773 markers, but 414 markers did not generate amplicons (Supplemental Table 3). The amplified markers were classified into single, double and multiple loci generators. When native polyacrylamide gels were used for electrophoresis, hetero-duplex bands were often observed, which made it difficult to distinguish between multiple and double loci. In cases where multiple bands of random size were generated, the markers were classified as multiple loci (e.g., AHS0003: see a picture of its electrophoresis on the web database). In situations where separate multiple bands were observed and these corresponded to expected and high molecular sizes, the markers were classified as double loci (e.g., AHS0007). Eventually, 1,927, 617 and 229 markers were classified into single, double and multiple loci generators, respectively. A total of 1,571 markers fulfilled the following conditions: (a) they generated single or double loci; (b) PCR amplicons were shorter than 500 bp; (c) polymorphisms were observed within the 24 Arachis accessions; and (d) weak or rare bands were not observed. These markers were then used for polymorphic analysis of the 16 Arachis accessions listed in Table 1.
Fluoro-fragment analysis indicated polymorphism in 81.5% (1,281/1,571) of the markers among all the tested lines (16 accessions), with 23.3% (366/1,571) in cultivated peanut (12 accessions), 44.5% (872/1,571) between the two diploid species with the AA genome, and 48.3% (759/1,571) between the two diploid species with the BB genome. The polymorphic markers, PIC, number of alleles, and sizes of PCR amplicons in each of the 16 accessions are listed in MOESM4.
With a total of 1,571 markers, 2,433 loci were identified in the set of 16 accessions and 2,366 were identified in the set of 12 tetraploid accessions. The number of alleles per locus ranged from one to six among the 16 accessions, with an average of 2.1 (Fig. 2a). The number of alleles per locus ranged from one to five among the 12 cultivated peanut accessions, with an average of 1.4. The SSR markers with the highest number of alleles per locus were AHS2568 (6 alleles/locus, in all tested accessions) and AHS0326 (5 alleles/locus, in the cultivated peanut accessions). Among the 16 accessions, PIC values ranged from 0.00 to 0.73 with an average of 0.19, whereas among the 12 cultivated peanuts accessions, they ranged from 0.00 to 0.72 with an average of 0.05 (Fig. 2b). In polymorphic markers, the mean PIC values were 0.23 and 0.19 for the 16 Arachis and 12 cultivated peanut accessions, respectively.

Distribution of the number of alleles per locus (a) and PIC (b) of 1571 EST-SSR markers investigated with a Fluoro-fragment Analyzer. Black and white bars indicate values for all 16 accessions and the 12 cultivated peanuts accessions, respectively
When considering the association between the number of polymorphic markers and SSR motifs, more markers possessed TNRs (1,243) than di- (137) or tetranucleotide (191) repeats (Table 2). The percentages of prescreened di-, tri-, and tetranucleotide repeat markers in all designed markers were 45.4, 48.9 and 55.3%, respectively. No significant differences were observed between the average PIC values of different SSR motifs in the markers. Among the 16 Arachis accessions, the PIC values of di-, tri-, and tetranucleotide SSR motifs were 0.22, 0.18 and 0.20, respectively, whereas among the 12 cultivated peanuts accessions, they were 0.05, 0.04 and 0.05, respectively. There were no clear correlations between the number of SSR motifs and PICs among the 16 Arachis accessions (R = 0.17) or the cultivated peanut accessions (R = 0.07; MOESM7).
Similarity coefficients were used to examine the genetic relationships between the 16 Arachis accessions. All possible genotypes showed similarity coefficients ranging from 0.37 to 0.97 (MOESM5). The lowest similarity coefficient of 0.37 was observed between a diploid accession with the BB genome (K30076) and two diploid accessions with the AA genome (V10309 and K7988). The highest similarity coefficient of 0.97 was found for two Japanese peanuts accessions, YI-0311 and Sachihomare.
From the UPGMA dendrogram, it is clear that there is one major cluster (Cluster1) that comprises all tetraploid and single BB genome diploid species of A. magna (K30097) (Fig. 3). In addition, the dendrogram suggests that diploid species containing the BB genome are genetically closer to cultivated peanuts than diploid species with the AA genome. In the cultivated peanut accessions, there was no clear clustering with respect to botanical type and agronomic status, i.e., varieties and landraces. As regards geographical origin, and with the exception of accession PI315608, the two cultivated peanut accessions derived from India were distinct from accessions derived from Japan and USA.

Dendrogram for 16 Arachis accessions based on cluster analysis of the 1,571 polymorphic EST-SSR markers. DICE similarity coefficient estimates of the UPGMA algorithmic module were used. Botanical types are indicated as colored squares around the accession name (purple, Hybrid; blue, Virginia; green, Spanish; light green, Valencia; red, diploids with an AA genome; and yellow, diploids with a BB genome). The origin of each accession is indicated with a letter following the accession name (J Japan, B Brazil, I India, U United States of America). Cluster1 represents a candidate cluster comprising cultivated peanuts and a diploid species with a BB genome
Discussion
In this study, a total of 1,254 SSR regions were identified in 10,102 potential non-redundant ESTs. Previously, Guo et al. (2009) reported the discovery of 856 SSR regions in 6888 non-redundant peanut ESTs, while Liang et al. (2009) identified 881 SSR regions in 11,431 non-redundant ESTs. In this study, the frequency of SSR identification was similar to these previous studies. The poly(AAG)n motif was the most abundant SSR pattern, followed by poly(AG)n, poly(AAT)n and poly(GGT)n. With respect to the number of nucleotide repeats, TNRs were more abundant than tetra- or dinucleotide repeats. Earlier reports have indicated that TNRs are generally the most common motif in both monocots and dicots (Eujayl et al. 2004; Varshney et al. 2005b). When there is variation in SSR length, the TNR abundance in ESTs can be attributed to maintaining an absence of frame shift mutations in the coding regions (Metzgar et al. 2000). In addition, selection pressure probably eliminates repeats that encode hydrophobic or basic amino acids, and TNR repeats corresponding to small hydrophilic amino acids are perhaps more easily tolerated (Katti et al. 2001). The frequency of SSR motifs was similar to the frequencies found for three other legume EST collections (G. max, L. japonicus and M. truncatula) (Jayashree et al. 2006). In the genic regions of the L. japonicus genome, (AAG)n and (GGT)n are enriched in coding regions, while (AG)n and (AAT)n are enriched in 5′ and 3′ untranslated regions (UTRs) (Sato et al. 2008). Therefore, the SSR motif frequency of peanut ESTs indicates that the peanut cDNA libraries were of sufficiently high quality to cover significant portions of 5′ UTRs.
In this study, a total of 3,187 primer pairs were designed as novel EST-SSR markers. The number of EST-SSR markers generated per non-redundant EST was 0.32, which was more than 7–12 times higher than in previous reports (0.042, for 290 markers/6888 ESTs by Guo et al. 2009; and 0.025, for 290 markers/11431ESTs by Liang et al. 2009). In previous studies, only perfect SSR regions were used for marker generation, whereas in this study, SSR regions containing one or two mismatches were also used for primer design. This strategy generated more SSR markers per unique EST.
A total of 1,571 EST-SSR markers were prescreened by 10% polyacrylamide gel electrophoresis and identified as polymorphic among the 24 Arachis accessions. Fluoro-fragment analysis was then used to confirm 1,281 markers as polymorphic among the 16 Arachis accessions. Changes to the Arachis accessions under investigation and to methods of polymorphism detection resulted in fewer polymorphic markers being used in the second analysis. The 1,281 confirmed polymorphic markers should be useful for genetic analyses of Arachis spp. Polymorphisms in the 12 cultivated peanut accessions were observed in 366 markers, which represent 11.5% (366/3,187) of the designed markers. This rate was higher than in some of the previous reports, which had polymorphism rates of 8.9% (Guo et al. 2009), 7.5% (Moretzsohn et al. 2005) and 10.3% (Liang et al. 2009), but less than that reported by Luo et al. (20%; 2005). Therefore, we concluded that the presence of one or two mismatches in SSR regions did not affect the quality of EST-SSR markers and provided the advantage of increasing the number of markers generated. In cultivated peanut, the ratio of polymorphic marker to designed marker was also higher than other marker systems such as RAPD (6.6%, Subramanian et al. 2000) or AFLP (6.7%; He and Prakash 1997), but was lower than for most genomic SSR markers, i.e., 44.2% (Cuc et al. 2008), 29.2% (He et al. 2005), 33.9% (He et al. 2003) and 57% (Ferguson et al. 2004). The lower levels of polymorphism in tetraploids may be attributed to their origins in highly-conserved portions of the genome (Varshney et al. 2005b).
In cultivated peanut, the low level of polymorphism in EST-SSR markers may be compensated by a higher potential for cross-species transferability to wild species. According to previous reports, the transferability of EST-SSRs from one species to another ranges from 40 to 89% (Gao et al. 2003; Saha et al. 2004; Eujayl et al. 2004; Yu et al. 2004). In this analysis of 1,571 pre-screened polymorphic markers, only three markers exhibited no amplification in the four diploid species (AHE0178, AHS1601 and AHS2313), which indicates a high level of transferability across Arachis spp.
In the 16 Arachis and cultivated peanut accessions, the mean PIC values for polymorphic markers were 0.23 and 0.19, respectively. These PIC values are lower than the values reported by Liang et al. (2009), which were 0.33 and 0.72 for cultivated peanut and wild Arachis species, respectively. They are also lower than the PIC values of EST-SSR markers reported for several self-crossing plant species, including soybean (0.40; Hisano et al. 2007) and barley (0.45; Thiel et al. 2003), and out-crossing plant species, such as sugarcane (0.23, Cordeiro et al. 2001; and 0.66, Pinto et al. 2004) and red clover (0.60; Sato et al. 2005). PIC values are generally higher in out-crossing species than in self-crossing species, but these values depend upon the accessions being tested. Cultivated peanut is considered to have low genetic diversity (Dwivedi et al. 2007) and the lower PIC values identified in this study appear to reflect this low diversity.
The diversity analysis indicated greater genetic diversity between diploid species with AA and BB genomes than between diploid species and cultivated peanut. Since the ancestors of the cultivated peanut are considered to be A. duranensis and A. ipaënsis (Halward et al. 1991; Krapovickas and Gregory 1994; Kochert et al. 1996; Seijo et al. 2004, 2007), this result appears to be reasonable. The UPGMA dendrogram suggested that diploid species with the BB genome were genetically closer to cultivated peanut than those with the AA genome. In Fluoro-fragment analysis, a larger number of null alleles were observed in the AA genome accessions (805/2,433, A. duranensis; 807, A. stenosperma) than BB genome accessions (191, A. ipaënsis; 715, A. ipaënsis, MOESM4). We considered that the result suggested two possibilities: (a) the genetic distance between cultivated peanut and the BB genome accessions was closer to that of cultivated peanut and the AA genome accessions or (b) gene expression in peanut was biased towards the B genome, such that a greater number of B-genome-specific EST-SSR markers were designed. Interestingly, the genetic distance between cultivated peanut and A. magna (K30097) was shorter than between cultivated peanut and A. ipaënsis (K30076), which was considered to be the foremost candidate for the B genome donor. In the 12 cultivated peanut accessions, there was no clear clustering with respect to botanical types and agronomic status, although slight genetic differences were observed with geographical origin, i.e., between two Indian accessions and other peanut accessions, with the exception of one landrace from the United States (PI315608). The two distinct Indian accessions, ICGV86031 and TAG24, were reported as F2 mapping parents of a linkage map that contained a total of 135 SSR marker loci. This result suggests that, in the future, it may be possible to generate a denser linkage map for cultivated peanut using more genetically diverse mapping populations from cultivated peanut.
It is difficult for conventional breeding strategies to select many agronomic traits in peanut, and marker-assisted selection offers an additional tool for genetic improvement of cultivars. Comparative mapping across legume species, including diploid Arachis species, should advance in the near future, especially with the availability of second and third generation sequencers and the development of additional ESTs from different tissues and genotypes. The EST-SSR markers identified in this study provide a valuable public genomic resource for the future development of functional and comparative genomics and MAS in peanut.
References
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME (1993) Optimizing parental selection for genetic linkage maps. Genome 36:181–186
Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408(6814):796–815
Asamizu E, Nakamura Y, Sato S, Fukuzawa H, Tabata S (1999) A large scale structural analysis of cDNAs in a unicellular green alga, Chlamydomonas reinhardtii. I. Generation of 3433 non-redundant expressed sequence tags. DNA Res 6:369–373
Botstein D, White RL, Skolnick M, Davis RE (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Gen 32:314–331
Bravo JP, Hoshino AA, Angelici CMLCD, Lopes CR, Gimenes MA (2006) Transferability and use of microsatellite markers for the genetic analysis of the germplasm of some Arachis section species of the genus Arachis. Gen Mol Biol 29:516–524
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WE, Wetter T, Suhai S (2004) Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14:1147–1159
Cordeiro GM, Casu R, McIntyre CL, Manners JM, Henry RJ (2001) Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci 160(6):1115–1123
Cuc LM, Mace ES, Crouch JH, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8:55
Dwivedi SL, Gurtu S, Chandra S, Yuejin W, Nigam SN (2001) Assessment of genetic diversity among selected groundnut germplasm I: RAPD analysis. Plant Breed 120:345–349
Dwivedi SL, Bertioli DJ, Crouch JH, Valls JF, Upadhyaya HD, Fávero F, Moretzohn M, Paterson AH (2007) Peanut. In: Kole C (ed) Genome mapping and molecular breeding in plants, vol 2 oilseeds. Springer, Berlin, pp 115–151
Eujayl I, Sledge MK, Wang L, May GD, Chekhovskiy K, Zwonitzer JC, Mian MA (2004) Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp. Theor Appl Genet 108:414–422
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
FAOSTAT (2006) FAO production yearbook 60. FAOSTAT, Rome, Italy
Ferguson ME, Burow MD, Schulze SR, Bramel PJ, Paterson AH, Kresovich S, Mitchell S (2004) Microsatellite identification and characterization of peanut (A. hypogaea L.). Theor Appl Genet 108:1064–1070
Foncéka D, Hodo-Abalo T, Rivallan R, Faye I, Sall MN, Ndoye O, Fávero AP, Bertioli DJ, Glaszmann JC, Courtois B, Rami JF (2009) Genetic mapping of wild introgressions into cultivated peanut: a way toward enlarging the genetic basis of a recent allotetraploid. BMC Plant Biol 9:103
Galgaro L, Lopes CR, Gimenes M, Valls JFM, Kochert G (1998) Genetic variation between several species of sections Extranervosae, Caulorhizae, Heteranthae and Triseminatae (genus Arachis) estimated by DNA polymorphism. Genome 41:445–454
Gao LF, Tang JF, Li HW, Jia JZ (2003) Analysis of microsatellites in major crops assessed by computational and experimental approaches. Mol Breed 12:245–261
Gimenes MA, Lopes CR, Valls JFM (2002) Genetic relationships among Arachis species based on AFLP. Genet Mol Biol 25:349–353
Gimenes MA, Hoshino AA, Barbosa AV, Palmieri DA, Lopes CR (2007) Characterization and transferability of microsatellite markers of the cultivated peanut (Arachis hypogaea L.). BMC Plant Biol 7:9
Guo B, Chen X, Hong Y, Liang X, Dang P, Brenneman T, Holbrook C, Culbreath A (2009) Analysis of gene expression profiles in leaf tissues of cultivated peanuts and development of EST- SSR markers and gene discovery. Int J Plant Genomics 2009:715605
Gupta PK, Rustgi S (2004) Molecular markers from the transcribed/expressed region of the genome in higher plants. Funct Integr Genomics 4:139–162
Halward TM, Stalker HT, Larue EA, Kochert G (1991) Genetic variation detectable with molecular markers among unadapted germplasm resources of cultivated peanut and related wild species. Genome 34:1013–1020
He GH, Prakash CS (1997) Identification of polymorphic DNA markers in cultivated peanut (Arachis hypogaea L.). Euphytica 97:143–149
He G, Meng RH, Newman M, Gao GQ, Pittman RN, Prakash CS (2003) Microsatellites as DNA markers in cultivated peanut (Arachis hypogaea L.). BMC Plant Biol 3:3
He G, Meng RH, Gao H, Guo B, Gao G, Newman M, Pittman RN, Prakash CS (2005) Simple sequence repeat markers for botanical varieties of cultivated peanut (Arachis hypogaea L.). Euphytica 142:131–136
Herselman L (2003) Genetic variation among Southern African cultivated peanut (Arachis hypogaea L.) genotypes as revealed by AFLP analysis. Euphytica 133:319–327
Hilu KW, Stalker HT (1995) Genetic relationships between peanut and wild species of Arachis sect Arachis (Fabaceae): evidence from RAPDs. Plant Syst Evol 198:167–178
Hisano H, Sato S, Isobe S, Sasamoto S, Wada T, Matsuno A, Fujishiro T, Yamada M, Nakayama S, Nakamura Y, Watanabe S, Harada K, Tabata S (2007) Characterization of the soybean genome using EST-derived microsatellite markers. DNA Res 14:271–281
Hong Y, Chen X, Liang X, Liu H, Zhou G, Li S, Wen S, Holbrook CC, Guo B (2010) A SSR-based composite genetic linkage map for the cultivated peanut (Arachis hypogaea L.) genome. BMC Plant Biol 10:17
Hopkins MS, Casa AM, Wang T, Mitchell SE, Dean RE, Kochert GD, Kresovich S (1999) Discovery and characterization of polymorphic simple sequence repeats (SSRs) in peanut. Crop Sci 39:1243–1247
Jayashree B, Punna R, Prasad P, Bantte K, Hash CT, Chandra S, Hoisington DA, Varshney RK (2006) A database of simple sequence repeats from cereal and legume expressed sequence tags mined in silico: survey and evaluation. In Silico Biol 6:607–620
Jinyan W, Lijuan P, Quingli Y, Shanlin Y (2009) Development and characterization of EST–SSR markers from NCBI and cDNA library in cultivated peanut (Arachis hypogaea L.). Mol Plant Breed 7:806–810
Kameswara Rao N, Reddy LJ, Bramel PJ (2003) Potential of wild species for genetic enhancement of some semi-arid food crops. Gen Res Crop Evol 50:707–721
Kantety RV, La Rota M, Matthews DE, Sorrells ME (2002) Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol 48:501–510
Katti MV, Ranjekar PK, Gupta V (2001) Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol Biol Evol 18:1161–1167
Kerridge PC, Hardy B (eds) (1994) Biology and economy of forage Arachis. CIAT, Cali, Colombia. CIAT Publication No. 240
Kochert G, Halward T, Branch WD, Simpson CE (1991) RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor Appl Genet 81:565–570
Kochert G, Stalker HT, Gimenes M, Galgaro L, Lopes CR, Moore K (1996) RFLP and cytogenetic evidence of the origin and evolution of allotetraploid domesticated peanut, Arachis hypogaea (Leguminosae). Am J Bot 83:1282–1291
Krapovickas A, Gregory WC (1994) Taxonomía del género Arachis (Leguminosae). Bonplandia 8:1–186
Liang X, Chen X, Hong Y, Liu H, Zhou G, Li S, Guo B (2009) Utility of EST-derived SSR in cultivated peanut (Arachis hypogaea L.) and Arachis wild species. BMC Plant Biol 9:35
Luo M, Dang P, Guo BZ, He G, Holbrook CC, Bausher MG, Lee RD (2005) Generation of expressed sequence tag (ESTs) for gene discovery and marker development in cultivated peanut. Crop Sci 45:346–353
Mace ES, Phong DT, Upadhyaya HD, Chandra S, Crouch JH (2006) SSR analysis of cultivated groundnut (Arachis hypogaea L.) germplasm resistant to rust and late leaf spot diseases. Euphytica 152:317–330
Metzgar D, Bytof J, Wills C (2000) Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res 10:72–80
Moretzsohn Mde C, Hopkins MS, Mitchell SE, Kresovich S, Valls JF, Ferreira ME (2004) Genetic diversity of peanut (Arachis hypogaea L.) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Plant Biol 4:11
Moretzsohn MC, Leoi L, Proite K, Guimaraes PM, Leal-Bertioli SC, Gimenes MA, Martins WS, Valls JF, Grattapaglia D, Bertioli DJ (2005) A microsatellite-based, gene-rich linkage map for the AA genome of Arachis (Fabaceae). Theor Appl Genet 111:1060–1071
Moretzsohn MC, Barbosa AVG, Alves-Freitas DMT, Teixeira C, Bertioli SCM M, Guimarães PM, Pereira RW, Lopes CR, Cavallari MM, Valls JFM, Bertioli DJ, Gimenes MA (2009) A linkage map for the B-genome of Arachis (Fabaceae) and its synteny to the A-genome. BMC Plant Biol 9:40
Naito Y, Suzuki S, Iwata Y, Kuboyama T (2008) Genetic diversity and relationship analysis of peanut germplasm using SSR markers. Breed Sci 58:293–300
Paik-Ro OG, Smith RL, Knauft DA (1992) Restriction fragment length polymorphism evaluation of six peanut species within the Arachis section. Theor Appl Genet 84:201–208
Palmieri DA, Hoshino AA, Bravo JP, Lopes CR, Gimenes MA (2002) Isolation and characterization of microsatellite loci from the forage species Arachis pintoi (Genus Arachis). Mol Ecol Notes 2:551–553
Palmieri DA, Bechara MD, Curi RA, Gimenes MA, Lopes CR (2005) Novel polymorphic microsatellite markers in section Caulorrhizae (Arachis, Fabaceae). Mol Ecol Notes 5:77–79
Pinto LR, Oliveira KM, Ulian EC, Garcia AA, de Souza AP (2004) Survey in the sugarcane expressed sequence tag database (SUCEST) for simple sequence repeats. Genome 47(5):795–804
Proite K, Leal-Bertioli SC, Bertioli DJ, Moretzsohn MC, da Silva FR, Martins NF, Guimaraes PM (2007) ESTs from a wild Arachis species for gene discovery and marker development. BMC Plant Biol 7:7
Rafalski JA, Vogel IM et al. (1996) Generating and using DNA markers in plants. In: Non mammalian, genic analysis: a practical guide. Academic, San Diego. ISBN: 0121012859, pp 75–135
Raina SN, Mukai Y (1999) Detection of a variable number of 18S–5.8S–26S and 5S ribosomal DNA loci by fluorescent in situ hybridization in diploid and tetraploid Arachis species. Genome 42:52–59
Raina SN, Rani V, Kojima T, Ogihara Y, Singh KP, Devarumath RM (2001) RAPD and ISSR fingerprints as useful genetic markers for analysis of genetic diversity, varietal identification, and phylogenetic relationships in peanut (Arachis hypogaea) cultivars and wild species. Genome 44:763–772
Ramsey J, Schemske DW (1998) Pathways, mechanisms and rates of polyploid formation in flowering plants. Annu Rev Ecol Syst 29:467–501
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277
Rohlf FJ (1998) NTSYSpc. Numerical taxonomy and multivariate analysis system. Applied Biostatistics, Inc., New York
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386
Saha MC, Mian MA, Eujayl I, Zwonitzer JC, Wang L, May GD (2004) Tall fescue EST-SSR markers with transferability across several grass species. Theor Appl Genet 109:783–791
Sato S, Isobe S, Asamizu E, Ohmido N, Kataoka R, Nakamura Y, Kaneko T, Sakurai N, Okumura K, Klimenko I, Sasamoto S, Wada T, Watanabe A, Kohara M, Fujishiro T, Tabata S (2005) Comprehensive structural analysis of the genome of red clover (Trifolium pretense L.). DNA Res 12:301–364
Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, Sasamoto S, Watanabe A, Ono A, Kawashima K, Fujishiro T, Katoh M, Kohara M, Kishida Y, Minami C, Nakayama S, Nakazaki N, Shimizu Y, Shinpo S, Takahashi C, Wada T, Yamada M, Ohmido N, Hayashi M, Fukui K, Baba T, Nakamichi T, Mori H, Tabata S (2008) Genome structure of the legume, Lotus japonicus. DNA Res 15:227–239
Savage GP, Keenen JL (1994) The composition and nutritive value of groundnut kernels. In: Smart J (ed) The groundnut crop: a scientific basis of improvement. Chapman and Hall, London, pp 173–213
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA (2010) Genome sequence of the palaeopolyploid soybean. Nature 14:178–183
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ (2000) Analysis of SSRs derived from grape ESTs. Theor Appl Genet 100:723–726
Seijo JG, Lavia GI, Fernandez A, Krapovickas A, Ducasse D, Moscone EA (2004) Physical mapping of the 5S and 18S–25S rRNA genes by FISH as evidence that Arachis duranensis and A. ipaënsis are the wild diploid progenitors of A. hypogaea (Leguminosae). Am J Bot 91:1294–1303
Seijo G, Lavia GI, Fernandez A, Krapovickas A, Ducasse DA, Bertioli DJ, Moscone EA (2007) Genomic relationships between the cultivated peanut (Arachis hypogaea, Leguminosae) and its close relatives revealed by double GISH. Am J Bot 94:1963–1971
Soltis PS, Soltis DE (2000) The role of genetic and genomic attributes in the success of polyploids. Proc Natl Acad Sci USA 97:7051–7057
Song GQ, Li MJ, Xiao H, Wang XJ, Tang RH, Xia H, Zhao CZ, Bi YP (2010) EST sequencing and SSR marker development from cultivated peanut (Arachis hypogaea L.) Elec J Biotech 13:1–9
Stalker HT (1997) Peanut (Arachis hypogaea L.). Field Crops Res 53:205–217
Stalker HT, Simpson CE (1995) Germplasm resources in Arachis. In: Pattee HE, Stalker HT (eds) Advances in peanut science. American Peanut Research and Education Society, Inc., Stillwater, pp 14–53
Subramanian V, Gurtu S, Nageswara Rao RC, Nigam SN (2000) Identification of DNA polymorphism in cultivated groundnut using random amplified polymorphic DNA (RAPD) assay. Genome 43:656–660
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA (2003) The COG database: an updated version includes eukaryotes. BMC Bioinform 4:41–54
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Varshney RK, Graner A, Sorrells ME (2005a) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23:48–55
Varshney RK, Sigmund R, Börner A, Korzun V, Stein N, Sorrells ME, Langridge P, Graner A (2005b) Interspecific transferability and comparative mapping of barley EST-SSR markers in wheat, rye and rice. Plant Sci 168:195–202
Varshney RK, Bertioli DJ, Moretzsohn MC, Vadez V, Krishnamurthy L, Aruna R, Nigam SN, Moss BJ, Seetha K, Ravi K, He G, Knapp SJ, Hoisington DA (2009a) The first SSR-based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor Appl Genet 118:729–739
Varshney RK, Mahendar T, Aruna R, Nigam SN, Neelima K, Vadez V (2009b) High level of natural variation in groundnut (Arachis hypogaea L.) germplasm collection assayed by selected informative SSR markers. Plant Breed 128:486–494
Yu JK, La Rota M, Kantety RV, Sorrells ME (2004) EST derived SSR markers for comparative mapping in wheat and rice. Mol Genet Genomics 271:742–751
Acknowledgments
We thank Dr. David J Bertioli of the Universidade Católica de Brasília and Dr. K. Rajeev Varshney at ICRISAT for providing DNA from different Arachis species. This work was supported by the Kazusa DNA Research Institute Foundation, a JSPS (Japanese Society for the Promotion of Science) Grant in Aid for Scientific Research B (ID: 20380008), and a postdoctoral fellowship for foreign researchers (ID: 08617).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11032_2011_9604_MOESM6_ESM.pdf
Comparison of the functional classification of non-redundant peanut EST sequences from four cDNA libraries. Non-redundant peanut EST sequences obtained from gynophore, root, leaf and seedling cDNA libraries were compared with the KOG sequence set using BLASTX and then classified into the KOG categories of most similar sequence. Columns represent the proportion of non-redundant ESTs assigned to the KOG classification. Supplementary material 6 (PDF 17 kb)
11032_2011_9604_MOESM7_ESM.pdf
Correlation between number of SSR repeats and PIC. Red and blue dots represent PICs investigated among all 16 Arachis accessions and 12 cultivated peanut accessions, respectively. Supplementary material 7 (PDF 842 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Koilkonda, P., Sato, S., Tabata, S. et al. Large-scale development of expressed sequence tag-derived simple sequence repeat markers and diversity analysis in Arachis spp.. Mol Breeding 30, 125–138 (2012). https://doi.org/10.1007/s11032-011-9604-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-011-9604-8