
Abstract
Late leaf spot (LLS) and rust have the greatest impact on yield losses worldwide in groundnut (Arachis hypogaea L.). With the objective of identifying tightly linked markers to these diseases, a total of 3,097 simple sequence repeats (SSRs) were screened on the parents of two recombinant inbred line (RIL) populations, namely TAG 24 × GPBD 4 (RIL-4) and TG 26 × GPBD 4 (RIL-5), and segregation data were obtained for 209 marker loci for each of the mapping populations. Linkage map analysis of the 209 loci resulted in the mapping of 188 and 181 loci in RIL-4 and RIL-5 respectively. Using 143 markers common to the two maps, a consensus map with 225 SSR loci and total map distance of 1,152.9 cM was developed. Comprehensive quantitative trait locus (QTL) analysis detected a total of 28 QTL for LLS and 15 QTL for rust. A major QTL for LLS, namely QTLLLS01 (GM1573/GM1009-pPGPseq8D09), with 10.27–62.34% phenotypic variance explained (PVE) was detected in all the six environments in the RIL-4 population. In the case of rust resistance, in addition to marker IPAHM103 identified earlier, four new markers (GM2009, GM1536, GM2301 and GM2079) showed significant association with the major QTL (82.96% PVE). Localization of 42 QTL for LLS and rust on the consensus map identified two candidate genomic regions conferring resistance to LLS and rust. One region present on linkage group AhXV contained three QTL each for LLS (up to 67.98% PVE) and rust (up to 82.96% PVE). The second candidate genomic region contained the major QTL with up to 62.34% PVE for LLS. Molecular markers associated with the major QTL for resistance to LLS and rust can be deployed in molecular breeding for developing groundnut varieties with enhanced resistance to foliar diseases.
Similar content being viewed by others

Introduction
Groundnut or peanut (Arachis hypogaea L.) is an important oilseed crop and also a major source of vegetable oil (48%) and protein (25%) in tropical and subtropical regions of the World. It is grown in more than 100 countries of Asia, Africa and America with a global production of 35.52 Mt from an area of 23.50 Mha (FAO 2009). Although India is a leading producer of groundnut, the productivity is very low (about 0.92 t ha−1) compared to the other major producers such as China (3.31 t ha−1), which can be attributed to the damage caused by biotic and abiotic stresses. Among the biotic stresses, two fungal diseases, viz. late leaf spot (LLS) caused by Cercosporidium personatum and rust caused by Puccinia arachidis, are widespread in most of the tropical countries and severely affect productivity. Moreover, co-occurrence of the two diseases leads to yield loss of up to 50–70% (Subrahmanyam et al. 1984) along with adverse effects on the quality of the produce. Despite the fact that several fungicides are available to control these diseases, host-plant resistance is considered to be the best approach to managing these diseases and overcoming the hazardous effect of fungicides. Classical genetic studies on LLS and rust resistance suggest that resistance to these fungal diseases is complex and polygenic in nature and most likely controlled by recessive genes (Nevill 1982; Green and Wynne 1986; Dwivedi et al. 2002).
Although in the past elite cultivars or varieties resistant to LLS and/or rust have been developed worldwide through conventional breeding, co-occurrence of these two diseases and the defoliating nature of LLS pose serious challenges to the breeding community in phenotypic selection. Recent years have witnessed momentous advances in the area of crop genomics and its integration with conventional breeding (Varshney et al. 2005). As a result, several studies, especially in the temperate cereals, have demonstrated the efficacy of molecular markers and marker-assisted selection (MAS) in enhancing the effectiveness of conventional breeding methods, particularly in the case of low-heritability traits, where phenotypic selection was complicated (Varshney et al. 2006). Among different types of marker systems, microsatellite or simple sequence repeat (SSR) markers are considered to be the markers of choice for application in breeding programs (Gupta and Varshney 2000). In the case of groundnut, a large number of SSR markers have become available as a result of the concerted efforts of several research groups worldwide (e.g., Hopkins et al. 1999; He et al. 2003; Ferguson et al. 2004; Moretzsohn et al. 2005; Mace et al. 2007; Cuc et al. 2008; Liang et al. 2009; Yuan et al. 2010; Pandey et al. 2011). Although these SSR markers have been used for construction of genetic maps and trait mapping in the cultivated groundnut, the number of marker loci integrated into a single genetic map is not very large (Varshney et al. 2009a; Khedikar et al. 2010; Sarvamangala et al. 2011). To date, the densest genetic map based on a single mapping population contains 191 marker loci (Ravi et al. 2011) and the composite map based on three mapping populations has just 175 marker loci (Hong et al. 2010).
In terms of mapping resistance to LLS and rust, so far only one study has been conducted in cultivated groundnut, based on a partial genetic map comprising 56 SSR loci for the TAG 24 × GPBD 4 recombinant inbred line (RIL) population (RIL-4) (Khedikar et al. 2010). This study reported a major quantitative trait locus (QTL) for rust and only minor QTL for LLS. This may be attributed to the low marker density on the genetic map used for QTL analysis and/or use of the single mapping population. It was therefore imperative to saturate the genetic map as well as to use new mapping populations. Development of genetic maps for new populations offer the possibility of developing the consensus map based on common markers mapped on genetic maps for different mapping populations (Ablett et al. 2003; Karakousis et al. 2003). In this context, one further RIL population (TG 26 × GPBD 4, referred as RIL-5) with a partial genetic map of 45 SSR loci (Sarvamangala et al. 2011) is available that segregates for resistance to LLS and rust.
With the objective of identifying major QTL for LLS and more closely linked markers for the major QTL for rust, the present study reports: (i) saturation of the genetic maps for TAG 24 × GPBD 4 (RIL-4) and TG 26 × GPBD 4 (RIL-5) populations, (ii) phenotyping of the RIL-4 population for LLS and rust under three additional environments and comprehensive QTL analysis based on a total of six environments, (iii) phenotyping of the RIL-5 population for LLS under six environments and for rust under seven environments and comprehensive QTL analysis, and (iv) construction of a consensus QTL map for LLS and rust.
Materials and methods
Mapping populations
Two RIL mapping populations of F6:7 generations were developed at the University of Agricultural Sciences, Dharwad, India, from the crosses TAG 24 × GPBD 4 (RIL-4; 266 RILs) and TG 26 × GPBD 4 (RIL-5, 146 RILs) using the single-seed descent (SSD) method. Both the susceptible parents (TAG 24 and TG 26) of these RIL populations were early maturing with high harvest index, better partitioning coefficient and tolerance to bud necrosis but were highly susceptible to LLS and rust. GPBD 4 is a highly resistant variety to LLS and rust and is used as a national resistance check for both diseases in field trials of the All India Coordinated Research Project (AICRP) on groundnut. This variety was derived from the cross KRG 1 × CS 16 (ICGV 86855) and is a second-cycle derivative of inter-specific hybridization with a desirable combination of early maturity, high yield, high pod growth rate, desirable pod and kernel features with high oil content.
Phenotyping for late leaf spot and rust
Phenotyping of both mapping populations was done during 2004 (F7), 2005 (F8), 2006 (F9), 2007 (F10), 2008 (F11), 2009 (F12) and 2010 (F13). The RIL-4 population, however, was not phenotyped for rust resistance in F7 and F8 generations and RIL-5 was not phenotyped in F9 generation for any disease. These RIL populations were sown in a randomized block design (RBD) with two replications at Dharwad. Seeds of these RILs were treated with seed protectant before sowing. Twenty-five seeds from each RIL were sown in 2.5 m rows with 45 and 10 cm inter- and intra-row spacing, respectively. The parental genotypes (TAG 24, TG 26 and GPBD 4) of both mapping populations were also sown after every 50 rows as controls. Artificial disease epiphytotics were created for both the diseases using the “spreader row technique”. Spreader rows of TMV 2 (national susceptibility check for both diseases) were sown at every tenth row as well as in a border around the field to maintain the effective inoculum load. The inoculums for LLS and rust were produced and maintained separately on VL-1 (resistant to rust but a highly susceptible variety to LLS) and M-110 (resistant to LLS but highly susceptible to rust) genotypes, respectively. The LLS conidia and rust urediniospores were isolated by soaking and rubbing infected leaves in water for 30 min and were used for inoculation. After 35 days of sowing, plants were inoculated uniformly in the evening with LLS or rust for a week as detailed in Khedikar et al. (2010). Disease scoring for LLS was done at 70 days (LLS-SI) and 90 days (LLS-SII) after sowing, and for rust at 80 days (Rust-SI) and 90 days (Rust-SII) after sowing in different seasons by using a modified 9-point scale (Subbarao et al. 1990). National susceptible (TMV 2) and resistant (GPBD 4) checks were used for comparing the disease reaction of RILs of the both populations.
Analysis of variance (ANOVA) at different stages and environments of disease scoring for LLS and rust was performed to test the significance of differences between RILs. Pooled analysis of variance was done for both diseases, considering the number of environments, replications and treatments (RIL) separately for two different stages. To assess and quantify the genetic variability among the RILs, phenotypic coefficient of variance, genotypic coefficient of variance (GCV) and heritability in the broad sense (h 2b.s) were estimated. The correlation coefficient (r) among the different stages and environments of LLS or rust was estimated. All above-mentioned statistical analyses were done with the software package Windostat ver. 8.5 (Indostat Services, Hyderabad, India, http://members.fortunecity.com/indostat/).
DNA extraction and SSR genotyping
DNA was extracted from fresh leaves of the parental genotypes and RILs of F7 and F8 generation in the case of RIL-4 and F10 generation in the case of the RIL-5 population using the modified cetyltrimethylammonium bromide (CTAB) extraction method, as described in Cuc et al. (2008). The DNA quality and quantity were checked on 0.8% agarose gels and DNA concentration was normalized to ~5 ng/μl for further genotyping work.
In addition to the markers reported in Khedikar et al. (2010) and Sarvamangala et al. (2011), a set of novel expressed sequence tag (EST)-SSR markers (2,098) developed at the University of Georgia (S. J. Knapp, unpublished) and genomic SSR markers (123) reported by Wang et al. (2007a) were used to screen parental genotypes of the respective mapping populations. Subsequently, polymorphic markers were identified and used to genotype the RILs of the respective populations. PCR and separation of amplicons for the SSR markers were done as described in Varshney et al. (2009a).
Construction of genetic maps
Genotyping data obtained for all the polymorphic marker loci on both the mapping populations (RIL-4 and RIL-5) were used for linkage map construction using Mapmaker/EXP v.3.0 (Lander et al. 1987; Lincoln et al. 1992). A minimum LOD score of 5.0 and maximum recombination fraction (∂) of 0.5 were set as threshold values for linkage group determination. Linkage groups were defined with the “Sequence All” command. The most likely order within each linkage group was estimated by using three-point analyses (“Three Point” command). Marker orders were confirmed by comparing the log likelihood of the possible orders using multipoint analysis (“Compare” command) and by permuting all the adjacent triple orders (“Ripple” command). In the second step, the LOD score was set to 3.0 in order to include new markers in the linkage groups. The “Try” and “Build” commands were used to determine the exact position and linkage group of the new marker orders. The new marker orders were again confirmed with the “Compare” and “Ripple” commands. Finally the best possible order in each linkage map was used for map construction. The recombination fraction was converted into map distances in centimorgans (cM) using the Kosambi mapping function (Kosambi 1944). The inter-marker distances calculated from Mapmaker were used to construct linkage map using MapChart version 2.2 (Voorrips 2006).
Quantitative trait loci (QTL) analysis
For identification of QTL, the composite interval mapping (CIM) approach (Zeng 1994) was employed using WinQTL Cartographer, version 2.5 (Wang et al. 2007b). CIM was performed using Model 6, scanning intervals of 2.0 cM between markers and putative QTL with a window size of 10.0 cM. The number of marker cofactors for the background control was set by forward–backward stepwise regression. Automatically “Locate QTLs” option was used with a minimum of 5 cM between QTL to define a QTL region and, if the peak’s distance was less than 5.0 cM, then the highest peak was considered to locate QTL. Permutations for 500 times were also done while determining the QTL using WinQTL Cartographer using the option “permutations times” with 0.05 significance level. Single marker analysis (SMA) was also performed using WinQTL Cartographer to confirm candidate SSR markers linked to the trait.
Development of a consensus map
A consensus map was developed with JoinMap 4.0 (Van Ooijen 2006) using two individual linkage maps constructed based on the RIL-4 and RIL-5 mapping populations. The “Locus Genotype Frequency” function was used to calculate Chi-squared (χ2) values for each marker to test for the expected 1:1 segregation ratio. Markers were placed into linkage groups with the “LOD Groupings” and “Create Groups for Mapping” command using the Kosambi mapping function (Kosambi 1944). Calculation parameters were set for a minimum LOD of 5 and recombination fraction of 0.45. Marker order in groups was established using the “Calculate Map” command. Linkage groups with common markers on individual maps were merged to create a composite map using the “Join-combine Groups for Map Integration” command and the order of the markers was fixed using the marker order from the framework map obtained from Mapmaker. Further marker order in groups was established using the “Calculate Map” command. Linkage maps were drawn using MapChart for Windows (Voorrips 2006).
Results
Phenotyping of mapping populations
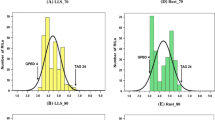
Two mapping populations, namely RIL-4 and RIL-5, have been phenotyped extensively for LLS and rust (Electronic Supplementary Material 1). For LLS, phenotyping was carried out in six environments/years during the rainy season for both RIL-4 (2004, 2005, 2006, 2008, 2009 and 2010) and RIL-5 (2004, 2005, 2007, 2008, 2009 and 2010) populations, whereas for rust, phenotyping of the RIL-4 population was conducted in six environments/years during the rainy seasons of 2006, 2007 (early sowing referred as E1 and late sowing referred as E2), 2008, 2009 and 2010. In order to estimate components of resistance to rust, viz. incubation period (IP), latent period (LP) and infection type (IT), an experiment was undertaken during 2008 under greenhouse conditions. Similarly phenotyping data for rust was generated across seven environments/years in the RIL-5 population during the rainy seasons of 2004, 2005, 2007 (E1 and E2), 2008, 2009 and 2010. However, part of these phenotyping data (e.g. LLS for three seasons, namely 2004, 2005 and 2006, rust for four seasons, namely 2006, 2007-E1, 2007-E2, 2008, and components of resistance, as mentioned above, during 2008) on RIL-4 was reported in Khedikar et al. (2010). Nevertheless, the ESM 1 summarises the entire dataset for both populations. The detailed analysis of phenotypic data showed lower disease incidence in GPBD 4 (resistant parent of RIL-4 and RIL-5) compared to TAG 24 (susceptible parent of RIL-4) and TG 26 (susceptible parent of RIL-5). Though the means of both the RILs were within the parental limits, few transgressive segregants were observed in both directions for LLS and rust. Analysis of variance and pooled analysis of variance of these phenotyping data revealed significant differences between RILs for LLS and rust at both stages and environments of phenotyping. The effect of genotype × environment (GE) interaction, however, was not significant. The genetic parameters estimated in both the populations revealed moderate to high phenotypic (PCV) and genotypic coefficient of variation (GCV) for LLS and rust at both stages; however PCV was always higher than GCV for both diseases (ESM 1). Similarly, broad-sense heritability (h 2b.s) was moderate to high at both stages for LLS and rust. The association analyses between stages in each environment showed highly significant and positive correlation for LLS (r = 0.189–0.932; P < 0.01) and rust (r = 0.166–0.983; P < 0.01). Significant positive correlation was observed even across the environments for LLS and rust (data not shown). However, the association between LLS and rust was towards the negative direction. The frequency distribution based on the pooled mean data of all the seasons for LLS and rust showed a normal distribution pattern for late leaf spot at both stages (LLS-SI and LLS-SII) in both RIL-4 and RIL-5 populations (Fig. 1). However, the frequency distribution for rust in both populations revealed a bimodal distribution at both stages (Rust-SI and Rust-SII) and skewed distribution towards susceptible and resistant parents.

Frequency distribution for LLS and rust resistance in RIL-4 (left) and RIL-5 (right) populations. The figure shows the frequency distribution of RIL populations against infestation on pooled mean disease scores in RIL-4 for LLS at stage I (a) and stage II (b); for rust at stage I (c) and stage II (d); in RIL-5 for LLS at stage I (e) and stage II (f); for rust at stage I (g) and stage II (h)
Marker analysis on parents and mapping populations
In addition to screening 1,089 SSR markers on the parents of RIL-4 (Khedikar et al. 2010) and 1,043 SSR markers on the parents of RIL-5 (Sarvamangala et al. 2011), new sets of 2,008 and 2,239 additional SSR markers were screened on the parents of the RIL-4 and RIL-5 populations, respectively. Of the new SSR markers screened, a total of 139 and 152 markers were found polymorphic between the parents of RIL-4 and RIL-5, respectively. After including the earlier polymorphic markers (Khedikar et al. 2010; Sarvamangala et al. 2011), a total of 206 and 205 markers were found polymorphic between the parents of RIL-4 and RIL-5, respectively (ESM 2). Subsequently, genotyping data were generated for 206 SSR markers on 182 lines of RIL-4 and for 205 markers on 138 lines of RIL-5. However, while genotyping the mapping populations, segregation data were scored at two loci for three markers (GM1971, GM2724 and pPGSseq18A05) in the case of RIL-4 and four markers (GM1971, GM2724, GM2589 and TC3H07) in RIL-5. As a result, segregation data were obtained for a total of 209 SSR loci each in RIL-4 (67 SSR loci from Khedikar et al. 2010 and 142 SSR loci in this study) and RIL-5 (53 SSR loci from Sarvamangala et al. 2011 and 156 SSR loci in this study).
Development of improved genetic maps
Genotyping data obtained for all 209 marker loci for both the mapping populations were subjected to Chi-squared (χ2) testing to estimate the segregation ratio. Out of 209 loci, a total of 165 (RIL-4) and 156 (RIL-5) marker loci showed the expected 1:1 segregation ratio (P < 0.05). The remaining 44 (20%) and 53 (28%) markers were found to be distorted in RIL-4 and RIL-5, respectively (ESM 3). Nevertheless, these markers were also used for constructing the genetic map.
As the earlier linkage maps developed based on the two populations under study had many fewer mapped loci, the linkage analysis was done on the entire marker dataset in each RIL population by using a minimum LOD score of 5.0 and a maximum recombination fraction (θ) of 0.37. As a result, an improved genetic map with 188 SSR loci onto 20 linkage groups (LGs) spanning 1,922.4 cM was developed based on the RIL-4 population. Similarly another genetic map comprising 181 loci onto 21 LGs covering 1,963 cM was developed based on the RIL-5 population (ESM 4, ESM 5, ESM 6).
Both genetic maps were compared with the reference genetic map for cultivated groundnut species developed based on the TAG 24 × ICGV 86031 (RIL-1) mapping population (Varshney et al. 2009a). A good congruence was found between the maps for common markers and for order of the markers, with few exceptions. Finally, names for the LGs of the newly developed genetic maps were assigned according to the LGs of the reference map (TAG24 × ICGV 86031) for cultivated groundnut based on the common markers observed, with the suffix “*” indicating LGs named by comparison with the reference genetic map (Varshney et al. 2009a). In the case of the genetic map based on the RIL-5 population, the LG AhIII* was split into three fragments and hence named AhIIIa*, AhIIIb* and AhIIIc* (ESM 6).
The number of markers mapped per LG ranged from two (AhXX) to 17 (AhIII*) and the length of the LGs ranged from 17.6 cM (AhXX) to 192.4 cM (AhXIV*) with an average map distance of 96.12 cM in the case of the genetic map based on RIL-4 (ESM 4, ESM 7). Similarly, in the case of the genetic map developed based on RIL-5, the number of markers mapped per LG ranged from two (AhXX and AhXXI) to 15 (AhXV and AhVII*) and the length of the LGs ranged from 14.7 cM (AhXIX) to 208.6 cM (AhXIV*) with an average map distance of 85.35 cM (ESM 4). The average map density ranged from 4.22 (AhVIII*) to 20.95 cM (AhXIX) in the genetic map based on RIL-4 and 4.54 (AhIIIc*) to 17.38 cM (AhXIV*) in the genetic map based on RIL-5 (ESM 7). The detailed features of these genetic maps are given in ESM 4 and ESM 7. Comparison of the two newly constructed genetic maps showed 143 common markers distributed on 18 LGs. The order of these common markers was similar in the majority of the cases (ESM 8).
Identification of QTL for LLS resistance
Phenotypic data obtained at both the stages (LLS-SI and LLS-SII) in all the environments for both the populations were analyzed together with mapping data for all mapped loci assigned to the genetic maps of the respective mapping populations using WinQTL Cartographer, version 2.5 (Wang et al. 2007b). As a result, a total of 13 QTL were identified for LLS in RIL-4 (ESM 9) and 15 QTL in RIL-5 (ESM 10). The 13 QTL identified in the RIL-4 population were distributed on nine LGs (AhXII*, AhXV, AhV*, AhXIII*, AhIX*, AhVIII*, AhXVIII*, AhVI* and AhVII*) (ESM 5). Among them, five were major QTL (QTLR4-LLS01, QTLR4-LLS02, QTLR4-LLS03, QTLR4-LLS04 and QTLR4-LLS05) with a phenotypic variance explained (PVE) ranging from 10.27 to 67.98% (Table 1). Similarly 15 QTL identified in the RIL-5 population were distributed on nine LGs (AhXV, AhVIII*, AhV*, AhI*, AhXIV*, AhXIII*, AhX*, AhVII* and AhIIIc*) (ESM 6). Among them, eight QTL (QTLR5-LLS01, QTLR5-LLS02, QTLR5-LLS03, QTLR5-LLS04 QTLR5-LLS05, QTLR5-LLS06, QTLR5-LLS07 and QTLR5-LLS08) were major with PVE ranging from 7.58 to 63.17% (Table 1). Though QTLR5-LLS01 and QTLR5-LLS02 were major QTL, the PVE was less than 10% in one of the environments, otherwise the PVE ranged from 7.58 to 49.64% and 9.79 to 22.46%, respectively. The remaining seven QTL in RIL-4 and eight QTL in RIL-5 were considered as minor QTL with <10% PVE (Collard et al. 2005).
In terms of stability of the QTL, the QTL identified in at least three seasons (years) were considered as the “consistent” QTL. Among the five major QTL identified in the RIL-4 population, one QTL, namely QTLR4-LLS01 with 10.27–62.34% PVE, was found consistently across both stages of all six seasons (Table 1). The other major QTL, namely QTLR4-LLS05 with 10.81–15.34% PVE, was identified in one or both stages of three seasons. On the other hand, only one (QTLR4-LLS09) out of eight minor QTL with 3.39–8.5% PVE was identified in three seasons. The remaining three major and seven minor QTL identified were found in less than three seasons and they were either specific to a particular stage or season of disease screening (Table 1, ESM 9). In the case of the RIL-5 population, of the 15 major and minor QTL identified, none of the QTL were present in more than three seasons (ESM 9). Only two major QTL, namely QTLR5-LLS01 (7.58–49.64% PVE) and QTLR5-LLS02 (9.79–22.46% PVE), were present in more than two seasons (Table 1). The remaining 13 QTL were either specific to one or both the stages of disease scoring in less than three seasons.
Identification of QTL for rust resistance
For identification of QTL controlling rust resistance, detailed QTL analysis was done in RIL-4 and RIL-5 using the corresponding genetic mapping data and extensive phenotyping data for rust obtained for the two scoring stages (80 days after sowing, referred as Rust-SI and 90 days after sowing referred to as Rust-SII) under six (in the case of RIL-4) or seven (in the case of RIL-5) seasons. As a result, a total of nine (RIL-4) and six (RIL-5) QTL for rust resistance were identified from both the populations with PVEs of 2.54–82.96% in RIL-4 and 2.89–78.96% in RIL-5 (ESM 10). The QTL were distributed on six (AhXV, AhXVI, AhII*, AhVII*, AhI* and AhXVIII*) and four (AhXV, AhV*, AhXX and AhXIV*) LGs in the RIL-4 and RIL-5 populations, respectively (ESM 5, ESM 6). Of these QTL, three QTL with 10.68–82.96% PVE in RIL-4 and four QTL with 15.04–78.96% PVE in RIL-5 were considered as major QTL (Table 2). The remaining six QTL in RIL-4 and two QTL in RIL-5 that explained <10% PV were considered as minor QTL (ESM 10).
In terms of identification of consistent QTL, all three major QTL (QTLR4-Rust01, QTLR4-Rust02 and QTLR4-Rust03) were consistently present in all the seasons, except for a few stages (viz. Rust-SII_2007E1 and Rust-SI_2010 for QTLR4-Rust01; Rust-SII_2007E1 and Incubation period for QTLR4-Rust02; Rust-SII_2009 and incubation period for QTLR4-Rust03) in the RIL-4 population (Table 2). However, in the case of the RIL-5 population, two QTL, namely QTLR5-Rust01 and QTLR5-Rust02, were present in both the stages of seven and six seasons, respectively. The remaining six minor QTL in the RIL-4 population and two each major and minor QTL in the RIL-5 population were either specific to one or both the stages of disease scoring in less than three seasons.
Development of a consensus map
A consensus map was developed by using 143 common markers distributed on 18 LGs on two genetic maps for the RIL-4 and RIL-5 populations. In this context, the newly constructed genetic map based on RIL-4 with the highest number (188) of mapped loci was used as a framework map. Subsequently, a consensus map was developed using the framework map as a fixed backbone onto which the unique loci of the genetic map developed based on RIL-5 were added following the “neighbors” map approach described by Cone et al. (2002). The markers from corresponding LGs of the genetic map based on RIL-5 were assigned onto LGs of the framework map (ESM 8). As a result, the consensus map developed here had 225 SSR loci onto 20 LGs, of which 18 LGs were integrated from both genetic maps using common markers and the remaining two LGs were adopted as such from the framework map (ESM 4). The number of mapped loci on the consensus map ranged from two (AhXX) to 17 (AhIII* and AhXVII*) (Fig. 2). The length of the LGs ranged from 17.52 cM (AhXX) to 116.16 cM (AhXIX) with an average map distance of 57.65 cM and average inter-locus gap distance of 5.15 cM (ESM 7). The average map density ranged from 2.40 (AhXVII*) to 29.04 (AhXIX) cM in the consensus map (ESM 7).

Consensus linkage map of cultivated groundnut showing relative QTL positions based on two mapping population for LLS and rust. The QTLs identified from the RIL-4 and RIL-5 populations are differentiated by different colors. The suffix “*” indicates the linkage groups named by comparison with the tetraploid reference map for cultivated groundnut (Varshney et al. 2009a)
Consensus QTL map for LLS and rust in cultivated groundnut
As the consensus map was developed based on RIL-4 and RIL-5 populations segregating for resistance to foliar diseases (LLS and rust), the QTL identified in RIL-4 and RIL-5 were placed in the respective position on the consensus map. In case of LLS, a total of 28 (13 major and 15 minor) QTL identified in the RIL-4 and RIL-5 populations were located on a total of 13 LGs of the consensus map (Fig. 2). In the case of rust, out of a total of 15 (seven major and eight minor) QTL identified in the RIL-4 and RIL-5 populations, 14 QTL (seven major and seven minor) were placed onto seven LGs of the consensus map. Interestingly, five LGs (AhIV*, AhXI*, AhXVII*, AhXIX and AhXX) did not have any QTL for LLS or rust. AhVIII* and AhXV LGs contained five QTL each and AhV* LG had four QTL for LLS. Interestingly, LG AhVIII* harbored the QTL identified for LLS from the RIL-5 population only.
Analysis also showed two candidate genomic regions containing the major as well as the consistent QTL for resistance to foliar diseases. One genomic region (GM2009–GM1954, 20.6 cM) present on LG AhXV contained three QTL each for LLS and rust. The second genomic region (29.3 cM) present on LG AhXII* and flanked by GM1573/GM2009 and pPGPseq8D09 markers harbored a major and consistent LLS resistance QTL that was expressed in both the stages of disease scoring of all the seasons and at both stages of disease scoring.
Discussion
QTL mapping is a prerequisite for identification of molecular markers associated with tolerance/resistance to abiotic/biotic stresses and agronomically important traits. In the case of groundnut, because of limited level polymorphism combined with the paucity of molecular markers, genetic mapping and QTL mapping has only just begun in recent years (Varshney et al. 2009a; Foncéka et al. 2009; Khedikar et al. 2010; Hong et al. 2010; Ravi et al. 2011; Sarvamangala et al. 2011). In the case of foliar disease resistance, only one QTL study in cultivated groundnut is available that reports identification of a major QTL for rust resistance (Khedikar et al. 2010). In the case of LLS, however, no major QTL was reported. The present study, therefore, attempted to identify major QTL for LLS and rust along with development of a consensus map which will accelerate genetics research and molecular breeding applications in cultivated groundnut.
Phenotypic evaluation for LLS and rust
To improve the accuracy of QTL mapping by reducing background noise, the utmost importance was given to the accuracy of phenotypic evaluation. As reliable phenotypic data is vital for identification of reliable QTL which will further help in potential use of the candidate QTL for MAS. Extensive phenotyping of both the populations for LLS and rust for 6–7 years was therefore done in the present study. The genetic estimates based on both the mapping populations exhibited considerable variation in resistance to LLS and rust. The magnitude of variation was moderate to high as revealed by PCV and GCV, accompanied by moderate to high broad-sense heritability. The near-normal to normal distribution revealed the quantitative nature of resistance for LLS. However, the frequency distribution for rust resistance at both the stages was skewed/bimodal towards the susceptible and resistant parents, indicating the possibility of a few major genes governing rust resistance.
Marker polymorphism, individual and consensus genetic maps
The availability of large-scale SSR markers for groundnut in recent years (Pandey et al. 2011) has allowed us to screen the parental genotypes of the RIL-4 and RIL-5 populations with 3,097 and 3,282 SSR markers, respectively. Very low levels of polymorphism (6.65 and 6.24%) were observed between the parents (TAG 24, TG 26 and GPBD 4) of the respective mapping populations. A low level of polymorphism was not unexpected, keeping in mind the low level of molecular diversity in cultivated groundnut and that the origin of the majority of SSRs used in this study were from highly conserved genic regions, i.e. EST-SSRs (Moretzsohn et al. 2004, 2005; Ferguson et al. 2004; Varshney et al. 2009a, b; Pandey et al. 2011).
In the present study, amplification of more than two loci was observed for three markers (GM1971, GM2724 and pPGSseq18A05) in the case of RIL-4 and four markers (GM1971, GM2724, GM2589 and TC3H07) in the RIL-5 population, which has also been observed in earlier studies (Varshney et al. 2009a; Ravi et al. 2011). Amplification of more than two loci could be attributed to amplification of a duplicated locus or a different locus due to the allotetraploid/polyploidy nature of the cultivated groundnut genome (Varshney et al. 2009a).
Of the 206 and 205 markers for which genotyping data were obtained, 44 (20%) and 53 (28%) markers showed segregation distortion in RIL-4 and RIL-5, respectively. The percentage of distorted markers in the present study was relatively less than in earlier mapping studies by Burow et al. (2001) (68%), Moretzsohn et al. (2005) (51%) and Varshney et al. (2009a) (35%). Generally, mapping populations developed from highly diverse genotypes with less genome similarity (e.g., cultivated × wild or synthetic genotypes) reveal higher segregation distortion. Since the number of polymorphic markers was limited in the present study, the distorted markers were also used for construction of genetic maps and these markers were placed in the most appropriate locations and orders using advanced functions of mapping programs. In brief, the present study presents two genetic maps with 188 and 181 SSR loci for the RIL-4 and RIL-5 populations, respectively. As expected, the map coverage was much higher (1922.4 cM for RIL-4 and 1963 cM for RIL-5) than the maps developed for the AA (1,230.9 cM, Moretzsohn et al. 2005) and BB (1,294.0 cM, Moretzsohn et al. 2009) genomes. Compared to the partial genetic maps developed earlier using RIL-4 by Khedikar et al. (2010) (462.24 cM; 56 loci mapped on 14 LGs) and RIL-5 by Sarvamangala et al. (2011) (657.90 cM; 45 loci mapped on eight LGs), the new genetic maps developed here are denser and improved and are comparable to the reference genetic map developed by Varshney et al. (2009a) (1,270.5 cM; 135 loci mapped on 22 LGs) and satuarated by Ravi et al. (2011) (1785.4 cM; 191 loci mapped on 22 LGs). It is also important to mention that marker orders in both the genetic maps developed here are in congruence for the majority of the mapped loci within themselves as well as with the reference map based on TAG 24 × ICGV 86031 (Varshney et al. 2009a; Ravi et al. 2011).
Based on 143 mapped loci common to the individual genetic maps for RIL-4 and RIL-5, a consensus map has been developed. This consensus map has 225 SSR loci mapped onto 20 LGs, of which 18 LGs were derived from the integration of LGs from the two genetic maps. The consensus map developed in the present study was comparable in terms of marker density and genome coverage to the earlier composite linkage map developed based on three populations by Hong et al. (2010). There are a few small LGs which could be artificial, and additional genetic markers can improve the linkage analysis to increase the density of markers in the future. The current consensus map has more marker loci (225 loci) mapped than the available published individual genetic map (191 loci, Ravi et al. 2011) or composite map (175 loci, Hong et al. 2010). Therefore, this consensus map will facilitate the selection of markers along the length of the LG/chromosome, which can be used for detecting recombinant individuals, fixing loci and recovering the recurrent parent genetic background, in addition to aligning future genetic maps (Somers et al. 2004; Varshney et al. 2007).
QTL for late leaf spot and rust
To date only one report is available on QTL identification for foliar disease in cultivated groundnut (Khedikar et al. 2010). This study, based on a partial genetic map for the RIL-4 population, reported a major QTL for rust and minor QTL for LLS. Due to the addition of more markers on genetic maps and QTL analysis based on multiple seasons’ phenotypic data on RIL-4 (Khedikar et al. 2010) and RIL-5 (Sarvamangala et al. 2011), this study reports a total of 28 QTL including 13 major QTL for resistance to LLS. Two of these 13 QTL were common to both populations. One such common QTL, namely QTLR4-LLS02 flanked by GM2009 and GM1536, is expressed with up to 67.98% PVE in one disease scoring stage of two seasons (LLS-SII_2008, LLS-SII_2009) in RIL-4 and with up to 49.64% PVE in both the stages of two seasons (LLS-SI_2008, LLS-SII_2008, LLS-SI_2009, LLS-SII_2009). Interestingly, it also appeared while performing pooled mean analysis of phenotypic data for the second (90 days after sowing) stage (LLS-SII_Pooled mean) in the case of the RIL-5 population. The second QTL, flanked by IPAHM103 and GM1954 located on AhXV LG, is expressed with up to 42.66% PVE in one season and at both the stages (LLS-SI_2008, LLS-SII_2008) in RIL-4 and with 63.10% PVE in one season (LLS-SII_2008) in RIL-5. These two QTL regions, therefore, seem to be the candidate QTL for introgression in the elite groundnut varieties through MAS.
Although two consistent QTL (appearing in at least three seasons), namely QTLR4-LLS01 (10.27–62.34% PVE) and QTLR4-LLS05 (10.81–15.34% PVE), were identifed in RIL-4, none of these QTL was validated in RIL-5. One of the reasons for not observing at least one consistent QTL (QTLR4-LLS01) in RIL-5 is the monomorphic nature of the diagnostic marker (GM1009) in RIL-5. Validation of these consistent QTL is therefore required before they are deployed in MAS.
For rust resistance, although 15 QTL were identified in the RIL-4 and RIL-5 populations, only two of seven major QTL were common between these populations. One such QTL, namely QTLR4-Rust01 flanked by GM2009 and GM1536, contributed up to 82.27% PVE in both stages of disease scoring in all the six seasons except for Rust-SI_2007E1 and Rust-SI_2010 as well as in pooled mean analysis at both the stages (Rust-SI_Pooled mean and Rust-SII_Pooled mean) and for the components of rust resistance, viz. incubation period, latent period and infection type in RIL-4. In the case of the RIL-5 population, this QTL with up to 66.05% PVE was detected in both stages of disease scoring of all seven seasons as well as for pooled means at both the stages. Similarly the second common QTL, namely QTLR4-Rust03 flanked by IPAHM103 and GM1954, consistently explained up to 82.96% PVE in both the stages (except Rust-SII_2009) of disease scoring in all the six seasons as well as for Rust-SI and Rust-SII_Pooled mean latent period and infection type analysis in the RIL-4 population. In the case of the RIL-5 population, too, this QTL explained up to 78.96% phenotypic variation in both stages (except Rust-SI_2005 and Rust-SII_2005) of the six seasons as well as for Rust-SI_Pooled mean and Rust-SII_Pooled mean. In addition, it is interesting to note that these two QTL regions for rust resistance also confer resistance to rust as well with higher phenotypic variation in both RIL-4 and RIL-5 populations. Therefore, this genomic region seems to be a promising target for MAS for introgression of resistance to both foliar diseases.
Among the population-specific major QTL identified for rust resistance in the RIL-4 population, one QTL, namely QTLR4-Rust02, was consistently expressed with up to 62.35% PVE in both the stages of disease scoring of all the six seasons except for Rust-SII_2007E1. It was also expressed for the components of rust resistance such as latent period and infection type. However, neither of the two major QTL specific to RIL-5 was found to be stable across the seasons.
While comparing the QTL identified in this study with the earlier reports, we observed that four QTL for resistance to LLS (QTLR4-LLS04, QTLR4-LLS07, QTLR4-LLS12 and QTLR4-LLS13) and three QTL for rust resistance (QTLR4-Rust03, QTLR4-Rust05 and QTLR4-Rust09) were also reported in our earlier study (Khedikar et al. 2010). It is important to mention here that though this study as well as Khedikar et al. (2010) used the RIL-4 population, a major QTL (QTLR4-LLS01) with up to 62.34% PVE was identified only in the present study and not in Khedikar et al. (2010). This became possible only because of saturating the genetic map of the RIL-4 population from 56 to 188 SSR loci. The LG AhXII on which this major QTL for LLS flanked by the markers GM1573/GM1009 and pGPseq8D09 is present has 10 SSR loci as compared to only three SSR loci in Khedikar et al. (2010). Moreover, the above-mentioned markers were not mapped in the RIL-4 population map of Khedikar et al. (2010). These observations therefore highlight the need to use good genetic maps for QTL analysis to avoid the possibility of losing the major QTL.
Candidate genomic regions conferring resistance to foliar diseases
Out of a total of 43 QTL identified for LLS and rust in the two populations, 42 QTL (20 major and 22 minor QTL for both LLs and rust) were mapped on the consensus map in the respective positions of the individual genetic maps for the RIL-4 and RIL-5 populations. The remaining minor QTL (QTLR5-Rust05) mapped onto LG AhXX of RIL-5 could not be located on the consensus map, as this linkage group was not merged into the consensus map due to the non-availability of common markers. A genomic region (20.6 cM) flanked by GM2009 and GM1954 on LG AhXV was found to contain the common QTL for LLS as well as rust and was expressed in one and/or both stages of many environments. The QTL for LLS and rust resistance present in this region contribute up to 67.98 and 82.96% phenotypic variation, respectively. Another interesting genomic region (29.3 cM) is on LG AhXII* and is flanked by GM1573/GM1009 and pPGPseq8D09. This region contains the consistent QTL for LLS that was expressed in both stages of all the six seasons in RIL-4 and contributes up to 62.3% phenotypic variation. These two genomic regions, therefore, seem to possess candidate genes that are either directly involved in or control the expression of the genes conferring resistance to these important fungal diseases. Although QTL cloning or sequencing of these regions will eventually reveal those genes, introgression of one or both of these candidate genomic regions through MAS seems to be the best approach to developing superior lines/varieties of groundnut with enhanced resistance to the foliar diseases.
Conclusion
The current study reports the construction of two genetic linkage maps with 188 and 181 loci on two RIL mapping populations and also the development of a consensus map for disease resistance studies with 225 loci mapped onto 20 LGs. These maps will be a useful resource and a tool from which potential SSR markers may be selected for future mapping projects in cultivated groundnut. Furthermore, a major QTL region for rust reported earlier has been saturated with five new additional markers, which enabled us to define properly the candidate genomic region with a smaller marker interval contributing up to a maximum of 82.96 and 66.05% phenotypic variation with respect to the populations for rust resistance. Furthermore, we report a major QTL for LLS with 10.27–62.34% PVE. Validation of the newly linked markers for rust resistance and the major QTL identified for LLS resistance is of prime importance for identifying the candidate genes as well as for their deployment in molecular breeding for resistance to foliar diseases in groundnut.
References
Ablett GA, Karakousis A, Banbury L, Cakir M, Holton TA, Langridge P, Henry RJ (2003) Application of SSR markers in the construction of Australian barley genetic maps. Aust J Agric Res 54:1187–1195
Burow MD, Simpson CE, Starr JL, Paterson AH (2001) Transmission genetics of chromatin from a synthetic amphidiploid to cultivated peanut (Arachis hypogaea L.): broadening the gene pool of a monophyletic polyploid species. Genetics 159:823–837
Collard BCY, Jahufer MZZ, Brouwer JB, Pang ECK (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica 142:169–196
Cone KC, McMullen MD, Bi IV, Davis GL, Yim Y-S, Gardiner JM, Pol-acco ML, Sanchez-Villeda H, Fang Z, Schroeder SG, Havermann SA, Bowers JE, Paterson AH, Soderlund CA, Engler FW, Wing RA, Coe EH (2002) Genetic, physical and informatics resources for maize: on the road to an integrated map. Plant Physiol 130:1598–1605
Cuc LM, Mace ES, Crouch JH, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8:55–65
Dwivedi SL, Pande S, Rao JN, Nigam SN (2002) Components of resistance to late leaf spot and rust among inter-specific derivatives and their significance in a foliar disease resistance breeding in groundnut (Arachis hypogaea L.). Euphytica 125:81–88
FAO (2009) Food and agricultural organization of the united nation, FAO statistical database. http://faostat.fao.org/faostat/collections?subset=agriculture
Ferguson ME, Burow MD, Schulze SR, Bramel PJ, Paterson AH, Kresovich S, Mitchell S (2004) Microsatellite identification and characterization in peanut (A. hypogaea L.). Theor Appl Genet 108:1064–1070
Foncéka D, Hodo-Abalo T, Rivallan R, Faye I, Sall MN, Ndoye O, Fávero AP, Bertioli DJ, Glaszmann J-C, Courtois B, Rami J-F (2009) Genetic mapping of wild introgressions into cultivated peanut: a way toward enlarging the genetic basis of a recent allotetraploid. BMC Plant Biol l9:103
Green CC, Wynne JC (1986) Diallel and generation means analyses for the components of resistance to Cercospora arachidicola in peanut. Theor Appl Genet 73:228–235
Gupta PK, Varshney RK (2000) The development and use of microsatellite markers for genetic analysis and plant breeding with emphasis on bread wheat. Euphytica 113:163–185
He G, Meng R, Newman M, Gao G, Pittman RN, Prakash CS (2003) Microsatellites as DNA markers in cultivated peanut (Arachis hypogaea L.). BMC Plant Biol 3:3–9
Hong Y, Chen X, Liang X, Liu H, Zhou G, Li S, Wen S, Holbrook CC, Guo B (2010) A SSR-based composite genetic linkage map for the cultivated peanut (Arachis hypogaea L.) genome. BMC Plant Biol 10:17
Hopkins MS, Casa AM, Wang T, Mitchell SE, Dean RE, Kochert GD, Kresovich S (1999) Discovery and characterization of polymorphic simple sequence repeats (SSRs) in peanut. Crop Sci 39:1243–1247
Karakousis A, Gustafson JP, Chalmers KJ, Barr AR, Langridge P (2003) A consensus map of barley integrating SSR, RFLP, and AFLP markers. Aust J Agric Res 54:1173–1185
Khedikar YP, Gowda MVC, Sarvamangala C, Patgar KV, Upadhyaya HD, Varshney RK (2010) A QTL study on late leaf spot and rust revealed one major QTL for molecular breeding for rust resistance in groundnut (Arachis hypogaea L.). Theor Appl Genet 121:71–984
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburn L (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Liang X, Chen X, Hong Y, Liu H, Zhou G, Li S, Guo B (2009) Utility of EST-derived SSR in cultivated peanut (Arachis hypogaea L.) and Arachis wild species. BMC Plant Biol 9:35
Lincoln S, Daly M, Lander E (1992) Construction of genetics maps with MAPMAKER/EXP 3.0, 3rd edn. Whitehead Institute Technical Report
Mace ES, Varshney RK, Mahalakshmi V, Seetha K, Gafoor A, Leeladevi Y, Crouch JH (2007) In silico development of simple sequence repeat markers within the aeshynomenoid/dalbergoid and genistoid clades of the Leguminosae family and their transferability to Arachis hypogaea, groundnut. Plant Sci 174:51–60
Moretzsohn MC, Hopkins MS, Mitchell SE, Kresovich S, Valls JFM, Ferreira ME (2004) Genetic diversity of peanut (Arachis hypogaea L.) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Plant Biol 4:11
Moretzsohn MC, Leoi L, Proite K, Guimara PM, Leal-Bertioli SCM, Gimenes MA, Martins WS, Valls JFM, Grattapaglia D, Bertioli DAJ (2005) Microsatellite-based gene-rich linkage map for the AA genome of Arachis (Fabaceae). Theor Appl Genet 111:1060–1071
Moretzsohn MC, Barbosa AVG, Alves DMT, Teixeira C, Leal-Bertioli SCM, Guimara PM, Pereira RW, Lopes CR, Cavallari MM, Valls JFM, Bertioli DJ, Gimenes MA (2009) A linkage map for the B-genome of Arachis (Fabaceae) and its synteny to the A-genome. BMC Plant Biol 9:40
Nevill DJ (1982) Inheritance of resistance to Cercosporidium personatum in groundnuts: a genetic model and its implications for selection. Oleagineux 37:355–362
Pandey MK, Gautami B, Jayakumar T, Sriswathi M, Upadhyaya HD, Gowda MVC, Radhakrishnan T, Bertioli DJ, Knapp SJ, Cook DR, Varshney RK (2011) Highly informative genic and genomic SSR markers to facilitate molecular breeding in cultivated groundnut (Arachis hypogaea). Plant Breed. doi:10.1111/j.1439-0523.2011.01911.x
Ravi K, Vadez V, Isobe S, Mir RR, Guo Y, Nigam SN, Gowda MVC, Radhakrishnan T, Bertioli DJ, Knapp SJ, Varshney RK (2011) Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (Arachis hypogaea L.) Theor Appl Genet 122:1119–1132
Sarvamangala C, Gowda MVC, Varshney RK (2011) Identification of quantitative trait loci for protein content oil content and oil quality for groundnut (Arachis hypogaea L.). Field Crops Res 122:49–59
Somers DJ, Isaac P, Edwards K (2004) A high-density microsatellite consensus map for bread wheat (Triticum aestivum L.). Theor Appl Genet 109:1105–1114
Subbarao PV, Subrahmanyam P, Reddy PM (1990) A modified nine point disease scale for assessment of rust and late leaf spot of groundnut. In: Second international congress of French phytopathological society. 28–30 November 1990, Montpellier, France, p 25
Subrahmanyam P, Williams JH, Mcdonald D, Gibbons RW (1984) The influence of foliar diseases and their control by selective fungicides on a range of groundnut (Arachis hypogaea L.) genotypes. Ann Appl Biol 104:467–476
Van Ooijen JW (2006) Joinmap® 4.0, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Varshney RK, Graner A, Sorrells ME (2005) Genomics assisted breeding for crop improvement. Trends Plant Sci 10:621–630
Varshney RK, Hoisington DA, Tyagi AK (2006) Advances in cereal genomics and applications in crop breeding. Trends Biotechnol 24:490–499
Varshney RK, Marcel TC, Ramsay L, Russel J, Roder MS, Stien N, Waugh R, Langridge P, Niks RE, Graner A (2007) A high density barley microsatellite consensus map with 775 SSR loci. Theor Appl Genet 114:1091–1103
Varshney RK, Bertioli DJ, Moretzsohn MC, Vadez V, Krishnamurty L, Aruna R, Nigam SN, Ravi K, He G, Knapp SJ, Hoisington DA (2009a) The first SSR based genetic linkage map for cultivated groundnut (Arachis hypogaea L.). Theor Appl Genet 118:729–739
Varshney RK, Mahendar T, Aruna R, Nigam SN, Vadez V, Hoisington DA (2009b) High level of natural variation in a groundnut (Arachis hypogaea L.) germplasm collection assayed by selected informative SSR markers. Plant Breed 128:486–494
Voorrips RE (2006) MapChart 2.2: software for the graphical presentation of linkage maps and QTLs. Plant Research International, Wageningen
Wang CT, Yang XD, Chen DX, Yu SL, Liu GZ, Tang YY, Xu JZ (2007a) Isolation of simple sequence repeats from groundnut. Electron J Biotechnol 10:473–480
Wang S, Basten CJ, Zeng ZB (2007b) Windows QTL cartographer 2.5. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm
Yuan M, Gong L, Meng R, Li S, Dang P, Guo B, He G (2010) Development of trinucleotide (GGC)n SSR markers in peanut (Arachis hypogaea L.). Electron J Biotechnol 13. http://dx.doi.org/10.2225/vol13-issue6-fulltext-6
Zeng ZB (1994) Precision mapping of quantitative trait loci. Genetics 136:1457–1468
Acknowledgments
The authors are grateful to the National Fund for Basic and Strategic Research in Agriculture (NFBSRA) of the Indian Council of Agriculture Research (ICAR), Department of Biotechnology (DBT) of the Government of India and Tropical Legume I—Objective 1 project of CGIAR Generation Challenge Programme (GCP), Mexico, and the Bill and Melinda Gates Foundation (BMGF), USA.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sujay, V., Gowda, M.V.C., Pandey, M.K. et al. Quantitative trait locus analysis and construction of consensus genetic map for foliar disease resistance based on two recombinant inbred line populations in cultivated groundnut (Arachis hypogaea L.). Mol Breeding 30, 773–788 (2012). https://doi.org/10.1007/s11032-011-9661-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-011-9661-z