
Abstract
Gene delivery vectors based on adeno-associated virus (AAV) are highly promising due to several desirable features of this parent virus, including a lack of pathogenicity, efficient infection of dividing and non-dividing cells, and sustained maintenance of the viral genome. However, several problems should be addressed to enhance the utility of AAV vectors, particularly those based on AAV2, the best characterized AAV serotype. First, altering viral tropism would be advantageous for broadening its utility in various tissue or cell types. In response to this need, vector pseudotyping, mosaic capsids, and targeting ligand insertion into the capsid have shown promise for altering AAV specificity. In addition, library selection and directed evolution have recently emerged as promising approaches to modulate AAV tropism despite limited knowledge of viral structure–function relationships. Second, pre-existing immunity to AAV must be addressed for successful clinical application of AAV vectors. “Shielding” polymers, site-directed mutagenesis, and alternative AAV serotypes have shown success in avoiding immune neutralization. Furthermore, directed evolution of the AAV capsid is a high throughput approach that has yielded vectors with substantial resistance to neutralizing antibodies. Molecular engineering and directed evolution of AAV vectors therefore offer promise for generating ‘designer’ gene delivery vectors with enhanced properties.
Similar content being viewed by others

INTRODUCTION
The ability to deliver genes with high efficiency and specificity can enable a broad range of biomedical efforts, from basic research to clinical translation. In particular, viral gene delivery vehicles or vectors offer the potential for safe and efficient gene delivery to various cell types in vitro and importantly in vivo. For example, vectors based upon adeno-associated virus (AAV) have several desirable features as gene delivery vectors. First, AAV is non-pathogenic (up to 90% of the human population has been exposed to AAV serotype 2) and therefore potentially safe for use as a vector (1,2). Furthermore, all viral genes can be deleted from recombinant AAV (rAAV) vectors, thereby yielding an efficient transgene delivery vehicle with enhanced safety and reduced immunogenicity. An additional attractive feature is that AAV can efficiently infect both dividing and non-dividing cells in muscle (3–7), liver (8–12), brain (13–16), lung (17–20), retina (21–25), heart (26–28), and pancreas (29–31). Fourth, although AAV serotype 2 is the best characterized, there are a number of other available AAV serotypes with differences in cellular tropism (32). Finally, although the wild-type AAV preferentially integrates into a specific locus of human chromosome 19 (33–36), recombinant virus has mechanisms for sustained episomal maintenance or integrates at a low rate semi-randomly (37).
Despite their advantageous properties, AAV vectors can and must be improved in numerous ways to enhance their utility from the lab to the clinic. Problems with AAV vectors include limited tissue tropism for serotypes that bind heparan sulfate (38,39); poor infection of refractory cell types such as stem cells (40,41); challenges with high-efficiency, targeted gene delivery to specific cell populations; preexisting immunity due to prior exposure of the majority of the human population with multiple AAV serotypes (2,42–47); and a limited transgene carrying capacity (48). This review will focus on recent advances in designing and engineering AAV vectors for altered tropism, enhanced gene delivery efficiency, and evasion of antibody neutralization.
AAV Biology
AAV has a single-stranded DNA genome approximately 4.7 kb long (49–51). The AAV genome contains two open reading frames (ORF) flanked by inverted terminal repeat elements (ITR) (52–54). These ITRs are the minimal cis-acting elements necessary for viral genome integration, replication, and packaging into the capsid shell. The first ORF (rep) encodes four Rep proteins that are involved in replication of the viral genome, whereas the second ORF (cap) encodes three structural proteins (VP1, VP2, and VP3) that are translated from one mRNA via alternative splicing and translation initiation at different start codons (1,55). Sixty total copies of these three structural proteins self-assemble to form the viral capsid, into which the viral genome then loads, and cap therefore plays a great role in the viral gene transduction properties. These properties are exploited by recombinant vectors, in which rep and cap are excised from between the ITRs, a gene of interest is inserted in their place, and the two viral ORFs are supplied as helper genes in trans to package the transgene inside the capsid (56).
AAV infection begins with viral binding to the cell surface, followed by viral uptake, intracellular trafficking, translocation to the nucleus, virion uncoating, synthesis of double stranded DNA, and viral gene expression (57–62). Although these steps collectively determine AAV tropism, viral binding to the cell surface is particularly important and has thus been a primary target for engineering to alter AAV tropism. AAV serotype 2 (AAV2) attaches to target cells by exploiting heparan sulfate proteoglycan (HSPG) as its primary cell surface receptor (62). In addition, fibroblast growth factor receptor 1 and αvβ5 integrin have been implicated as co-receptors (59,60).
AAV is an inherently defective virus, such that upon its arrival in the nucleus, viral replication requires functions supplied by co-infection with helper viruses, such as adenovirus and herpesvirus (63–66). In the absence of helper viruses or helper functions, the viral DNA can become integrated into the host chromosomal genome to establish a latent infection (67,68). Importantly, rAAV, which lacks both viral genes and is thus replication incompetent even in the presence of helper virus, can form a latent infection after entry.
MODULATING TROPISM AND ENHANCING TRANDUCTION
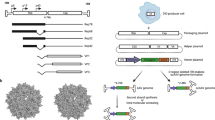
rAAV was first generated for transgene delivery in the 1980s (52,69). Vectors based on AAV2 (rAAV2) have been the most studied and are currently used in clinical trials for numerous diseases including cystic fibrosis, hemophilia B, prostate and melanoma cancers, Canavan disease, Alzheimer’s, Parkinson’s, muscular dystrophy, rheumatoid arthritis, and HIV vaccines (70). This vector has been shown in animal models to deliver genes to broad range of cells in muscle, brain, retina, liver, and lung (4,12,13,15,57,71–73). However, this broad tropism is not always beneficial for targeted gene delivery to specific types of tissues or organs. In addition, some highly desirable gene delivery targets, including stem cells, are refractory to vectors based on AAV2 (40). Therefore, a number of efforts have focused on manipulating capsid proteins to alter tropism and enhance transduction efficiency (Fig. 1).

Molecular engineering and evolution approaches have been applied to design AAV viral vectors for tropism change (A–D) and evasion of neutralizing antibodies (NABs) (C–F). A Mosaic capsids, composed of a mixture of capsid subunits from different serotypes, can possess the beneficial features of different serotypes. B Decoration of AAV capsids with either an antibody or a peptide targeting a cellular receptor can yield a targeted vector. C Pseudotyping the AAV2 genome with a capsid from a different serotype can “transfer” the desirable properties of the different serotype to AAV2. D Directed evolution of the AAV capsid, distinct from single round library generation and selection, involves multiple rounds of genetic diversification and selection. AAV libraries containing capsid variants are generated and subjected to selection for novel, diverse properties. Specific variants with desirable properties will be enriched and used for library generation of a next round of selection (denoted by the solid, curved arrows). E Disruption of the epitopes of AAV2 NABs allows evasion of antibody neutralization. F Conjugation of a synthetic polymer to AAV capsid can shield the AAV vector from NABs.
Isolation of Novel AAV Serotypes and Pseudotyping
The majority of capsid engineering work has been conducted with AAV2, the best characterized AAV serotype. However, over 100 AAV serotypes from different animal species have been isolated in recent years (5,6,74–81). AAV1, AAV2, AAV3, and AAV4 were isolated as contaminants in a simian Adenovirus type 15 stock (SV15), an Adenovirus type 12 stock, an Adenovirus type 7 stock, and African green monkeys infected with SV15, respectively (74,77,79). AAV5 was isolated from a human penile condylomatous wart (75) and AAV6 was found as a contaminant in a laboratory adenovirus stock. Gao et al. isolated AAV7 and AAV8 serotypes from rhesus monkeys, and AAV9 was isolated from three humans (6). AAV10 and AAV11 were isolated from cynomolgus monkeys (78), and Schimidt et al. isolated two new AAV serotypes, AAV (VR-195) and AAV (VR-355) from simian adenovirus stocks (81). Finally, Gao et al. found 86 AAV serotypes in nonhuman primates (6).
Many in vitro and in vivo studies have shown that these various natural AAV serotypes exhibit different tissue or cell tropisms. Furthermore, cross-packaging an AAV genome of one serotype into the capsid of another serotype, i.e. vector pseudotyping, can result in infectious vector with the tropism of the new capsid. For example, pseudotyped vector consisting of AAV2 genome and AAV4 capsid (AAV2/4) exhibited ependymal cell-specific transduction, whereas AAV2 vector (AAV2/2) showed transduction biased to neurons in the central nervous system (38). Since the biology and genetics of the AAV2 genome have been broadly studied, pseudotyping a AAV2 genome serves as a convenient technique to isolate and characterize the capsid properties of newly identified serotypes. Recently, Grimm et al. have examined effect of viral DNA packaging sequences on AAV vector transduction to liver. Based on the observed gene transfer to the liver by various pseudotyped vectors, they concluded that AAV tropism is determined by the AAV cap gene and not the rep gene (82), which supports validity of pseudotyping methods.
Chao et al. have reported that AAV2 genomes packaged by AAV1, AAV3, and AAV4 capsids showed 900-, 30-, and 3-fold enhanced transgene expression in skeletal muscle compared to AAV2/2 (83). In addition, AAV2/5 mediated efficient gene transfer to murine cerebellar neurons (84), and AAV2/6 efficiently transduced skeletal muscle (85). Gao et al. have shown that pseudotyped AAV2/7 exhibited gene transfer to skeletal muscle with efficiency comparable to that of AAV1, at that time the most efficient serotype for muscular delivery. Gao et al. also found that AAV2/8 showed 10- to 100-fold higher transgene expression upon delivery to the murine liver compared to other serotypes (5). In addition, AAV2/9 showed very efficient gene transfer in the liver, lung, muscle, and extracardiac tissue of mice in vivo (6,86). The capsid proteins of AAV10 and AAV11 showed 84 and 65% similarity, respectively, with the AAV2 capsid protein. Systemic injection of AAV2/10 led to persistent transduction in the murine liver, heart, muscle, lung, kidney, and uterus, whereas AAV2/11 resulted in efficient transduction of the muscle, kidney, spleen, lung, heart, and stomach. These results are distinct from AAV2/2, which yields persistent transduction only of liver and spleen (78). In addition, intravenously administered AAV2/10 and AAV2/11 mediated efficient delivery of a transgene to the lymphoid tissues in Cynomolgus monkeys (87). AAV (VR-195) and AAV (VR-355) showed distinct tropisms from AAV6 in a panel of human cancer cell lines, though they share 96% sequence identity with AAV6 (81).
Although the isolation of novel AAV serotypes has extended rAAV delivery to previously refractory cell types, it remains unlikely that a serotype with specificity for a single cell type or tissue will emerge. As a result, various efforts to apply rational or combinatorial rAAV design strategies are still being developed to generate novel capsid mutants for transduction of specific cells.
Mosaic Capsids
Mosaic virions, composed of a mixture of VP capsid subunits from different serotypes, are an alternative approach to modify the properties of AAV vectors. In particular, this method has the potential to combine beneficial features from different serotypes. For instance, AAV2 is readily purified by heparin affinity chromatography and has moderate infectivity for liver, whereas AAV1 shows efficient transduction of muscle. To combine the advantages of both AAV2 and AAV1, Hauck et al. generated mosaic capsid vectors, by mixing the two helper plasmids during vector packaging, that can efficiently transduce both muscle and liver (88). Furthermore, Rabinowitz et al. examined properties of mosaic capsids made from pairwise combinations of AAV serotype 1 to 5. Mixing AAV3 and AAV5 at the 3:1 ratio exhibited dual binding properties to heparin and mucin agarose, which mimic the primary binding receptors for AAV3 and AAV5, respectively. An unexpected, synergistic transduction effect was observed for some cell lines when AAV1 capsid subunits were mixed with AAV2 or AAV3, which suggests the potential utility of this approach to produce new and novel AAV vector tropisms (89).
Gigout et al. extended the definition of a mosaic capsid by generating vectors with a mixture of wild-type capsid protein and mutant capsid containing the IgG binding Z34C fragment of protein A, building upon earlier work discussed below [receptor targeting via genetic engineering (90)]. These AAV2 mosaic capsid vectors were able to selectively and efficiently transduce MO7e and Jurkat cells in the presence of targeting antibodies against either the stem cell factor receptor kit (CD117) or β1 integrins (CD29), whereas AAV2 vector with a IgG binding domain alone exhibited 20- and 100-fold lower infectivity, respectively, than that of wild-type AAV2 vector (91). More recently, Stachler and Bartlett have produced mosaic vectors composed of AAV1 capsid proteins containing an RGD4C peptide to target integrins present on vasculature, and AAV1 capsid proteins containing a peptide that can be metabolically biotinylated for efficient purification by avidin affinity chromatography. The resulting mosaic AAV1 vectors exhibited 50- to 100-fold enhanced transduction of endothelial cells and could be efficiently purified without reduction in transduction efficiency (92). Despite this impressive promise, the mosaic capsid approach has two potential shortcomings. First, the ratios between two serotypes may not be reproducible from packaging run to run or scaleup. Second, a mosaic capsid can potentially be inactivated by NABs directed against either serotype. Additional investigation is required to address these issues (88).
Receptor Targeting via Chemical Engineering
AAV2 vectors bind to HSPG, a receptor present in many tissues and cell types (62). This broad distribution therefore restricts this receptor’s utility for targeted transduction in vivo. In order to overcome this limitation, several research groups have developed novel conjugate-based targeting methods to bridge interactions between the AAV vector and specific cell receptors, thereby allowing AAV transduction of nonpermissive cell lines (93,94). Bartlett et al. have shown that binding of bispecific F(ab’γ)2 antibody, with specificity for the AAV2 capsid and the surface receptor αIIbβ3, to AAV vectors enables transduction of normally nonpermissive megakaryocyte cell lines (93). However, the reversible and potentially transient nature of virus-bispecific antibody complexes in vivo may be a limiting factor for achieving of efficient virus uptake and correct intracellular trafficking in vivo (43).
As an alternative to bispecific antibodies, Ponnazhagan et al. used a bispecific avidin-human epidermal growth factor (EGF) fusion protein in conjunction with biotinylated rAAV2 to target cells via the EGF receptor. This approach significantly increased transduction of EGF receptor-positive SKOV3.ip1 cells. Similar to EGF, human fibroblast growth factor 1 (FGF1) has been fused to avidin to make a bridge between biotinylated rAAV and FGF receptor-positive M07e cells (94). Recently, Arnold et al. have described an alternative to chemical biotinylation of AAV: metabolic biotinylation of an AAV variant with a BirA biotin ligase substrate peptide genetically inserted into the capsid. Co-expression of Escherichia coli BirA yields biotinylated vector during production (95).
Receptor Targeting via Genetic Engineering
The majority of AAV targeting studies have involved direct genetic insertion of targeting ligands into the cap gene. In early work, Yang et al. created an AAV vector with a single-chain antibody against human CD34 genetically inserted at the N-terminal region of capsid proteins in an effort to target hematopoietic progenitor cells (96). This approach significantly increased the rAAV selectivity towards a CD34+ human myoleukemia cell line, which is refractory to infection of wild-type AAV vector; however, titer was significantly compromised, a recurring theme in capsid engineering. In a subsequent landmark study, Girod et al. generated AAV2 capsid mutant (I-587) vector by inserting a 14-amino-acid peptide, L14 (QAGTFALRGDNPQG), at position 587 of the AAV2 capsid protein, an important and subsequently often used position chosen via homology mapping to the known crystal structure of canine parvovirus [the AAV2 structure was solved several years later (97)]. L14, which contains an RGD motif that binds several cellular integrin receptors, mediated efficient transduction of cell lines presenting appropriate integrins on their surface. Furthermore, the genetically modified virus packaged at titers comparable to wild-type AAV2 vector (98).
Wu et al. used a similar approach to enhance AAV infectivity. Insertion of the serpin receptor ligand in the N-terminal regions of VP2 led to 15-fold enhanced infectivity to IB3 lung epithelial cells (99). Loiler et al. have also shown that display of an ApoE ligand at the VP2 N-terminus of rAAV2 resulted in 220- and 4-fold higher infection of both islet cells ex vivo and murine hepatocytes in vivo compared to wild-type rAAV, respectively (100).
Although a given peptide insertion allows targeting of a single receptor, targeting a new receptor will then require a new genetic modification of the capsid proteins. In order to facilitate targeting new surface receptors, Ried et al. developed a versatile rAAV vector targeting system that is readily capable of redirecting rAAV binding specificity. The Z34C immunoglobulin (IgG) binding domain was inserted at position 587 of the capsid protein to enable rAAV to bind different antibodies via their Fc regions. The Fab region of bound IgGs thus remains free to function as a ligand directed against a specific cell surface receptor. rAAV2-Z34C vectors coupled to antibodies against CD29, CD117, and CXCR4 efficiently infected human hematopoietic cell lines, which are ordinarily refractory to infection of wild-type rAAV2 vector (90). While highly versatile, this approach may also involve reversible association of the AAV and IgG, which could pose a challenge in vivo.
Library Selection and Directed Evolution Approaches
AAV vectors based on naturally occurring novel serotypes, mosaic vectors, and rationally modified capsids have shown some successes in altering AAV vector tropism or enhancing infectivity. However, these approaches may have limited potential for generating AAV tropism beyond those already present in natural serotypes or in known, defined targeting peptides. Although antibody conjugation to AAV results in re-targeting of the vector to non-permissive cells to some extent, this approach has several challenges including improving packaging efficiency and improving the efficiency of intracellular trafficking (101).
Advances in molecular cloning techniques and the development of diverse high-throughput screening/selection methods have enabled researchers to explore library-based approaches to improve protein properties and to isolate novel proteins/peptides. Library-based approaches have also been extended to modify viral vectors for gene delivery. This section will focus three different approaches: isolation of novel peptides from phage libraries and subsequent insertion into AAV, direct selection of targeting peptide libraries in context of the AAV capsid, and the directed evolution of novel capsid proteins.
Insertion of Phage Library-Derived Peptide at Capsid Proteins
M13 phage has long been used as a powerful platform for the surface display of foreign proteins or peptides via insertion into the phage coat proteins (102). Peptide libraries displayed on the surface of the phage have been broadly employed to select biologically active peptides that bind to many different targets, including antibodies, receptors, enzymes, and cultured cells (103). In addition, Pasqualini et al. have successfully used phage display peptide library to select peptides capable of mediating selective localization of phage to murine brain and kidney vasculature (104). Several investigators have first isolated cell and tissue targeting peptides via phage display, then inserted the selected peptides into AAV in order to alter vector tropism (105–107). Grifman et al. have incorporated a tumor-targeting peptide previously identified from phage display into rAAV2, which successfully shifted vector tropism. However, peptides selected via phage often suffer a reduction in targeting capacity when transferred to the different protein context of the AAV capsid (105). Analogously, Nicklin et al. have selected a peptide from a phage display library that binds selectively and efficiently to human umbilical vein endothelial cells (HUVECs). The selected peptide SIGYPLP when inserted at position 587 of the capsid protein mediated enhanced transduction of HUVEC cells compared with wild-type AAV vector. Additionally, enhanced transduction was not observed in other cell lines including primary human vascular smooth muscle cells and human hepatocytes, indicating that the modified AAV vector exhibited a degree of specificity (106). More recently, Work et al. have shown that in vivo selection of a phage library resulted in isolation of peptides that mediated phage localization to rat brain and lung. Subsequent insertion of these peptides into the V3 region of the AAV capsid protein yielded AAV vectors that exhibited preferential localization to rat brain and lung (108). However, these studies collectively showed that transferring targeting peptides from phage to AAV resulted in reduction in transduction efficiency, posing a limitation of this approach.
Random Peptide Library Displayed Directly on AAV
In order to overcome the challenge of selecting a targeting peptide in a different context from that in which it is ultimately used, peptides can be directly screened or selected in the context of AAV. Perabo et al. have utilized a novel AAV library, which displayed random peptides on the AAV capsid, to select variants with altered tropism (109). Specifically, random 7 amino acid peptide sequences were inserted at position 587 of the capsid, previously identified by Girod et al. for its ability to tolerate insertions (98). The resulting capsid mutant library was subjected to repeated cycles of infection and harvesting on two cell lines, a human megakaryocytic and a B-cell chronic lymphocytic leukemia cell line, which are refractory to infection by wild-type AAV. The resulting capsid mutant clones selected from the library transduced the same cells with 100-fold increased efficiency compared to wild-type AAV vector (109). Muller et al. used an analogous approach to construct a random peptide library displayed on AAV, which was then subjected to selection on human coronary artery endothelial cells. The selected peptides led to 10- to 630-fold enhanced replicative titers and 4- to 40-fold enhanced expression of luciferase in human coronary artery endothelial cells (110). This creative and promising approach can improve interactions of virus with a cellular target protein, though it may be unable to overcome multiple extracellular and intracellular barriers to transduction, which may require engineering multiple regions on the viral surface.
Directed Evolution of the Capsid Protein
Directed evolution has been used to generate protein pharmaceuticals with enhanced biological activities, antibodies with enhanced binding affinity, new vaccines, and retroviral vectors with improved properties (111–115). Maheshri et al. have applied this powerful approach to create AAV capsids with novel and enhanced properties, including altered receptor binding (116). A library of capsid mutants was prepared by error-prone PCR followed by the staggered extension process to distribute random point mutations throughout the primary sequence of the capsid protein (116). After several rounds of heparin affinity fractionation and passaging on cells, AAV2 mutants with affinities either higher and lower than wild-type virus were isolated. This method has also been successful in enhancing AAV infectivity of nonpermissive cells, such as human astrocytes and other cell types (Koerber and Schaffer, unpublished data). This approach can be further extended to address many additional challenges in the development of viral gene delivery vectors.
EVADING IMMUNE RESPONSES
Although AAV vectors have many desirable properties, another challenge they face is pre-existing humoral and cellular immunity. For example, seroepidemiological studies have shown that 30–80% of the human population is seropositive against AAV2 (2), and antibodies that neutralize AAV2 infectivity been found in 20 to 67% of the human population (2,42–44). Similar statistics exist for other human AAV serotypes, as well as non-human serotypes that share significant sequence similarity with human serotypes. Numerous animal studies have shown that pre-exposure to AAV is a major barrier to gene delivery. For example, Maning et al. found that NABs directed against the AAV2 capsid prevents transgene expression after readministration of rAAV vectors to muscle in a murine model (117). Recently, Riviere et al. also found that repeated administration of rAAV2 vector to skeletal muscle of immunocompetent mice resulted in 10- to 100-fold reduction in transgene expression (118). In addition, Peden et al. have shown that preimmunization by administration of AAV to rat brains prevents trangene expression following administration of rAAV (45).
NABs pose the primary defense against initial viral infection. Burton and colleagues have proposed the “occupancy”, or the “antibody coating” theory that posits that the number of antibody required to neutralize the infectivity has linear correlation with the surface area of particle (119–121). This theory also implies that neutralization of viruses results from steric hindrance of a portion of the viral surface hampering interactions between virus and cell surface or receptors (120,122). Considering the relative sizes of an antibody, IgG (14–17 nm) (123) and an AAV2 virion (25 nm) (124), the occupancy theory seems plausible for AAV neutralization.
Solutions to Antibody Neutralization
There are several approaches to overcome the problem of pre-existing immunity of AAV2 vector. First, transient immunosuppression during initial AAV vector administration has successfully circumvented the issue of anti-AAV immune response in animal models (18,125,126). However, while it has promise in attenuating cellular immune responses, this approach does not solve the problem of pre-exiting, circulating antibodies in the human population. Accordingly, there have been several attempts to directly modify the AAV capsid to evade antibody neutralization.
Chemical Engineering
Synthetic polymers have been conjugated to AAV in attempts to shield the vector from antibody neutralization. Conjugation of synthetic polymers, in particular biocompatible polyethylene glycol (PEG), to protein therapeutics has been widely used to enhance their half-life in serum by reducing proteolysis and immune responses (127–132). Furthermore, covalent attachment of PEG to the surface (PEGylation) of adenovirus has been successfully used to shield the virus from NABs in vitro and in vivo (133–140). For example, Croyle et al. have shown that PEGylated adenovirus exhibited 10-fold enhanced transduction efficiency in the presence of NABs (139). Analogously, conjugation of PEG to AAV vectors resulted in 2.3- to 5-fold enhanced protection of the vectors from antibody neutralization without substantial loss of infectivity (141,142). However, several limitations of this approach include difficulty in controlling the number and sites of PEG conjugation, incomplete protection of the full viral surface, and loss of viral infectivity at high levels of PEGylation.
Isolation of Novel Serotypes and Pseudotyping
A new serotype is operationally defined as a virus that does not efficiently crossreact with NABs for all other characterized serotypes. Therefore, developing AAV vectors using serotypes other than AAV2 seems a straightforward approach to overcome pre-exiting immunity of AAV2. Although extensive serological studies of all serotypes must still be conducted, several research groups have explored the utility of novel serotypes to achieve evasion of NABs directed against AAV2 and other human serotypes. Xiao et al. have shown that antibodies generated against AAV2 did not cross-neutralize AAV1 vectors administered to murine liver and skeletal muscle (7). Halbert et al. have shown that AAV2 vectors pseudotyped with the AAV6 capsid (AAV2/6) efficiently transduced mouse lung even after administration of AAV2 (20), while prior administration of AAV2 completely blocked transduction of a subsequent AAV2 vector. Hildinger et al. and Sandalon et al. showed that AAV2/5 was not neutralized by antibodies raised against AAV2 and exhibited efficient transduction to murine smooth muscle cell or lung, respectively (143,144). Likewise, Peden et al. have shown that AAV5 vector can transduce rat brain pre-immunized against AAV2 vector (45). More recently, Riviere et al. have examined cross-administration of AAV1, AAV2, and AAV5 vectors to mouse skeletal muscle. Prior immunization with various serotypes did not significantly reduce transgene expression from a second vector of a different serotype (118), whereas readministration of the same serotype resulted in reduced transgene expression. Although AAV1, AAV3, and AAV5 serotypes showed some success in solving the problem of pre-existing immunity in animal models, application of this approach to humans will be limited by substantial pre-existing immunity directed against these serotypes (1,72).
Other recently discovered serotypes offer more promise, as for example Gao et al. have reported that only 6% of human sera are positive against AAV7 and AAV8 (5). In addition, pseudotyped AAV2/7 and AAV2/8 exhibited negligible neutralization following pre-administration of AAV2 vector (5). Although AAV7 and AAV8 may circumvent pre-existing B cell immunity in human, these vectors face other limitations. First, AAV also elicits a cytotoxic T lymphocyte response (CTL). Recent studies have shown that AAV2 and AAV8 capsids induce a CD8+ CTL response in a mouse model (145,146). Importantly, although AAV2 and AAV8 may not share B cell epitopes, prior administration of AAV2 activated T cells that could mount an immune response against AAV8 due to similar T cell epitopes present in both capsids. In an importat recent study, Mingozzi et al. extended this concept to human subjects. They showed that human T cells stimulated by exposure to rAAV2 in a clinical trial activate and proliferate upon exposure to alternate AAV serotypes (AAV1 and AAV8), which could greatly limit the utility of alternate AAV serotypes in overcoming pre-existing immunity to human AAV serotypes (147). Transient immunosuppression offers the potential to overcome pre-existing T cell immunity. However, finding immunologically “orthogonal” AAV serotypes that can evade pre-existing B cell and T cell immunity, as well as offer the possibility for repeat administration, remains a challenge.
Disruption of Epitopes
Progress has been made in engineering AAV to overcome antibody neutralization, in particular by mutating the viral capsid to alter neutralizing epitopes and thereby reduce antibody affinity for the vector. However, rational design of vector mutants for reduced neutralization requires extensive knowledge of the locations of neutralizing epitopes, as well as how to mutate the virus without compromising viral infectivity. Several studies have mapped epitopes of AAV2 NABs. First, Moskalenko et al. identified seven regions of the capsid as epitopes for human polyclonal NABs, using enzyme-linked immunosorbent assay (ELISA) and a capsid peptide scan library (44). Second, Wobus et al. mapped several linear or conformational eptitopes of AAV2 for three AAV2-specific murine neutralizing monoclonal antibodies by using a gene fragment phage display library, ELISA, peptide scans, and peptide competition experiments (47).
In order to circumvent antibody neutralization, AAV must be mutated to disrupt these and likely other epitopes while maintaining the numerous viral functions required for cellular infection and gene delivery, including receptor binding. Opie et al. found that residues 484, 487, 532, in particular, 585, and 588 are important for AAV binding to its primary receptor, HSPG, on surface of cells (148). Wu et al. have also shown that HSPG binding is greatly affected by peptide insertion or mutation at five regions of capsid protein: positions 509, 522, 561–565, 585–588, and 591 (99). These residues do not overlap with most of identified NAB epitopes (44,47). Therefore, guided by knowledge of resides to preserve to maintain receptor binding, several research groups have attempted to disrupt epitopes via capsid mutation, though such mutations may compromise other aspects of AAV transduction. Recently, Lochrie et al. have identified positions of AAV2 capsid proteins that affect transduction and antibody neutralization by using site-directed point mutagenesis (149). Two point mutations, S264A and V708K, exhibited 60-, and 220-fold enhanced resistance to neutralization by a monoclonal antibody, respectively, with capsid formation and transduction efficiency comparable to wild-type AAV2. Residues 264 and 708 are inside or very close to epitopes previously implicated in neutralization by the monoclonal antibody A20 (44,47), but they are distant from the residues critical for HSPG binding in primary capsid sequence (148). Huttner et al. found that insertion of L14 peptide at position 534 and 573 of AAV2 capsid protein, variants previously generated for targeted gene delivery (98), serendipitously also resulted in up to 70% lower neutralization by AAV NAB as compared to wild-type AAV (150). However, these mutant AAV2s exhibited significantly lower infectious titers.
Library Selection and Directed Evolution Approaches
Epitope mapping and the determination of the AAV crystal structure (44,47,97) could be very helpful to aid in rational modification of capsid proteins to address the problem of pre-exiting immunity. However, rational design of capsid proteins is still challenging due to a) numerous epitopes that must be modified, coupled with b) complex mechanisms of viral infection that can readily be disrupted by capsid modification. Therefore, high-throughput screening/selection of capsid mutants from AAV library is a promising strategy to address the issue of NAB evasion.
Maheshri et al. performed directed evolution of AAV to create vectors that evade antibody neutralization (116). Directed evolution is defined by iterative rounds of library generation, selection, generation of a new library, selection, etc. Directed evolution, unlike a single round of library generation and selection, therefore allows for iterative improvement of function, similar to the process by which nature evolves new biological entities and functionalities. In this report, large mutant AAV capsid library was prepared by using error-prone PCR followed by the staggered extension process. The AAV library was then incubated with pre-immunized rabbit sera, and viruses that evaded serum neutralization and were thus able to infect cells in culture were amplified. After two rounds of mutagenesis and selection, several resulting AAV capsid mutants packaged at titers equal to wild-type AAV but exhibited 1–3 orders of magnitude higher resistance to NABs in vivo compared to the wild-type capsid (116). The most effective mutant r2.15 has five mutations: E12A, K258N, T567S, N587I, and T716A. K258 and T716 overlap with epitopes for polyclonal antibodies directed against AAV2 (44). T567 is inside an epitope for the neutralizing monoclonal antibody A20 (47), and N587 lies in the HSPG binding domain, into which insertion of L14 peptide previously provided moderate resistance to antibody neutralization (150).
Perabo et al. also constructed a AAV capsid mutant library, which was subjected to selection against human sera neutralizing AAV2 (151). They performed a single round of library generation and selection and yielded a clone carrying two mutations (R459D and N551D) that exhibited 5.5-fold higher resistance against pre-immunized human sera compared to the wild-type AAV capsid. Residues 459 and 551 are located in close proximity in the three-dimensional structure of the viral particle and are exposed on the capsid surface. Residue 459 overlaps with a NAB epitope. Interestingly, the N551D mutation may be related to the T550K mutation, which was found by site-directed mutations on capsid surface and results in 3-fold resistance to intravenous IgG neutralization compared to wild-type AAV2 (149). These approaches show promise for the creation of ‘designer’ gene delivery vectors with enhanced properties via directed evolution (116) or single round library selection (151).
FUTURE DIRECTIONS
AAV has been extensively investigated as a promising gene delivery vector. In this review, we focused on recent advances in designing AAV vectors to change tropism, enhance delivery efficiency, and evade antibody neutralization. However, there are still several problems to be addressed. First, targeted gene delivery to specific cell types or even organs is still challenging. Recently, targeted gene delivery to vascular tissue in vivo was attempted via insertion of vascular endothelial cell targeting ligands into the AAV capsid (108,152). More recently, AAV2 libraries displaying random peptides were intravenously injected into mice, and the resulting distribution of AAV variants in the vasculature of different organs was evaluated (107). In the future, further molecular engineering of more selective and efficient AAV vectors to target organs or cell types can be anticipated. Second, the transgene carrying capacity of AAV vector is limited. Dong et al. reported that the optimum size of AAV2 vector genomes is between 4.1 and 4.9 kb (48), which restricts delivery of larger transgenes. Grieger et al. found out that proteasomal degradation after cell entry is responsible for poor infectivity of AAV carrying larger genomes (153). Therefore, addition of proteasome inhibitors led to efficient transduction of AAVs containing genomes as large as 6 kb. As an alternative approach, trans-splicing of AAV vectors have been developed (154–156). This approach requires splitting a large gene up to 10 kb into two segments that contain either intron donor or acceptor and coadministration of two vectors into target cells, ultimately leading to functional mRNA formation inside cells. Although this approach shows promise for doubling transgene packaging capacity, it suffers from low trans-splicing efficiency (157). Finally, directed evolution promises to provide novel approaches to overcome many challenges with AAV. It can address the problem of antibody neutralization; however, the challenge of pre-existing T cell immunity remains (147). However, advances in knowledge of AAV infection mechanisms and in novel molecular engineering approaches for AAV vectors promise to yield new, enhanced AAV gene delivery vectors.
References
K. I. Bernsand and R. M. Linden. The cryptic life style of adeno-associated virus. Bioessays 17:237–245 (1995).
K. Erles, P. Sebokova, and J. R. Schlehofer. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV). J. Med. Virol. 59:406–411 (1999).
X. A. Xiao, J. A. Li, and R. J. Samulski. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol. 70:8098–8108 (1996).
K. J. Fisher, K. Jooss, J. Alston, et al. Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med. 3:306–312 (1997).
G. P. Gao, M. R. Alvira, L. L. Wang, et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U. S. A. 99:11854–11859 (2002).
G. P. Gao, L. H. Vandenberghe, M. R. Alvira, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78:6381–6388 (2004).
W. D. Xiao, N. Chirmule, S. C. Berta, et al. Gene therapy vectors based on adeno-associated virus type 1. J. Virol. 73:3994–4003 (1999).
T. Vandendriessche, L. Thorrez, A. Acosta-Sanchez, et al. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J. Thromb. Haemost. 5:16–24 (2007).
S. F. C. Vaessen, R. J. Veldman, E. M. Comijn, et al. Optimization of adeno-associated virus mediated apolipoprotein A-1 gene delivery to the murine liver to assess effects on atherosclerosis. Atheroscler. Suppl. 7:95–95 (2006).
J. Y. N. Lauand and N. Muzyczka. Adeno-associated virus (AAV) as a gene delivery vector for liver cells. Hepatology 26:273–273 (1997).
S. Ponnazhagan, P. Mukherjee, M. C. Yoder, et al. Adeno-associated virus 2-mediated gene transfer in vivo: organ-tropism and expression of transduced sequences in mice. Gene 190:203–210 (1997).
D. D. Koeberl, I. E. Alexander, C. L. Halbert, et al. Persistent expression of human clotting factor IX from mouse liver after intravenous injection of adeno-associated virus vectors. Proc. Natl. Acad. Sci. U. S. A. 94:1426–1431 (1997).
T. J. McCown, X. Xiao, J. Li, et al. Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res. 713:99–107 (1996).
B. Du, P. Wu, D. M. BoldtHoule, et al. Efficient transduction of human neurons with an adeno-associated virus vector. Gene Ther. 3:254–261 (1996).
M. G. Kaplitt, P. Leone, R. J. Samulski, et al. Long-term gene-expression and phenotypic correction using adenoassociated virus vectors in the mammalian brain. Nat. Genet. 8:148–154 (1994).
C. M. Defiebre, P. Wu, D. Notabartolo, et al. Differential adenoassociated virus vector-driven expression of a neuropeptide-y gene in primary rat-brain astroglial cultures after transfection with sendai virosomes versus lipofectin(Tm). Neurochem. Res. 19:643-648 (1994).
C. K. Conrad, S. S. Allen, S. A. Afione, et al. Safety of single-dose administration of an adenoassociated virus (AAV)-CFTR vector in the primate lung. Gene Ther. 3:658–668 (1996).
C. L. Halbert, T. A. Standaert, C. B. Wilson, et al. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J. Virol. 72:9795–9805 (1998).
C.L. Halbert, T.A. Standaert, M.L. Aitken, et al. Transduction by adeno-associated virus vectors in the rabbit airway: Efficiency, persistence, and readministration. J. Virol. 71:5932-5941 (1997).
C. L. Halbert, J. M. Allen, A. D. Miller. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J. Virol. 75:6615–6624 (2001).
C. A. Grant, S. Ponnazhagan, X. S. Wang, et al. Evaluation of recombinant adeno-associated virus as a gene transfer vector for the retina. Curr. Eye Res. 16:949–956 (1997).
R. R. Ali, M. B. Reichel, A. J. Thrasher, et al. Gene transfer into the mouse retina mediated by an adeno-associated viral vector. Hum. Mol. Genet. 5:591–594 (1996).
R. R. Ali, M. B. Reichel, M. De Alwis, et al. Adeno-associated virus gene transfer to mouse retina. Hum. Gene Ther. 9:81–86 (1998).
J. Bennett, D. S. Duan, J. F. Engelhardt, et al. Real-time, noninvasive in vivo assessment of adeno-associated virus-mediated retinal transduction. Invest. Ophthalmol. Vis. Sci. 38:2857–2863 (1997).
M. Allocca, A. Tessitore, G. Cotugno, et al. AAV-mediated gene transfer for retinal diseases. Expert Opin. Biol. Ther. 6:1279–1294 (2006).
D. Chu, C. C. Sullivan, M. D. Weitzman, et al. Direct comparison of efficiency and stability of gene transfer into the mammalian heart using adeno-associated virus versus adenovirus vectors. J. Thorac. Cardiovasc. Surg. 126:671–679 (2003).
H. C. Champion, D. Georgakopoulos, S. Haldar, et al. Robust adenoviral and adeno-associated viral gene transfer to the in vivo murine heart—Application to study of phospholamban physiology. Circulation 108:2790–2797 (2003).
Z. Wang, T. Zhu, C. P. Qiao, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 23:321–328 (2005).
Z. Wang, T. Zhu, K. K. Rehman, et al. Widespread and stable pancreatic gene transfer by adeno-associated virus vectors via different routes. Diabetes 55:875–884 (2006).
S. A. Loiler, Q. S. Tang, T. Clarke, et al. Localized gene expression following administration of adeno-associated viral vectors via pancreatic ducts. Mol. Ther. 12:519–527 (2005).
A. Y. Wang, P. D. Peng, A. Ehrhardt, et al. Comparison of adenoviral and adeno-associated viral vectors for pancreatic gene delivery in vivo. Hum. Gene Ther. 15:405–413 (2004).
Z. J. Wu, A. Asokan, R. J. Samulski. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 14:316–327 (2006).
B. C. Schnepp, K. R. Clark, D. L. Klemanski, et al. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J. Virol. 77:3495–3504 (2003).
N. Dutheil, F. Shi, T. Dupressoir, et al. Adeno-associated virus site-specifically integrates into a muscle-specific DNA region. Proc. Natl. Acad. Sci. U. S. A. 97:4862–4866 (2000).
C. Balague, M. Kalla, W. W. Zhang. Adeno-associated virus Rep78 protein and terminal repeats enhance integration of DNA sequences into the cellular genome. J. Virol. 71:3299–3306 (1997).
R. T. Surosky, M. Urabe, S. G. Godwin, et al. Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J. Virol. 71:7951–7959 (1997).
H. Nakai, X. L. Wu, S. Fuess, et al. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J. Virol. 79:3606–3614 (2005).
B. L. Davidson, C. S. Stein, J. A. Heth, et al. Recombinant adeno-associated virus type 2, 4, and 5 vectors: Transduction of variant cell types and regions in the mammalian central nervous system. Proc. Natl. Acad. Sci. U. S. A. 97:3428–3432 (2000).
J. B. Nguyen, R. Sanchez-Pernaute, J. Cunningham, et al. Convection-enhanced delivery of AAV-2 combined with heparin increases TK gene transfer in the rat brain. Neuroreport 12:1961–1964 (2001).
J. R. Smith-Arica, A. J. Thomson, R. Ansell, et al. Infection efficiency of human and mouse embryonic stem cells using adenoviral and adeno-associated viral vectors. Cloning Stem Cells 5:51–62 (2003).
S. M. Hughes, F. Moussavi-Harami, S. L. Sauter, et al. Viral-mediated gene transfer to mouse primary neural progenitor cells. Mol. Ther. 5:16–24 (2002).
V. Cottard, C. Valvason, G. Falgarone, et al. Immune response against gene therapy vectors: Influence of synovial fluid on adeno-associated virus mediated gene transfer to chondrocytes. J. Clin. Immunol. 24:162–169 (2004).
H. Buning, M. U. Ried, L. Perabo, et al. Receptor targeting of adeno-associated virus vectors. Gene Ther. 10:1142–1151 (2003).
M. Moskalenko, L. L. Chen, M. van Roey, et al. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: Implications for gene therapy and virus structure. J. Virol. 74:1761–1766 (2000).
C. S. Peden, C. Burger, N. Muzyczka, et al. Circulating anti-wild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene transfer in the brain. J. Virol. 78:6344–6359 (2004).
J. Y. Sun, V. Anand-Jawa, S. Chatterjee, et al. Immune responses to adeno-associated virus and its recombinant vectors. Gene Ther. 10:964–976 (2003).
C. E. Wobus, B. Hugle-Dorr, A. Girod, et al. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: Epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 74:9281–9293 (2000).
J. Y. Dong, P. D. Fan, and R. A. Frizzell. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 7:2101–2112 (1996).
A. Srivastava, E. W. Lusby, K. I. Berns. Nucleotide-sequence and organization of the adeno-associated virus-2 genome. J. Virol. 45:555–564 (1983).
X. Xiao, J. Li, T. J. McCown, et al. Gene transfer by adeno-associated virus vectors into the central nervous system. Exp. Neurol. 144:113–124 (1997).
H. Bueler. Adeno associated viral vectors for gene transfer and gene therapy. Biol. Chem. 380:613–622 (1999).
P. L. Hermonat, M. A. Labow, R. Wright, et al. Genetics of adeno-associated virus—isolation and preliminary characterization of adeno-associated virus type-2 mutants. J. Virol. 51:329–339 (1984).
J. D. Tratschin, I. L. Miller, and B. J. Carter. Genetic-analysis of adeno-associated virus—properties of deletion mutants constructed invitro and evidence for an adeno-associated virus-replication function. J. Virol. 51:611–619 (1984).
Q. C. Yang, A. Kadam, and J. P. Trempe. Mutational analysis of the adenoassociated virus rep gene. J. Virol. 66:6058–6069 (1992).
C. W. Li, D. E. Bowles, T. van Dyke, et al. Adeno-associated virus vectors: potential applications for cancer gene therapy. Cancer Gene Ther. 12:913–925 (2005).
J. C. Grieger, V. W. Choi, and J. Samulski. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 1:1412–1428 (2006).
J. S. Bartlett, R. Wilcher, R. J. Samulski. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 74:2777–2785 (2000).
D. S. Duan, Q. Li, A. W. Kao, et al. Dynamin is required for recombinant adeno-associated virus type 2 infection. J. Virol. 73:10371–10376 (1999).
K. Qing, C. Mah, J. Hansen, et al. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 5:71–77 (1999).
C. Summerford, J. S. Bartlett, R. J. Samulski. alpha V beta 5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat. Med. 5:78–82 (1999).
J. S. Bartlett, R. J. Samulski, T. J. McCown. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum. Gene Ther. 9:1181–1186 (1998).
C. Summerfordand, and R. J. Samulski. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72:1438–1445 (1998).
T. Matsushita, S. Elliger, C. Elliger, et al. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 5:938–945 (1998).
F. W. Weindlerand, and R. Heilbronn. A subset of herpes-simplex virus-replication genes provides helper functions for productive adenoassociated virus-replication. J. Virol. 65:2476–2483 (1991).
W. D. Richardsonand and H. Westphal. Requirement for either early region-1a or early region-1b adenovirus gene-products in the helper effect for adeno-associated virus. J. Virol. 51:404–410 (1984).
J. E. Conway, S. Zolotukhin, N. Muzyczka, et al. Recombinant adeno-associated virus type 2 replication and packaging is entirely supported by a herpes simplex virus type 1 amplicon expressing Rep and Cap. J. Virol. 71:8780–8789 (1997).
K. I. Berns, T. C. Pinkerton, G. F. Thomas, et al. Detection of adeno-associated virus (Aav)-specific nucleotide-sequences in DNA isolated from latently infected detroit 6 cells. Virology 68:556–560 (1975).
R. M. Kotin, M. Siniscalco, R. J. Samulski, et al. Site-specific integration by adenoassociated virus. Proc. Natl. Acad. Sci. U. S. A. 87:2211–2215 (1990).
R. J. Samulski, L. S. Chang, and T. Shenk. Helper-free stocks of recombinant adeno-associated viruses: normal integration does not require viral gene expression. J. Virol. 63:3822–3828 (1989).
B. J. Carter. Adeno-associated virus vectors in clinical trials. Hum. Gene Ther. 16:541–550 (2005).
J. G. Flannery, S. Zolotukhin, M. I. Vaquero, et al. Efficient photoreceptor-targeted gene expression in vivo by recombinant adeno-associated virus. Proc. Natl. Acad. Sci. U. S. A. 94:6916–6921 (1997).
C. L. Halbert, E. A. Rutledge, J. M. Allen, et al. Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J. Virol. 74:1524–1532 (2000).
M. C. Senut, S. T. Suhr, B. Kaspar, et al. Intraneuronal aggregate formation and cell death after viral expression of expanded polyglutamine tracts in the adult rat brain. J. Neurosci. 20:219–229 (2000).
R. W. Atchison, B. C. Casto, W. M. Hammon. Adenovirus-associated defective virus particles. Science 149:754–756 (1965).
U. Bantelschaaland, and H. Z. Hausen. Characterization of the DNA of a defective human parvovirus isolated from a genital site. Virology 134:52–63 (1984).
D. Grimm, M. A. Kay, J. A. Kleinschmidt. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 7:839–850 (2003).
M. D. Hoggan, N. R. Blacklow, and W. P. Rowe. Studies of small DNA viruses found in various adenovirus preparations—physical biological and immunological characteristics. Proc. Natl. Acad. Sci. U. S. A. 55:1467–1474 (1966).
S. Mori, L. Wang, T. Takeuchi, et al. Two novel adeno-associated viruses from cynomolgus monkey: pseudotyping characterization of capsid protein. Virology 330:375–383 (2004).
W. P. Parks, M. Green, M. Pina, et al. Physicochemical characterization of adeno-associated satellite virus type 4 and its nucleic acid. Am. Soc. Microbiol. 1:980–987 (1967).
E. A. Rutledge, C. L. Halbert, and D. W. Russell. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J. Virol. 72:309–319 (1998).
M. Schmidt, E. Grot, P. Cervenka, et al. Identification and characterization of novel adeno-associated virus isolates in ATCC virus stocks. J. Virol. 80:5082–5085 (2006).
D. Grimm, K. Pandey, H. Nakai, et al. Liver transduction with recombinant adeno-associated virus is primarily restricted by capsid serotype not vector genotype. J. Virol. 80:426–439 (2006).
H. J. Chao, Y. B. Liu, J. Rabinowitz, et al. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol. Ther. 2:619–623 (2000).
J. M. Alisky, S. M. Hughes, S. L. Sauter, et al. Transduction of murine cerebellar neurons with recombinant FIV and AAV5 vectors. Neuroreport 11:2669–2673 (2000).
M. J. Blankinship, P. Gregorevic, J. M. Allen, et al. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol. Ther. 10:671–678 (2004).
C. A. Pacak, C. S. Mah, B. D. Thattaliyath, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res. 99:E3–E9 (2006).
S. Mori, T. Takeuchi, Y. Enomoto, et al. Biodistribution of a low dose of intravenously administered AAV-2, 10, and 11 vectors to cynomolgus monkeys. Jpn. J. Infect. Dis. 59:285–293 (2006).
B. Hauck, L. Chen, and W. D. Xiao. Generation and characterization of chimeric recombinant AAV vectors. Mol. Ther. 7:419–425 (2003).
J. E. Rabinowitz, D. E. Bowles, S. M. Faust, et al. Cross-dressing the virion: the transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J. Virol. 78:4421–4432 (2004).
M. U. Ried, A. Girod, K. Leike, et al. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J. Virol. 76:4559–4566 (2002).
L. Gigout, P. Rebollo, N. Clement, et al. Altering AAV tropism with mosaic viral capsids. Mol. Ther. 11:856–865 (2005).
M. D. Stachler, and J. S. Bartlett. Mosaic vectors comprised of modified AAV1 capsid proteins for efficient vector purification and targeting to vascular endothelial cells. Gene Ther. 13:926–931 (2006).
J. S. Bartlett, J. Kleinschmidt, R. C. Boucher, et al. Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab’gamma)2 antibody. Nat. Biotechnol. 17:181–186 (1999).
S. Ponnazhagan, G. Mahendra, S. Kumar, et al. Conjugate-based targeting of recombinant adeno-associated virus type 2 vectors by using avidin-linked ligands. J. Virol. 76:12900–12907 (2002).
G. S. Arnold, A. K. Sasser, M. D. Stachler, et al. Metabolic biotinylation provides a unique platform for the purification and targeting of multiple AAV vector serotypes. Mol. Ther. 14:97–106 (2006).
Q. C. Yang, M. Mamounas, G. Yu, et al. Development of novel cell surface CD34-targeted recombinant adenoassociated virus vectors for gene therapy. Hum. Gene Ther. 9:1929–1937 (1998).
Q. Xie, W. Bu, S. Bhatia, et al. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. U. S. A. 99:10405–10410 (2002).
A. Girod, M. Ried, C. Wobus, et al. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 5:1052–1056 (1999).
P. Wu, W. Xiao, T. Conlon, et al. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 74:8635–8647 (2000).
S. A. Loiler, T. J. Conlon, S. Song, et al. Targeting recombinant adeno-associated virus vectors to enhance gene transfer to pancreatic islets and liver. Gene Ther. 10:1551–1558 (2003).
G. A. Pieterszand, and I. F. C. McKenzie. Antibody conjugates for the treatment of cancer. Immunol. Rev. 129:57–80 (1992).
G. P. Smith. Filamentous fusion phage—novel expression vectors that display cloned antigens on the virion surface. Science 228:1315–1317 (1985).
R. Cortese, P. Monaci, A. Luzzago, et al. Selection of biologically active peptides by phage display of random peptide libraries. Curr. Opin. Biotechnol. 7:616–621 (1996).
R. Pasqualiniand, and E. Ruoslahti. Organ targeting in vivo using phage display peptide libraries. Nature 380:364–366 (1996).
M. Grifman, M. Trepel, P. Speece, et al. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol. Ther. 3:964–975 (2001).
S. A. Nicklin, H. Buening, K. L. Dishart, et al. Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells. Mol. Ther. 4:174–181 (2001).
L. Perabo, D. Goldnau, K. White, et al. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J. Virol. 80:7265–7269 (2006).
L. M. Work, H. Buning, E. Hunt, et al. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol. Ther. 13:683–693 (2006).
L. Perabo, H. Buning, D. M. Kofler, et al. In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Mol. Ther. 8:151–157 (2003).
O. J. Muller, F. Kaul, M. D. Weitzman, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 21:1040–1046 (2003).
A. Rothe, R. J. Hosse, and B. E. Power. In vitro display technologies reveal novel biopharmaceutics. FASEB J. 20:1599–1610 (2006).
H. R. Hoogenboom. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 23:1105–1116 (2005).
L. Yuan, I. Kurek, J. English, et al. Laboratory-directed protein evolution. Microbiol. Mol. Biol. Rev. 69:373–392 (2005).
K. Bupp, A. Sarangi, and M. J. Roth. Selection of feline leukemia virus envelope proteins from a library by functional association with a murine leukemia virus envelope. Virology 351:340–348 (2006).
J. H. Yuand, and D. V. Schaffer. High-throughput, library-based selection of a murine leukemia virus variant to infect nondividing cells. J. Virol. 80:8981–8988 (2006).
N. Maheshri, J. T. Koerber, B. K. Kaspar, et al. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat. Biotechnol. 24:198–204 (2006).
W. C. Manning, S. Zhou, M. P. Bland, et al. Transient immunosuppression allows transgene expression following readministration of adeno-associated viral vectors. Hum. Gene Ther. 9:477–485 (1998).
C. Riviere, O. Danos, and A. M. Douar. Long-term expression and repeated administration of AAV type 1, 2 and 5 vectors in skeletal muscle of immunocompetent adult mice. Gene Ther. 13:1300–1308 (2006).
D. R. Burton, R. A. Williamson, and P. Parren. Antibody and virus: Binding and neutralization. Virology 270:1–3 (2000).
D. R. Burton, E. O. Saphire, and P. Parren. A model for neutralization of viruses based on antibody coating of the virion surface. Antib. Viral Infect. 260:109–143 (2001).
P. Parren, P. A. Marx, A. J. Hessell, et al. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J. Virol. 75:8340–8347 (2001).
P. J. Klasseand, and Q. J. Sattentau. Mechanisms of virus neutralization by antibody. Antib. Viral Infect. 260:87–108 (2001).
E. O. Saphire, R. L. Stanfield, M. D. M. Crispin, et al. Contrasting IgG structures reveal extreme asymmetry and flexibility. J. Mol. Biol. 319:9–18 (2002).
M. Marquart, J. Deisenhofer, R. Huber, et al. Crystallographic refinement and atomic models of the intact immunoglobulin molecule kol and its antigen-binding fragment at 3.0-a and 1.9-a resolution. J. Mol. Biol. 141:369–391 (1980).
N. Chirmule, W. D. Xiao, A. Truneh, et al. Humoral immunity to adeno-associated virus type 2 vectors following administration to murine and nonhuman primate muscle. J. Virol. 74:2420–2425 (2000).
H. Y. Jiang, L. B. Couto, S. Patarroyo-White, et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood 108:3321–3328 (2006).
D. H. Doherty, M. S. Rosendahl, D. J. Smith, et al. Site-specific PEGylation of engineered cysteine analogues of recombinant human granulocyte-macrophage colony-stimulating factor. Bioconjug. Chem. 16:1291–1298 (2005).
G. Molineux. The design and development of pegfilgrastim (PEG-rmetHuG-CSF, neulasta((R))). Curr. Pharm. Des. 10:1235–1244 (2004).
C. K. Edwards, S. W. Martin, J. Seely, et al. Design of PEGylated soluble tumor necrosis factor receptor type I (PEG sTNF-RI) for chronic inflammatory diseases. Adv. Drug Deliv. Rev. 55:1315–1336 (2003).
B. A. Luxon, M. Grace, D. Brassard, et al. Pegylated interferons for the treatment of chronic hepatitis C infection. Clin. Ther. 24:1363–1383 (2002).
G. Molineux. Pegylation: engineering improved pharmaceuticals for enhanced therapy. Cancer Treat. Rev. 28:13–16 (2002).
F. Malik, C. Delgado, C. Knusli, et al. Polyethylene-glycol (Peg)-modified granulocyte–macrophage colony-stimulating factor (Gm-Csf) with conserved biological-activity. Exp. Hematol. 20:1028–1035 (1992).
F. Kreppel, J. Gackowski, E. Schmidt, et al. Combined genetic and chemical capsid modifications enable flexible and efficient de- and retargeting of adenovirus vectors. Mol. Ther. 12:107–117 (2005).
Y. Eto, J. Q. Gao, F. Sekiguchi, et al. PEGylated adenovirus vectors containing RGD peptides on the tip of PEG show high transduction efficiency and antibody evasion ability. J. Gene Med. 7:604–612 (2005).
K. I. Ogawara, M. G. Rots, R. J. Kok, et al. A novel strategy to modify adenovirus tropism and enhance transgene delivery to activated vascular endothelial cells in vitro and in vivo. Hum. Gene Ther. 15:433–443 (2004).
G. P. Tang, J. M. Zeng, S. J. Gao, et al. Polyethylene glycol modified polyethylenimine for improved CNS gene transfer: effects of PEGylation extent. Biomaterials 24:2351–2362 (2003).
M. A. Croyle, N. Chirmule, Y. Zhang, et al. PEGylation of E1-deleted adenovirus vectors allows significant gene expression on readministration to liver. Hum. Gene Ther. 13:1887–1900 (2002).
R. Alemany, K. Suzuki, and D. T. Curiel. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 81:2605–2609 (2000).
M. A. Croyle, Q. C. Yu, and J. M. Wilson. Development of a rapid method for the PEGylation of adenoviruses with enhanced transduction and improved stability under harsh storage conditions. Hum. Gene Ther. 11:1713–1722 (2000).
C. R. O’Riordan, A. Lachapelle, C. Delgado, et al. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum. Gene Ther. 10:1349–1358 (1999).
H. T. Le, Q. C. Yu, J. M. Wilson, et al. Utility of PEGylated recombinant adeno-associated viruses for gene transfer. J. Control. Release 108:161–177 (2005).
G. K. Lee, N. Maheshri, B. Kaspar, et al. PEG conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol. Bioeng. 92:24–34 (2005).
M. Hildinger, A. Auricchio, G. Gao, et al. Hybrid vectors based on adeno-associated virus serotypes 2 and 5 for muscle-directed gene transfer. J. Virol. 75:6199–6203 (2001).
Z. Sandalon, E. M. Bruckheimer, K. H. Lustig, et al. Secretion of a TNFR : Fc fusion protein following pulmonary administration of pseudotyped adeno-associated virus vectors. J. Virol. 78:12355–12365 (2004).
J. Chen, Q. Wu, P. Yang, et al. Determination of specific CD4 and CD8 T cell epitopes after AAV2-and AAV8-hF.IX gene therapy. Mol. Ther. 13:260–269 (2006).
D. E. Sabatino, F. Mingozzi, D. J. Hui, et al. Identification of mouse AAV capsid-specific CD8+ T cell epitopes. Mol. Ther. 12:1023–1033 (2005).
F. Mingozzi, M. V. Maus, D. J. Hui, et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13:419–422 (2007).
S. R. Opie, K. H. Warrington, M. Aqbandje-McKenna, et al. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 77:6995–7006 (2003).
M. A. Lochrie, G. P. Tatsuno, B. Christie, et al. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J. Virol. 80:821–834 (2006).
N. A. Huttner, A. Girod, L. Perabo, et al. Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene Ther. 10:2139–2147 (2003).
L. Perabo, J. Endell, S. King, et al. Combinatorial engineering of a gene therapy vector: directed evolution of adeno-associated virus. J. Gene Med. 8:155–162 (2006).
S. J. White, S. A. Nicklin, H. Buning, et al. Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation 109:513–519 (2004).
J. C. Grieger, and R. J. Samulski. Packaging capacity of adeno-associated virus serotypes: Impact of larger genomes on infectivity and postentry steps. J. Virol. 79:9933–9944 (2005).
Z. Y. Yan, Y. L. Zhang, D. S. Duan, et al. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc. Natl. Acad. Sci. U. S. A. 97:6716–6721 (2000).
H. Nakai, T. A. Storm, and M. A. Kay. Increasing the size of rAAV-mediated expression cassettes in vivo by intermolecular joining of two complementary vectors. Nat. Biotechnol. 18:527–532 (2000).
L. W. Sun, J. Li, and X. Xiao. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat. Med. 6:599–602 (2000).
Y. Lai, Y. P. Yue, M. J. Liu, et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat. Biotechnol. 23:1435–1439 (2005).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kwon, I., Schaffer, D.V. Designer Gene Delivery Vectors: Molecular Engineering and Evolution of Adeno-Associated Viral Vectors for Enhanced Gene Transfer. Pharm Res 25, 489–499 (2008). https://doi.org/10.1007/s11095-007-9431-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-007-9431-0