
Abstract
Click chemistry refers to a group of reactions that are fast, simple to use, easy to purify, versatile, regiospecific, and give high product yields. While there are a number of reactions that fulfill the criteria, the Huisgen 1,3-dipolar cycloaddition of azides and terminal alkynes has emerged as the frontrunner. It has found applications in a wide variety of research areas, including materials sciences, polymer chemistry, and pharmaceutical sciences. In this manuscript, important aspects of the Huisgen cycloaddition will be reviewed, along with some of its many pharmaceutical applications. Bioconjugation, nanoparticle surface modification, and pharmaceutical-related polymer chemistry will all be covered. Limitations of the reaction will also be discussed.
Similar content being viewed by others
INTRODUCTION
In the field of pharmaceutical science, researchers are constantly seeking for new molecules and constructs that exhibit specific properties. While one could easily come up with a structure design that would fit the needs, the real struggle lies in its synthesis and purification. If only molecular synthesis were as simple as building a LEGO® castle or assembling an IKEA® desk. If only readily available structure units could be easily linked together to form numerous molecules of desire in just a few crisp steps. While it still remains a far-reaching dream, the new concept of click chemistry seems to offer a glimpse of hope.
Since being first introduced by Dr. Barry Sharpless’ group in 1999 at the 217th American Chemical Society annual meeting, click chemistry has become a very popular topic, as evidenced by a nearly exponential growth in the amount of related publications. A literature search via SciFinder Scholar®, performed on December 31st of 2007, revealed a total of 788 publications containing the keywords “click chemistry” or “click reaction”, which included journal articles, reviews, preprints, abstracts, patents, and dissertations. As shown in Fig. 1, publications in this area have quickly increased over the past 4 years.

Number of publications containing the key words “click chemistry” or “click reaction” from 1999–2007. The literature search was performed via SciFinder Scholar® on Dec. 31, 2007 and included journal articles, abstracts, preprints, dissertations, patents, and reviews. The years 1999 and 2000 each contain one publication.
In his landmark review in 2001, Dr. Sharpless defined click chemistry as a group of reactions that “…must be modular, wide in scope, give very high yields, generate only inoffensive byproducts that can be removed by nonchromatographic methods, and be stereospecific (but not necessarily enantioselective). The required process characteristics include simple reaction conditions (ideally, the process should be insensitive to oxygen and water), readily available starting materials and reagents, the use of no solvent or a solvent that is benign (such as water) or easily removed, and simple product isolation. Purification, if required, must be by nonchromatographic methods, such as crystallization or distillation, and the product must be stable under physiological conditions” (1). The identification of this group of reactions was a direct result of Mother Nature’s strategy of achieving astonishing biological diversity from a very limited number of monomers (i.e. proteins from amino acids, nucleic acids from nucleotides, etc.). Puzzled by the slowness and inefficiency of the conventional drug discovery process, Dr. Sharpless proposed a new tactic, one in where large combinatorial libraries could be easily prepared by linking available building blocks via click reactions. The rule of thumb for this approach was that “…all searches must be restricted to molecules that are easy to make” (1).
Although this novel philosophy for drug discovery is very appealing, the medicinal chemists in this field seem to be hesitant. As shown in Fig. 2, among all the publications identified through SciFinder Scholar®, only 14% are drug discovery related. Interestingly, applications of click reactions in polymer sciences are warmly welcomed and considered as an immediate success. Biomedical applications of click chemistry, especially in pharmaceutical sciences, are also emerging as a field of great interest. One reason is in many areas of research, such as drug delivery and nanomedicine, linker chemistry plays a pivotal role. Biological therapeutics needs to be tagged with probes for the convenience of detection and evaluation. Nanoscale delivery vehicles need to be assembled to provide drug transportation in vivo. Targeting moieties and drug(s) need to be attached or loaded onto delivery systems. These are very tedious tasks as there are many constraints on performing successful linker chemistry. For instance, they are typically performed in water. Furthermore, the reaction conditions are preferred to be mild so that biologicals and other fragile structures present will not loss their functions. Protections and deprotections also need to be considered to avoid unwanted side reactions. Click chemistry, through its unique features, easily satisfies all of these constraints. It has repeatedly shown to serve the needs of the pharmaceutical community exceedingly well, despite the fact that it was intended to serve an entirely different purpose. In this review, important aspects of click chemistry will be discussed, along with its emerging applications in drug delivery and nanomedicine. We will also provide some of our concerns and vision for its future development in pharmaceutical sciences. Several reviews on click chemistry have already been published (1–3). The readers may find them helpful in understanding other aspects of click chemistry or in obtaining more background information.

Major classifications of the applications of click chemistry. Analysis was performed based on a literature search via SciFinder Scholar® on Dec. 31, 2007. The search included journal articles, abstracts, preprints, dissertations, patents, and reviews. The field of drug discovery was considered separate from pharmaceutical and the category “other” contains miscellaneous applications that cannot be grouped into the other three categories, such as applications in materials sciences, certain reviews, and novel methods for improved catalysts. Many applications are subjective in that they can fall into more than one category (i.e. the synthesis of certain block copolymers could be considered both pharmaceutical and polymer-related). In these cases, the applications were categorized as pharmaceutical if and only if their corresponding abstracts specifically mentioned that the involved compound(s) could play a role in a pharmaceutical setting. It is intended for this chart to represent a general overview and should not be taken literally.
CLASSIFICATION OF CLICK REACTIONS
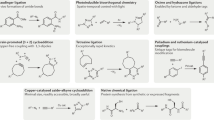
As already implicated, click chemistry encompasses a group of powerful linking reactions that are simple to perform, have high yields, require no or minimal purification, and are versatile in joining diverse structures without the prerequisite of protection steps. To date, four major classifications of click reactions have been identified (Fig. 3).
-
Cycloadditions—these primarily refer to 1,3-dipolar cycloadditions, but also include hetero-Diels-Alder cycloadditions (2).
-
Nucleophilic ring-openings—these refer to the openings of strained heterocyclic electrophiles, such as aziridines, epoxides, cyclic sulfates, aziridinium ions, episulfonium ions, etc. (2).
-
Carbonyl chemistry of the non-aldol type—examples include the formations of ureas, thioureas, hydrazones, oxime ethers, amides, aromatic heterocycles, etc. (1). Carbonyl reactions of the aldol type generally have low thermodynamic driving forces, hence they have longer reaction times and give side products, and therefore cannot be considered click reactions (1).
-
Additions to carbon-carbon multiple bonds—examples include epoxidations, aziridinations, dihydroxylations, sulfenyl halide additions, nitrosyl halide additions, and certain Michael additions (1,2).

Major classifications of click chemistry reactions, along with corresponding examples. Nu Nucleophile; EWG electron withdrawing group.
Among the four major classifications, cycloadditions, particularly the CuI-catalyzed Huisgen 1,3-dipolar cycloaddition (HDC) of azides and terminal alkynes to form 1,2,3-triazoles (4), are the most widely used. Based on the literature search mentioned earlier, nearly 100% of the publications referred to this click reaction, which has found applications across many diverse research areas. In the following sections, the potential of this click reaction and its pharmaceutical applications will be reviewed.
CuI-CATALYZED HUISGEN 1,3-DIPOLAR CYCLOADDITION OF AZIDES AND TERMINAL ALKYNES
The CuI-catalyzed Huisgen 1,3-dipolar cycloaddition of azides and terminal alkynes to form 1,2,3-triazoles is the model example of a click reaction (Fig. 3). It fulfills all of the criteria of click chemistry perfectly, no matter how subjective they may be, and is therefore extremely reliable and easy to use. This reaction exclusively forms 1,4-substituted products, making it regiospecific. It typically does not require temperature elevation but can be performed over a wide range of temperatures (0–160°C), in a variety of solvents (including water), and over a wide range of pH values (5 through 12). It proceeds as much as 107 times faster than the uncatalyzed version, and purification essentially consists of product filtration (3,5–7). Furthermore, it is unaffected by steric factors. “Variously substituted primary, secondary, tertiary, and aromatic azides readily participate in this transformation. Tolerance for variations in the acetylene component is also excellent” (6). All of these characteristics make this cycloaddition particularly popular among the other click reactions described above.
Two additional reasons for the popularity of this cycloaddition are azides and terminal alkynes are fairly easy to install and they are extremely stable at standard conditions (2,8). They both can tolerate oxygen, water, common organic synthesis conditions, biological molecules, a large range of solvents and pH’s, and the reaction conditions of living systems (reducing environment, hydrolysis, etc.) (2,3,9). Even though the decomposition of aliphatic azides is thermodynamically favored, a kinetic barrier exists that allows them to be stable in the aforementioned conditions (3). They will essentially remain “invisible” in solution until a dipolarophile, such as an alkyne, comes into contact (3).
Mechanism of HDC Reaction
In general, cycloadditions proceed through a concerted mechanism. However, experimental kinetic data (10) and molecular modeling (7) performed on the HDC reaction seem to favor a stepwise reaction pathway (3,5). It has been calculated that the activation barrier for a catalyzed concerted HDC reaction is actually greater than that for an uncatalyzed concerted reaction (27.8 kcal/mol vs. 26 kcal/mol in one particular reaction using density functional theory calculations) (7). Furthermore, a stepwise-catalyzed HDC reaction has an activation barrier 11 kcal/mol lower than a concerted catalyzed reaction (3).
Based on experimental evidence (5,6) and the fact that CuI can readily insert itself into terminal alkynes [Sonogashira coupling (11)], it is envisioned that the first step of the reaction involves π complexation of a CuI dimer to the alkyne (1 in Fig. 4). Thereafter, deprotonation of the terminal hydrogen occurs to form a Cu-acetylide (5). There are actually several different kinds of Cu-acetylide complexes that can form, depending on the reaction conditions utilized; 2 represents just one possibility (7). The π complexation of CuI lowers the pKa of the terminal alkyne by as much as 9.8 pH units, allowing deprotonation to occur in an aqueous solvent without the addition of a base (3). If a non-basic solvent such as acetonitrile was to be used, then a base, such as 2,6-lutidine or N,N′-diisopropylethylamine (DIPEA), would have to be added (12).

Proposed mechanism for the HDC reaction. Ligands are represented by “L” and symbolize a wide variety of possible compounds, depending on the catalyst used. As an example, if CuBr was used as the catalyst then the ligand would be bromide. Figure adapted from reference 3 with permission from Wiley-VCH.
In the following step, N(1) displaces one of the ligands from the second Cu in the Cu-acetylide complex to form 3. In turn, this “activates” the azide for nucleophilic attack C(5). Due to proximity and electronic factors, N(3) can now easily attack C(4) of the alkyne, leading to a metallocycle (not shown for simplicity). The metallocycle then contracts when the lone pair of electrons of N(1) attacks C(5) to form the respective triazole 4. Once 4 forms, the attached Cu dimer immediately complexes to a second terminal alkyne. However, this second alkyne cannot undergo a cycloaddition due to the unfavorable structure of the complex, and it dissociates upon protonation to reform 4. One final protonation releases the CuI catalyst from the 1,2,3-triazole product 5, to undergo a second catalytic cycle with different substrates (3). Both of these protonations are most likely the result of interactions with protonated external base and/or solvent, but further studies are needed to conclusively confirm (3).
Catalysts
There are a number of methods to generate the active catalyst for the HDC reaction. One of the most common techniques is to reduce CuII salts, such as CuSO4·5H2O, in situ to form CuI salts. Sodium ascorbate is typically used as the reducing agent in a 3- to 10-fold excess (3), but other reducing agents, including hydrazine (13) and tris(2-carboxyethyl)phosphine (TCEP) (9), have been used with reasonable success. The advantages of this strategy are it is cheap, it can be performed in water, and it does not require deoxygenated atmosphere (3,14). Not only does an aqueous solvent remove the need for a base, as previously explained, but it also eliminates the need for protecting groups (O–H and N–H functional groups essentially remain “invisible” in aqueous solutions) and it is environmentally safe (1). The main disadvantage is the reducing agent might reduce CuII down to Cu0. This can generally be prevented, though, by using a proper ratio of reducing agent to catalyst and/or adding a copper-stabilizing agent, such as tris-(hydroxypropyltriazolylmethyl)amine (THPTA) (3).
A second way to create the catalyst is to directly add CuI salts. Many such compounds have been utilized over the past few years, including CuBr, CuI, CuOTf·C6H6 (OTf = trifluoromethanesulfonate), [Cu(NCCH3)4][PF6], etc. (6). This method does not require a reducing agent, but it has to be done in a deoxygenated environment and in an organic solvent (or a mixed solvent), meaning that protection groups will probably be needed along with a base (14). It has been shown that using excess amounts of both the bases 2,6-lutidine and DIPEA produce the best results, causing the least amount of side products (6,12). Still, CuI salts are not as reliable as the CuII procedure (6).
Oxidizing copper metal with an amine salt is another way to generate the catalyst (7,14). There are a considerable number of disadvantages with this strategy. Longer reaction times are needed, as well as larger amounts of copper, it is more expensive, and requires a slightly acidic environment to dissolve the metal, which could be damaging to any acidic-sensitive functional groups present in the reactants (3). For more information on this method or any of the others mentioned thus far, refer to reference 3 for an exquisite review.
Recently, CuI-modified zeolites were reported as catalysts for the HDC reaction (15). “Zeolites” refers to a family of aluminosilicate minerals, occurring both naturally and synthetically, that are highly porous and therefore have large surface areas (16–18). They are particularly desired as catalysts because of their “…high concentration of active acid sites, their high thermal/hydrothermal stability, and high size selectivity” (18). In one particular HDC reaction, the zeolite USY, modified with CuI, exhibited far better results than CuCl. Its product yield was thirteen percent higher and its reaction time was roughly three times as short (15). Pending more research, this approach could prove to be very valuable.
Another research group has attempted to find a replacement for copper as the catalyst. Knowing that other transition metals can insert into alkynes (19,20), Golas et al. reacted propargyl ether with azide-terminated polystyrene to make polymers using three different catalyst: NiCl2, PtCl2, and PdCl2 (13). Although none of them displayed catalytic activity as high or as fast as CuBr, they all produced polymerization products. PtCl2 produced polymers with the highest molecular weight, followed by PdCl2, followed by NiCl2.
In 2005, pentamethyl cyclopentadienyl ruthenium (II) complexes (Cp*Ru), such as Cp*RuCl(PPh3)2, were discovered as novel catalysts for click chemistry. Contrary to all previously mentioned catalysts, Cp*Ru complexes afford only 1,5-substituted 1,2,3-triazoles (21). Furthermore, they can work on both terminal and internal alkynes alike (22). An example of such a reaction is shown in Fig. 5. Due to their recent discovery, though, limited information is available detailing the role of ruthenium(II) complexes in click chemistry, as evidenced by a search via SciFinder Scholar®.

Using a Cp*Ru catalyst in the click reaction exclusively forms 1,5-substituted 1,2,3-triazoles. These catalysts can also work on internal alkynes (not shown), contrary to all other known catalysts.
Finally, studies have shown that catalysts are not always required for the cycloaddition to proceed. By using electron deficient alkynes, the reaction can proceed readily at ambient conditions (23). However, electron deficient alkynes are very reactive toward nucleophiles and can lead to side products (24), which has kept these types of reactions estranged from the field of click chemistry. Other studies have shown that the cycloaddition can occur rapidly if the alkyne is first incorporated into an eight-member ring, forming a cyclooctyne (24–26). Cyclooctynes are very unstable, due to a high degree of ring strain (18 kcal/mol), which causes them to react readily with azides (24). While this method leaves no side products and requires no cytotoxic catalysts, it requires the prerequisite of connecting the alkyne of interest to an eight-member ring. This can be a very challenging task and is much more complicated than simply adding an acetylene functional group, as in traditional click reactions. An additional concern with the cyclooctyne method is it produces a racemic mixture of regioisomers (Fig. 6) (24). This stipulation is typically inconsequential to pharmaceutical applications, but it defies the very definition of click chemistry, which requires click reactions to be regiospecific.

Huisgen 1,3-dipolar cycloadditions of azides and alkynes without CuI catalyst. If an alkyne is first incorporated into an eight-member ring then a 1,3-dipolar cycloaddition of azides and alkynes can proceed rapidly without the aid of a catalyst. However, a racemic mixture of regioisomers is obtained.
APPLICATIONS OF CLICK CHEMISTRY IN PHARMACEUTICAL SCIENCES
Since its debut in 1999, click chemistry has stimulated enormous amount of interests in many different research fields, being utilized in everything from microelectronics to virus labeling to treatments for cancer. In the following sections, an in-depth look will be taken at some of its applications pertaining to the field of pharmaceutical sciences. The area of drug discovery will be excluded since multiple review papers on this subject already exist (2,27–29).
Polymer Therapeutics and Click Chemistry
“‘Polymer therapeutics’ is an umbrella term used to describe polymeric drugs, polymer-drug conjugates, polymer-protein conjugates, polymeric micelles to which drug is covalently bound, and multi-component polyplexes that are being developed as non-viral vectors. All subclasses use specific water-soluble polymers, either as the bioactive itself or as an inert functional part of a multifaceted construct for improved drug, protein, or gene delivery” (30). While polymer therapeutics have been used to treat numerous diseases, they are particularly effective against those that have an enhanced permeability and retention (EPR) effect (31), such as rheumatoid arthritis (32) and solid tumors (33). Non-viral gene therapy formulations based on synthetic polymers have been extensively investigated and have shown better safety profiles than viral gene vehicles, though their gene transfection efficacy is relatively low (34). Conjugation of biocompatible polymers, such as poly(ethylene glycol) (PEG), to therapeutic proteins and peptides has proved to be a very effective strategy in reducing immunogenicity and increasing bioavailability (35,36). Clearly, biocompatible polymers have played vital roles in modern pharmaceutical sciences, especially in drug delivery and formulations. Nevertheless, simpler and more efficient polymer chemistry is needed to meet the ever-increasing demand for highly diverse pharmaceutical properties.
As explained in previous sections, click chemistry was initially developed as a drug discovery tool. However, its most successful applications thus far have been in the field of polymer chemistry. As seen in Fig. 2, over one third of all publications containing the keywords “click chemistry” or “click reaction” are related to polymer synthesis and/or modification. Generally, these publications can be classified into five broad categories: block copolymer synthesis, linear multifunctional copolymer synthesis, dendrimer synthesis, polymer network synthesis, and polymer analogous modification. In addition to significantly improving product yields, most of these click chemistry applications drastically simplified the synthetic routes and purification procedures. Therefore, it is the belief of the authors that this new “tool in the box” may shift the paradigm of polymer synthesis and lead to new strategies of polymer therapeutics development.
Synthesis of Block Copolymers with Click Reaction
Biocompatible amphiphilic block copolymers have many pharmaceutical applications. They have been extensively used in the formulation of various nanoparticulate structures, such as micelles, nanospheres, nanocapsules, polymersomes, etc. (37–39). Typically, block copolymers are synthesized via two routes: (A) sequential addition of different monomers into polymerizations containing living reaction centers (40,41). Living ionic polymerizations, atom transfer free radical polymerizations (ATRP), reversible addition fragmentation chain transfer (RAFT) polymerizations, ring-opening polymerizations (ROP), or their combination have all been utilized to obtain well-defined block copolymers of different components. (B) Linking different linear polymer chains via their terminal functionalities. While the latter method allows the combination of polymer blocks that may not be compatible with the first, the lack of efficient linker chemistry has made this route rarely used.
The emergence of click chemistry drastically changed the scientific community’s views on block copolymer synthesis. Because of its extremely high reaction efficiency and tolerance to a variety of functional groups, click chemistry has become the hallmark of linker chemistry. It is one of the most efficient ways to join two substances together and has thus been used repeatedly to link well-defined homopolymers to form block copolymers. Recently, Van Camp et al. reported a synthetic strategy for diverse amphiphilic copolymer structures by combination of ATRP and the HDC reaction. Using a modular approach, polymers with alkyne functionalities as well as polymers with azide functionalities [e.g. poly(1-ethoxyethyl acrylate) and poly(acrylic acid)], were first synthesized via ATRP. They were then subsequently “clicked” together to yield block copolymers (42). Similarly, Opsteen et al. described the synthesis of polystyrene (PS), poly(tert-butyl acrylate) (PtBA), poly(methyl acrylate) (PMA) block copolymers using click chemistry (43). Using an initiator containing a triisopropylsilyl (TIPS) protected acetylene, the three homopolymer blocks were obtained via ATRP and the terminal bromides were then converted to azides. Following TIPS deprotection, the heterotelechelic homopolymers were joined together via HDC reactions. When RAFT polymerization was employed to obtain the homopolymer blocks, however, specially functionalized chain transfer agents had to be synthesized to allow the introduction of terminal azides or acetylenes (44,45). Additionally, this modular strategy of clicking different homopolymer blocks together has also been exploited by numerous other research teams (46–49). Among these works, one report focused on the synthesis of well-defined block copolymers composed of a rigid polypeptide sequence poly(g-benzyl-l-glutamate), or PBLG, and a poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) block (46). The azide and terminal alkyne functionalities were introduced into the α-positions of both the PBLG and PDMAEMA precursors by using appropriate α-ω-functionalized initiators. The PBLG block was obtained by ring-opening polymerization of γ-benzyl-l-glutamate N-carboxyanhydride (NCA), and the PDMAEMA blocks from ATRP of 2-(dimethylamino)ethyl methacrylate. Although this approach of incorporating a polypeptide block into a copolymer is truly innovative, the need of column chromatography for purification violates Sharpless’ definition of a click reaction. Hence, one could make the argument that this copolymer application does not actually utilize click chemistry.
Clearly, click chemistry has revitalized the second strategy of block copolymer synthesis. Many monomers that cannot be used to produce block copolymers via living polymerizations (due to extremely disparate reactivities or solubility differences) can now be easily incorporated through the second strategy. Quite literally, with click chemistry, any two homopolymer blocks can be joined together to form block copolymers. This opens the door for combinatorial block copolymer synthesis, allowing diverse copolymers with very unique properties to be synthesized quickly and easily, which could potentially lead to great strides in the field of pharmaceutical sciences.
Click Chemistry and Polymeric Micelles
One of the main pharmaceutical applications of amphiphilic block copolymers is the construction of polymeric micelles as delivery vehicles for therapeutic, imaging, or diagnostic agents (50–52). Although polymeric micelles are simple and effective delivery systems that have been evaluated in clinical treatments for various cancers, they still face several challenges such as stability and control of drug release. To improve these problem areas, many different methodologies have been explored (53,54). One of the most promising methods was developed by Wooley’s group at Washington University.
As a novel method for stabilizing polymeric micelles, the group reported the synthesis of well-defined core crosslinked polymeric micelles, utilizing multi-functional dendritic crosslinkers (55). Amphiphilic diblock copolymers of poly(acrylic acid)-b-poly(styrene) (PAA-b-PS) were first functionalized with terminal alkynes throughout the hydrophobic polystyrene block segment to form micelles that were click-readied for crosslinking. Azide functionalized 1st generation dendrimers were then “clicked” with the alkynyl groups of the micelles to form core crosslinked micelles. It was estimated, on average, that 3.4 polymer chains were conjugated to each dendrimer. The remaining click-readied functionalities (both terminal alkynes and azides) could be used as handles for further chemical modifications, such as conjugating dyes.
In addition to crosslinking micelles, the same group was able to introduce click-readied functionalities selectively throughout either the core or shell of micelles (56). After obtaining a poly(acrylic acid)-b-polystyrene copolymer micelle, it was modified so that poly(acrylic acid) within the micelle shell displayed either azido or alkynyl functional groups via amidation chemistry. Using amidation chemistry once more, the unreacted acrylic acids were linked together via a diamine linker, crosslinking the shell of the micelle in an intramicellar fashion. Following similar chemistry, a shell-crosslinked micelle containing click-readied functional groups in the polystyrene core was also prepared. The availability and reactivity of the functional groups in both of these crosslinked micelles towards click chemistry was demonstrated by reactions with complementary click-functionalized fluorescent dyes. Studies by analytical ultracentrifugation sedimentation confirmed the covalent attachment of the fluorescent tags in the core or shell regions of the crosslinked micelles.
While the strategies used by this research team are very appealing, they are still in their early stages of development for pharmaceutical applications. The polymers used in their studies are not biocompatible. Therefore, it would be of immense importance to see if the same chemistry is applicable to biocompatible block copolymers such as polyester/polyethylene glycol block copolymer systems, which are often used in drug delivery.
Click Chemistry and Linear Multifunctional Polymeric Delivery Systems
The most commonly used biocompatible water-soluble polymers for drug delivery include N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers, polyglutamate, and PEG. Among them, HPMA copolymer is the most extensively studied drug carrier, with more than half of all polymeric drug conjugates in clinical evaluations based on this polymer (57,58). Polyglutamate-drug conjugates, on the other hand, are the most advanced in clinical evaluation with polyglutamate-paclitaxel in phase III clinical trial (59,60). As for PEG, its main clinical applications are PEGylations of protein therapeutics to alter their pharmacokinetics and render them nonimmunogeneic (61). It has also been used as a carrier for chemotherapeutic agents (62).
While the field of water-soluble polymeric drug conjugates has gradually matured over the past few decades, challenges still remain in many aspects of its development: (1) compared to the construction of other drug delivery systems (e.g. liposome, micelles and nanoparticles), the synthesis of polymer-drug conjugate is relatively complex. For carriers based on vinyl polymers such as HPMA copolymer, the introduction of a new drug to the conjugate often involves multi-step synthesis of a new drug-containing vinyl monomer and its subsequent copolymerization with HPMA and other co-monomers. (2) Another issue is the nondegradable nature of vinyl polymers and PEG. While a polymer carrier with high molecular weight (MW) would render a long half-life for the conjugated drug in circulation, the MW must be set lower than the threshold of the renal glomerular filtration to avoid chronic accumulation of the polymer carrier in vivo and its potential negative impact. (3) Due to the recent advancement of living free radical polymerizations, the MW and polydispersity index (PDI) of vinyl polymer–drug conjugates may be properly controlled. Nevertheless, the distribution of functionality along the polymer backbone still cannot be controlled. (4) Different from vinyl polymers and polyglutamate, PEG has limited sites for functionalization. Only the two chain termini can be used for drug conjugation, which significantly lowers the drug loading capacity of PEG as a carrier.
As one of the potential solutions to these challenges, a linear multifunctional PEG design has been proposed as a polymeric drug carrier (63). The concept is to join short telechelic PEGs with tri-functional linkers. While two functionalities will be used to link the PEGs, the third one will be used for drug conjugation. Different from traditional PEG designs, it has multiple functionalities pendent to the polymer backbone. Short peptide sequences can also be introduced into the polymer main chain to allow biodegradation of the polymer carrier into short PEG segments. The length of the short PEG predetermines the distance between adjacent functionalities. With this method vinyl monomer synthesis is no longer necessary. While several approaches have been made to synthesize the linear multifunctional PEG construct (63–66), further improvements are desperately needed. In particular, the chemistry that joins the short PEG segments should be much more efficient, and tolerant of pendent functionality conjugation.
Recently, Liu et al. (67) used click chemistry to synthesize a linear multifunctional PEG as a novel drug delivery system. PEG oligomer diol was first activated with phosgene and capped with propargyl amine so that it was acetylene terminated. This was to serve as the polymer building block. A diazide monomer, 2,2-bis(azidomethyl)propane-1,3-diol, and various functional derivatives were synthesized as the functionality building blocks. All of these building blocks were dissolved in water and copolymerized in a step-growth fashion via the HDC reaction. Due to the self-catalyzing feature that has been observed in click reactions using 2,2-bis(azidomethyl)propane-1,3-diol (5), this click copolymerization was shown to be highly efficient. The strategy was used to synthesize a prototype bone-targeting PEG conjugate that used alendronate (a bisphosphonate) as the targeting moiety. The content of alendronate could be easily adjusted since the copolymerization was performed in water, a good solvent for bisphosphonates. Compared to traditional synthetic polymeric delivery systems, the advantages of this approach are that the entire synthetic procedure is modular, simple, free from vinyl chemistry, and produces high yields; no special reaction condition(s) or protection steps are needed; oligopeptide sequences can be easily inserted between PEG and the acetylene functionality to allow biodegradability of the delivery system. Furthermore, by adding some monoacetylene-functionalized PEG to the system, Liu et al. have been able to control the average molecular weight. More work needs to be done, however, to improve the wide polydispersity of the copolymer.
Other Polymer-Related Applications of Click Chemistry
Due to their concentrated surface functionality and narrow polydispersity, dendrimers have been actively investigated as potential drug delivery carriers (68,69). They are typically synthesized via two strategies: by the divergent approach or by the convergent approach. Without elaboration, both approaches are often complicated, involving multiple reaction steps, and both require extensive purification. Click chemistry can greatly simplify the synthesis of dendrimers, making them more applicable and affordable.
As an example of a dendrimer used for drug delivery, Gopin et al. (70) reported the synthesis and evaluation of a single-triggered disassemble dendrimer as a potential platform for a multi-prodrug. “These unique structural dendrimers can release all of their tail units through a self-immolative chain fragmentation that is initiated by a single cleavage at the dendrimer’s core” (70). To allow enzymatic activation of the second-generation of self-immolative dendrimers, azide-monoterminated PEG was conjugated to the dendritic platform via click chemistry, which prevented aggregate formation of the hydrophobic drugs.
Polymer networks such as hydrogels are also often used in drug delivery (71,72). A few attempts have been made to use click chemistry to synthesize polymer networks with high efficiency and reduced defects (73–75). The problem with this approach, however, is the difficulty of removing copper catalyst from the network.
As discussed above, polymer analogous reactions are not very efficient due to the steric hindrance caused by the polymer backbone. There are often unreacted pendent functional groups leftover. Because of its high reaction yield and mild reaction conditions, people have tried to introduce azides or acetylenes as novel pendent functionalities to the polymer backbone so that it can be used for further click modifications (76,77). One concern, however, is that this strategy will make sense only when the click functionalities are introduced by copolymerization, not polymer analogous reaction.
Click Chemistry and Nanoparticular Delivery Systems
Nanoparticular delivery systems are the carriers ranging in size from 10 to 1000 nm. Over the past few decades, delivery systems such as quantum dots, magnetic nanoparticles, gold nanoparticles, micelles, and liposomes have all been extensively investigated for imaging and drug/gene delivery applications (78,79). Due to the small size, they are able to penetrate through fenestrated vasculature, allowing efficient drug accumulation at target sites. Moreover, drug targeting by nanoparticular delivery systems offers several important advantages: they reduce drug dose, minimize side-effects, protect drugs against degradation, and enhance drug stability (80).
Surface modifications of these carriers can have significant impacts on their physical-chemical properties and therapeutic efficacy. They can alter nanoparticle zeta potentials, hydrophobicities, and targeting capabilities. These modifications can be performed using a variety of techniques, including physical adsorption, electrostatic binding, specific recognition, and covalent coupling, each of which has its own advantages and disadvantages (79). In this part of the review, the application of click chemistry towards the modification of nanoparticular carrier surfaces will be examined.
Gold and Magnetic Nanoparticles
The use of gold nanoparticles in pharmaceutical research has become a well-established practice, as evidenced by the ever-growing plethora of scientific literature. Due to their unique physical properties (81), they have been used to label antibodies (82), target polynucleotides (83), in cancer cell diagnostics (84), in drug delivery systems (85), etc. Many of these methods, however, rely on the non-covalent attachments of biological molecules, which have important drawbacks (86). For instance, non-covalent interactions are much weaker than covalent bonds, meaning that it is much easier to break apart the nanoparticle-biological molecule conjugate. For long-term conjugation, covalent bonds must be used. One study performed by Brennan et al. showed that bioconjugations to gold nanoparticles via covalent bonds could occur easily and rapidly with click chemistry (86).
Gold nanoparticles were first prepared and functionalized with azide groups at the surface. A lipase enzyme (a 30 kDa globular recombinant protein) was also modified to express a single terminal alkyne and was allowed to react with the nanoparticles. Gel electrophoresis verified that several lipases had covalently attached to each nanoparticle, without any nonspecific binding, and a lipase assay showed that they still retained their enzymatic activity. Following a similar procedure, Fleming et al. (87) were able to conjugate several different alkynyl derivatives, including ferrocene, aniline, and PEG, to gold nanoparticles via the click reaction.
Magnetic nanoparticles are of great interest in pharmaceutical sciences because of their good biocompatibility, injectability, and highly specific accumulation in target tissues under a local magnetic field (88). Several methods have been investigated to functionalize magnetic nanoparticles (89), but Lin et al. (90) were one of the first to take advantage of click chemistry. They successfully attached several different organic molecules to magnetic particles via the HDC reaction. These molecules included Tn antigen, flag peptide, biotin, 2,4-dinitrophenol (DNP), and maltose binding protein (MBP), each of which had been functionalized to contain a terminal alkyne. The magnetic nanoparticles, on the other hand, were azide functionalized. Fluorescent tests showed that each of the bioconjugations was a success and that there was no nonspecific binding of the 1,2,3-triazole or unreacted azide. Furthermore, it was observed that the azide linker length of the magnetic nanoparticles drastically affects how well MBP, and most likely other proteins, can bind. As linker length increased, so did binding efficiency, which was attributed to steric hindrance effects between the bulky nanoparticle and MBP.
Different from Lin et al., White et al. chose ligands containing either a phosphonic acid or a carboxylic acid group at one terminus to bind strongly to the surface of a γ-Fe2O3 nanoparticle, and either an azide or acetylene group at the other terminus to provide a chemical handle for modification (91). Both benzyl azide and 5-chloropentyne were then successfully conjugated through click chemistry. To demonstrate the versatility of the strategy, an acetylene terminated polymeric ligand was also attached to the nanoparticles.
Liposomes
Since the pioneering work of Bangham and co-workers in the late 1950s (92), liposomes have stimulated ongoing interest in pharmaceutical sciences. Several liposomal formulations have been approved by the US Food and Drug Administration (FDA) for clinical use, especially for the treatment of cancers, over the past decade (93). However, this particular drug carrier still have not attained its full potential; there are still some aspects that need optimization. Their surfaces have to be modified to avoid uptake by the phagocytic cells of the reticular endothelial system (RES), drug release mechanisms need to be improved so that the drug is completely and exclusively deposited at the disease target site, and more efficient strategies for conjugating targeting moieties (such as peptides and antibodies) to liposomal surfaces are required. Although great strides have been taken to resolve each of these issues (94–96), the latter still remains a problem. Many strategies are highly dependent upon chemical reactions that are not well-controlled, which often lead to unwanted products. One of the most popular methods involves the reaction between functionalized carboxylic acid groups and primary amines to form amide bonds (97). This method does not require prior ligand modification, thus reduces the risk of losing ligand bioactivity, but there is often a lack of specificity, resulting in an uncontrolled number of covalent bonds between the liposome and the targeting ligands. A much more efficient conjugation method relies on the reaction of thiol and maleimide functional groups to produce stable thioether bonds (98). However, in many cases, native thiol functional groups are either absent or present in insufficient amounts. Recently, hydrazone linkages have been studied for conjugation, which involves a mild oxidation of hydroxyl groups to aldehydes (99). It is obvious that this oxidation reaction can cause the ligand to loss its activity.
As already mentioned in previous sections, the HDC reaction is the epitome of linker chemistry and has proven to be a highly useful conjugation method. Despite this, few papers can be found relating click chemistry to liposomal conjugations. Two of these articles will now be discussed.
In a recent report, Hassane et al. (100) developed a novel strategy for conjugating mannose ligands to the surfaces of preformed liposomes using the click reaction. The resulting mannosylated liposomes can serve as vehicles to target specific cells, such as human dendritic cells (101). In this strategy, an alkyne terminated lipid anchor was first incorporated into liposomes. Then an unprotected mannosyl derivative was functionalized with an azide group and conjugated to the surface of the liposome in one single step. The group found that catalytic quantities of the ascorbate/CuSO4 system, however, were not enough to drive the click reaction to completion. A stabilizing agent that protects the Cu(I) oxidation state from degradation pathways had to be added (for more information on the role of Cu(I)-stabilizing agents in click chemistry refer to reference 3), which was found to greatly accelerate the reaction with high yields and reasonable reaction times (1 h). This conjugation reaction did not damage the integrity of the bilayers and did not significantly change the particle size. Residual copper in the conjugated liposome was not measurable (<0.03 μg/ml), meaning that the oxidation of unsaturated phospholipids by copper ions is avoided (102).
Cavalli et al. (103) investigated the facile in-situ surface modification of liposomes using the click reaction. Firstly, a mixture of alkyne-terminated 1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine (DOPE), 1,2-dioleoyl-sn-glycerol-3-phosphocholine (DOPC), and a lissamine rhodamine derivative of DOPE (DOPE-LR) were sonicated together to prepare unilamellar liposomes bearing terminal alkyne groups at their surface. Then an azide-functionalized nitro-benzoxadiazole (NBD) derivative, N3-Lys(NBD)-NH2, and CuBr were added to the vesicle solution. Using a method based on fluorescence resonance energy transfer (FRET), the reaction was followed in time, showing that the chemical modifications truly did occur at the surfaces of the liposomes. The reaction was completed within 4 h, with no dramatic size changes of the liposomes. These two studies clearly indicate the speed and versatility of the HDC reaction, allowing both carbohydrates and biologicals to be linked to liposome surfaces, regardless of the other functional groups present.
Bioconjugation
The use of bioconjugation and biopharmaceuticals are quickly becoming common practice. As of May 1st, 2007, an estimated 30 biopharmaceuticals are currently in clinical trials (104). This popularity stems from the fact that bioconjugation reactions can serve many purposes and satisfy many functional requirements. They are frequently employed to increase aqueous solubility, reduce immunogenicity, increase circulation time, and improve stability. They can also be used for radiolabeling and to attach fluorescent tags.
Excluding drug discovery and polymer chemistry, the most common pharmaceutical applications of click chemistry are bioconjugation reactions. One reason for this is 1,2,3-triazoles make ideal linkers. They are extremely water soluble, making in vivo administration much easier. In fact, there electronic properties are very similar to amide bonds, but they are not subject to the same hydrolysis reactions (3). They are also stable in typical biological conditions, which tend to be aqueous and mildly reducing in nature (2). Another advantageous property is they are extremely rigid, ensuring that the two linked substances are not interacting with each other (3). If the linker were flexible then the substances could easily aggregate and/or react with each other. Furthermore, side reactions at unintended sites of the substances are a major problem in bioconjugation reactions. The fact that azides and alkynes are relatively unreactive towards most functional groups further makes this click reaction sought after; it ensures that bioconjugation will only occur at the desired location(s). Finally, as previously mentioned, this reaction tolerates many different types of azides and alkynes. Primary, secondary, tertiary, and aromatic azides will all undergo click chemistry; the steric bulkiness of the attached group seems to be insignificant. This is important since many biological molecules, such as globular proteins, are sterically bulky. Following now is an overview of some bioconjugation studies that have been performed via click chemistry.
Radiolabeling
Radiolabeling has become a powerful tool in the pharmaceutical scientist’s arsenal. By connecting a radionuclide to a drug of interest, one can track its in vivo distribution (105), what specific receptors it attaches to (106), its metabolic pathway (105), etc. All that is required is a radionuclide that has desirable decay characteristics and an instrument that can detect the amount of radiation present. One particular radionuclide that is commonly used is technetium-99 m, or 99mTc. This radioisotope has an ideal half-life of 6 h and emits readily detectable 140 keV gamma rays (107). It currently has several applications in the medical field, ranging from identifying angiogenesis via attachment to monoclonal antibodies to evaluating ventricular function by conjugation to red blood cells (108,109).
A major obstacle with 99mTc, however, is it can be difficult to attach to organic molecules. One of the most common methods used is to take an organometallic precursor, such as the tricarbonyl molecule [99mTc(OH2)3(CO)3]+ (110), and complex it to a tridentate ligand that is already attached to the molecule of interest (107). This ligand is typically based on the amino acids cysteine, lysine, and especially histidine (111). The problems associated with this method, though, are numerous. The synthesis of such ligands requires multiple steps, which can be quite challenging, they are not always easily incorporated into the molecules of interest, and they can cross react with other functional groups present (107,111).
To avoid these problems, Mindt et al. (111) turned to click chemistry. Realizing that 1,2,3-triazoles share similar electronic properties to the imidazoles in histidines and that they too might be able to complex with organometallic precursors, the HDC reaction was employed. The facts that triazoles can be made in one easy step and azides can be easily incorporated into organic molecules made this strategy further attractive. By reacting various alkynes and azides, allowing each to complex with either [99mTc(OH2)3(CO)3]+ or [ReBr3(CO)3]2-, and determining the complexation efficiencies, EC50 values as high as 2 to 3 × 10−7 M were obtained. These numbers are almost as high as that for NЄ-methyl histidine (∼1 × 10−7 M), proving that 1,2,3-triazoles can chelate organometallic precursors just as well as histidine. To prove the versatility of the reaction, biomolecules from every major classification were used (a peptide [bombesin], a nucleic acid [thymidine], a carbohydrate, and a phospholipid) as click reaction substrates and subsequently radiolabeled. The bombesin triazole derivative, in particular, showed in vivo stability and receptor affinity similar to that of a bombesin histidine derivative. This further suggests that triazoles could be a beneficial alternative to histidine when it comes to complexation with radionuclides.
Tagging E. coli
The proteins expressed on the surfaces of microbial cells can be used and manipulated for various applications. They have proved quite useful in a broad range of fields, including protein engineering, screening for antibody fragments, whole cell catalysts, adsorbents for bioremediation, etc. (112,113). A major limitation, however, is there are only 20 naturally-occurring amino acids. If one could expand the set of amino acids expressed or specifically replace certain amino acids with synthetic derivatives then surface proteins could be further manipulated and the number of potential applications would be increased exponentially. In this direction, some research groups have already demonstrated remarkable success (113).
Capitalizing on the high reliability of the HDC reaction, Link and Tirrell (113) incorporated an azide-containing synthetic amino acid into the surface proteins of E. coli. The amino acid they chose was azidohomoalanine (AHA). It had been shown previously that AHA can replace methionine residues in proteins (114) and the outer membrane protein C (OmpC) of E. coli contains three such residues (115). A mutant E. coli containing nine methionine residues per OmpC was also created for comparison. Once AHA had been incorporated into OmpC for both strains, click reactions using a biotinylated alkyne reagent were performed. Both cells were successfully biotinylated, but only the mutant E. coli would bind with the large molecule avidin, presumably due to less steric crowding surrounding the additional methionine residues.
Tagging CPMV
When wanting to test the bioconjugation abilities of one or more compounds, Cowpea mosaic virus (CPMV) is frequently used as the scaffold. It can be made inexpensively on the gram scale and it is easy to separate from small unreacted reagents. Furthermore, it contains 60 identical copies of an asymmetric two-protein unit, each of which contains one cysteine and one lysine (9,116). These 60 cysteines and lysines can be manipulated and attached to various functional groups and biomolecules, as evidenced by countless studies (117–119).
In one study, conducted by Wang et al., fluorescein dye derivatives were attached to both azide-functionalized and alkyne-functionalized CPMV using click chemistry (116). Not only were all of the reactions successful, but also their product yield was as high as 100%. Two years later, in 2005, they were able to attach three more hemicyanine dyes to CPMV using the same reaction (9). Purification can be simply performed by dialysis or ultrafiltration.
Labeling DNA
Oligonucleotides represent a very important class of biomolecules. Although they have been “shunned” by the pharmaceutical community in the past due to lack of knowledge, they have recently found many applications and are considered to be the most important tools to many areas of research (120). They have been used for gene therapy (121), as antisense agents to treat diseases like leukemia (122), as molecular probes (123), etc. Adding different functional groups further increases their versatility, especially when one considers that functionalization can be introduced at either the 3′-end, 5′-end, or an internal position. The newly added functional groups can serve as handles for bioconjugation with a wide variety of biological molecules. Current methods for DNA bioconjugation, however, are inefficient. The procedure must be able to tolerate aqueous conditions, give high yields, and the resulting linkage must be stable in biological conditions (124). This is the perfect situation for click chemistry.
Utilizing the HDC reaction, Seo et al. were able to tag a fluorophore to the 5′-end of single-stranded DNA (124). The oligonucleotide was modified, through several reactions, to display a terminal alkyne at its 5′-end and the fluorophore contained an azido functional group. A 91% product yield was obtained, but no catalyst was used in the reaction, leading to a mixture of 1,4-substituted and 1,5-substituted 1,2,3-triazole products. If a copper catalyst were used then it is highly likely that only 1,4-substituted triazoles would be produced and that the reaction would proceed much faster. Seela et al. took DNA labeling one step further and synthesized nucleosides that each contained a single terminal alkyne on their aromatic nucleobase (125). Modified deoxyadenosine (dA), deoxyguanosine (dG), deoxycytidine (dC), and deoxythymidine (dT) were all included. Using solid phase synthesis, several oligonucleotides were subsequently synthesized, which either contained one modified nucleoside, two, or none at all (to serve as controls). The properties of the oligonucleotides and their duplexes were not significantly affected by the modified nucleosides, as evidenced by the similar melting temperatures when compared to the controls. Reporter molecules containing azido functional groups were then conjugated to the modified bases through click chemistry. Upon conjugation the reporter molecules began to fluoresce, indicating that the oligonucleotides had been successfully labeled. If made commercially available, Seela’s modified nucleosides could make oligonucleotide labeling trivial.
Polysaccharides
Polysaccharides are another major class of macromolecules that have received limited attention. Carbohydrates make up 5% to 10% of plasma membrane mass, in the form of glycoconjugates, and mediate a variety of events, such as cell-cell recognition, metastasis, fertilization, and immunological response (126,127). Alterations to cell surface oligosaccharides have been linked to a number of different diseases, including cancer and tuberculosis (127). Despite these pivotal biological functions, however, carbohydrates are rarely found in pharmaceutics. Their synthesis typically involves multiple reaction steps and many stereoisomers, making it challenging to even the most skilled chemists. Furthermore, their moderate affinity towards target receptors and enzymes and poor pharmacological properties make them seldom used as targeting moieties/lead compounds (2). Physical modifications of the carbohydrates can improve both of these issues, but chemical handles are restricted to alcohol functional groups and the steric bulkiness of polysaccharides often results in low product yields.
Due to the easy introduction of azide functional groups and high reliability of the HDC reaction towards the bulkiest of reactants, Liebert et al. used click chemistry to modify the surface of cellulose (128). It was thought that the 1,2,3-triazoles themselves would interact with biological targets through hydrogen bonding and dipole interactions, increasing the overall affinity of cellulose towards its desired target. The team first introduced azide functional groups into cellulose through two simple steps: tosylation of the secondary alcohol groups followed by azidation with sodium azide. The overall percent yield ranged from 60% to 99%. Three different terminal alkyne-containing low molecular weight compounds (methylpropiolate, 2-ethynylaniline, and 3-ethynylthiophene) were then mixed with three different strands of azide-celluloses in separate reactions, using a copper(II) salt catalyst and sodium ascorbate. All reactions were successful with product yields ranging from 75% to 98%, depending on the reaction conditions utilized.
In that same year, Hafrén et al. (129) modified cellulose with click chemistry but took a slightly different approach. In one single step, terminal alkynes were introduced through the secondary alcohols of cellulose using 5-hexynoic acid in a neat reaction. 3-Azidocoumarin and copper catalyst were then added. Upon reaction, the coumarin-cellulose compound began to fluoresce a bright blue color, indicating that the reaction was successful, but no product yield was calculated. The work of the Liebert and Hafrén research teams clearly show that polysaccharides can be modified via click chemistry. Both azide and terminal alkyne functional groups can be easily introduced and the HDC reaction still occurs in high yields despite steric hindrance.
THE PITFALLS OF CLICK CHEMISTRY
Up until now, this review may have given the impression that the Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition of terminal alkynes and azides is an “invincible” reaction, always giving high yields no matter what the situation. However, the HDC reaction has a number of limitations, each of which will be discussed. Firstly, like with any cycloaddition, if the diene (the azide in the case) is too electron deficient then it will not undergo the reaction. The energy of its ground state configuration is far too low for it to interact with a dienophile (the terminal alkyne). Likewise, the dienophile cannot be too electron rich. These situations are highly unlikely to occur, though, and require functional groups that are not commonly seen in biological systems or administered drugs.
A more common problem is alkyne homocoupling. This occurs when an alkyne reacts with a second alkyne instead of the azide. As shown in Fig. 7, there are several alkyne homocoupling side reactions that can occur, three of which include Glaser (130), Straus (131), and Eglinton couplings (132,133). Some of these require a CuI catalyst (Glaser and Straus), while others require CuII (Eglinton) (133). Some need the presence of oxygen to react (Glaser) while others can continue in inert atmosphere (Straus) (133). Most of these reactions, though, can be minimized by using a sterically bulky base (3). It has been shown that the smaller the base, the more it stabilizes the reactive intermediates of the homocoupling reactions (not shown), and the lower the percent yield of the cycloaddition (3,134,135). The small bases tetramethylethylenediamine (TMEDA), pyridine, and triethylamine are frequently the culprits when these side reactions occur (133,134).

Three types of alkyne homocouplings that can lower the percent yield of the HDC reaction: Glaser (top), Straus (middle) and Eglinton (bottom).
A less common problem is CuI saturation. In order for the click reaction to take place, the CuI-acetylide complex intermediate has to have physical contact with the azide (see section 3.1 for the mechanism). If the complex is closely surrounded by terminal alkynes, however, then there is a chance that the alkynes will chelate with the complex, thereby “saturating” it (3). This effectively prevents any azide functional groups from reaching the complex and performing displacement. CuI saturation is rare, though, as it requires a dienophile that contains multiple terminal alkynes that can coordinate to a single location. One notable example came through the work of Zhao et al. (136). A substrate containing four terminal alkynes in close proximity was unable to undergo an HDC reaction. However, when the alkynes were replaced with azide functional groups, the substrate readily reacted.
The stability of some azides may also be a limitation. It is known that if the ratio of nitrogen atoms to carbon atoms in an organic molecule exceeds, or is equal to, one then the molecule should be considered explosive and very dangerous. As an example, methyl azide often decomposes explosively and heavy-metal azides are frequently used as detonators (8). All that is required is a slight input of external energy, such as heat or pressure (8). Fortunately, this is generally not a major issue for pharmaceutical research, which tends to focus on larger molecules with high carbon contents.
The click reaction also has challenges pertaining specifically to the field of pharmaceutical sciences. One of the most obvious disadvantages is it requires a copper catalyst. Although the human body requires copper in order to function, excessive intake can lead to drastic consequences. Some associated side effects include hepatitis, neurological disorders, kidney diseases, and Alzheimer’s disease (137). The reason for this toxicity stems from the fact that copper can easily accept and donate single electrons to change oxidation states, thereby allowing it to catalyze toxic reactions, such as the in vivo reduction of hydrogen peroxide to form hydroxyl free radicals (138). Therefore, in order for the click reaction to find in vivo applications, the copper catalyst must be completely removed. This may not always be an easy task, but a few research groups have demonstrated some success. As an example, in the click-PEG delivery system developed by Liu et al. (67), about 98% of the copper was effectively removed by incubation with ethylenediaminetetraacetic acid (EDTA), followed by dialysis (unpublished data). However, this approach can only be applied to large molecular weight structures. Another group, Veinot et al., has used oxide-capped metallic iron nanoparticles (139) as Cu sequester. After performing a click reaction with phenyl azide and benzyl acetylene, the crude product was incubated with the nanoparticles twice and filtered twice. Analysis revealed that the copper concentration had been significantly reduced from 2,026 ppm to 4.6 ppm.
Another challenge involves biocompatibility of 1,2,3-triazoles. Despite the fact that they were first identified over a century ago, not much is known about their biological pathways. The individual toxicities of some 1,2,3-triazole-containing compounds have been extensively scrutinized, but no generalities have been established. As evidenced by a literature search via SciFinder Scholar on November 12th, 2007 there are no reports reviewing the in vivo metabolism of 1,2,3-triazoles. This is very surprising since many drug candidates have been synthesized via click chemistry and 1,2,3-triazoles have long been used as bioisosteric replacements of amide functional groups. In fact, the very intent of click chemistry is for drug discovery.
The final limitation discussed in this paper is, at the present, many “click-readied” building units and “click products” are not yet commercially available. This means it is the responsibility of the pharmaceutical scientist to synthesize his/her own compounds or to collaborate with others who can. Even though click reactions are some of the easiest and high-yielding reactions known, this task can seem daunting to even the most experienced pharmaceutical scientist, whose typical background is not in organic synthesis. Fortunately, click chemistry is still a relatively new idea. As time progresses and this methodology gains increased acceptance, more and more products may be made available on the market.
CONCLUSIONS
Despite the fact it debuted less than 10 years ago, click chemistry is quickly revolutionizing the way scientists view their research. Although all click reactions are easy to use, give high reaction yields, do not require long reaction times, are regiospecific, and are easy to purify, the CuI-catalyzed Huisgen 1,3-dipolar cycloaddition of azides and terminal alkynes has emerged as the most popular click reaction by far. It has found numerous applications across a wide variety of disciplines, including materials research, polymer chemistry, and the pharmaceutical sciences. It is particularly sought after by the pharmaceutical community for its tolerance of typical biological conditions, tolerance of most functional groups, and for the high aqueous solubility of 1,2,3-triazoles. Specifically, the HDC reaction has been used to synthesize novel polymeric delivery systems, dendrimers, to cross-link micelles, and to modify the surfaces of various nanoparticular delivery systems. The rigidity of 1,2,3-triazoles also makes the reaction ideal for bioconjugations. Oligonucleotides, proteins, polysaccharides, viruses, and bacteria such as E. coli have all been successfully conjugated to various substances using this approach.
The HDC reaction, however, is not perfect. There are a few important limitations that need to be considered. The most significant is it requires a copper catalyst. High level of copper in the body can lead to serious, even deadly, consequences. It is also important to realize that systemic studies need to be conducted to confirm the biocompatibility of 1,2,3-triazoles. Other less serious problems include CuI saturation and alkyne homocoupling.
Though the HDC reaction has several limitations, it is still one of the most versatile and beneficial chemistry tools for pharmaceutical applications. We believe that as the industry recognizes the importance and large potential of this reaction and provides all of the basic building blocks, it will continue to grow in popularity and contribute to many major advances in pharmaceutical sciences and other research fields. Hopefully as time progresses, other click reactions will also gain momentum and add additional items to our chemistry “tool box.”
References
H. C. Kolb, M. G. Finn, and K. B. Sharpless. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 40:2004–2021 (2001).
H. C. Kolb, and K. B. Sharpless. The growing impact of click chemistry on drug discovery. Drug Discov. Today. 8:1128–1137 (2003).
V. D. Bock, H. Hiemstra, and J. H.-V. Maarseveen. CuI-Catalyzed alkyne-azide “click” cycloadditions from a mechanistic and synthetic perspective. Eur. J. Org. Chem. 2006:51–68 (2006).
R. Huisgen. 1,3-Dipolar cycloadditions. Angew. Chem. 75:604–637 (1963).
V. O. Rodionov, V. V. Fokin, and M. G. Finn. Mechanism of the ligand-free CuI-catalyzed azide-alkyne cycloaddition reaction. Angew. Chem., Int. Ed. Engl. 44:2210–2215 (2005).
V. V. Rostovtsev, L. G. Green, V. V. Fokin, and K. B. Sharpless. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem., Int. Ed. Engl. 41:2596–2599 (2002).
F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless, and V. V. Fokin. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity intermediates. J. Am. Chem. Soc. 127:210–216 (2005).
S. Brase, C. Gil, K. Knepper, and V. Zimmermann. Organic azides: an exploding diversity of a unique class of compounds. Angew. Chem., Int. Ed., Engl. 44:5518–5240 (2005).
W. H. Zhan, H. N. Barnhill, K. Sivakumar, H. Tian, and Q. Wang. Synthesis of hemicyanine dyes for ‘click’ bioconjugation. Tetrahedron Lett. 46:1691–1695 (2005).
B. M. Mykhalichko, O. N. Temkin, and M. G. Mys’kiv. Polynuclear complexes of copper(I) halides: coordination chemistry and catalytic transformations of alkynes. Russ. Chem. Rev. 69:957–984 (2000).
R. Chinchilla, and C. Najera. The sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem. Rev. 107:874–922 (2007).
W. S. Horne, C. D. Stout, and M. R. Ghadiri. A heterocyclic polymer nanotube. J. Am. Chem. Soc. 125:9372–9376 (2003).
P. L. Golas, N. V. Tsarevsky, B. S. Sumerlin, and K. Matyjaszewski. Catalyst performance in “click” coupling reactions of polymers prepared by ARTP: ligand and metal effects. Macromolecules. 39:6451–6457 (2006).
H. A. Orgueria, D. Fokas, Y. Isome, P. C.-M. Chane, and C. M. Baldino. Regioselective synthesis of [1,2,3]-triazoles catalyzes by Cu(I) generated in situ from Cu(0) nanosize activated powder and amine hydrochloride salts. Tetrahedron Lett. 46:2911–2914 (2005).
S. Chassaing, M. Kumarraja, A. S.-S. Sido, P. Pale, and J. Sommer. Click chemistry in CuI-zeolites: the huisgen [3 + 2]-cycloaddition. Org. Lett. 9:883–886 (2007).
K. Klier. Transition-metal ions in zeolites: the perfect surface sites. Langmuir. 4:13–25 (1988).
B. K. Marcus, and W. E. Cormier. Going green with zeolites. Chem. Eng. Prog. 95:47–53 (1999).
Wittingham, S. Synthesis and characterization of the zeolite ZSM-5: a size and shape selective catalyst for xylene isomerization. http://imr.chem.binghamton.edu/labs/zeolite/zeolite.html, part of Binghamton University’s Institute for Materials Research website. http://imr.chem.binghamton.edu. Accessed August 9, 2007.
M. A. Bennett. Olefin and acetylene complexes of transition metals. Chem. Rev. 62:611–652 (1962).
F. R. Hartley. Thermodynamic data for olefin and acetylene complexes of transition metals. Chem. Rev. 73:163–190 (1973).
L. L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin, and G. Jia. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 127:15998–15999 (2005).
G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba, and A. A. Genazzani. Click chemistry reactions in medicinal chemistry: applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 28:278–308 (2007).
Z. Li, T. S. Seo, and J. Ju. 1,3-Dipolar cycloaddition of azides with electron-deficient alkynes under mild condition in water. Tetrahedron Lett. 45:3143–3146 (2004).
N. J. Agard, J. A. Prescher, and C. B. Bertozzi. A strain-promoted [3+2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126:15046–15047 (2004).
G. Wittig, and A. Krebs. On the existence of low-membered cycloalkynes. I. Chem. Ber. 94:3260–3275 (1961).
J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli, and C. R. Bertozzi. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A. 104:16793–16797 (2007).
L. Weber. In vitro combinatorial chemistry to create drug candidates. Drug Discov. Today: Tech. 1:261–267 (2004).
K. B. Sharpless, and R. Manetsch. In situ click chemistry: a powerful means for lead discovery. Exp. Opin. Drug Discov. 1:525–538 (2006).
S. Roper, and H. C. Kolb. Click chemistry for drug discovery. Meth. Princpl. Med. Chem. 34:313–339 (2006).
R. Duncan. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2:347–360 (2003).
Y. Matsumura, and H. Maeda. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 46:6387–6392 (1986).
D. Wang, S. C. Miller, X. M. Liu, B. Anderson, X. S. Wang, and S. R. Goldring. Novel dexamethasone-HPMA copolymer conjugate and its potential application in treatment of rheumatoid arthritis. Arthritis Res. Ther. 9:R2 (2007).
J. Kopeček, P. Kopečková, T. Minko, Z. R. Lu, and C. M. Peterson. Water soluble polymers in tumor targeted delivery. J. Controlled Release. 74:147–158 (2001).
T. G. Park, J. H. Jeong, and S. W. Kim. Current status of polymeric gene delivery systems. Adv. Drug Delivery Rev. 58:467–486 (2006).
M. J. Roberts, M. D. Bentley, and J. M. Harris. Chemistry for peptide and protein PEGylation. Adv. Drug Delivery Rev. 54:459–476 (2002).
F. M. Veronese, and J. M. Harris. Peptide and protein PEGylation III: advances in chemistry and clinical applications. Adv. Drug Delivery Rev. 60:1–2 (2008).
Y. Kakizawa, and K. Kataoka. Block copolymer micelles for delivery of gene and related compounds. Adv. Drug Delivery Rev. 54:203–222 (2002).
R. K. O’Reilly, C. J. Hawker, and K. L. Wooley. Cross-linked block copolymer micelles: functional nanostructures of great potential and versatility. Chem. Soc. Rev. 35:1068–1083 (2006).
K. Letchford, and H. Burt. A review of the formation and classification of amphiphilic block copolymer nanoparticulate structures: micelles, nanospheres, nanocapsules and polymersomes. Eur. J. Pharm. Biopharm. 65:259–269 (2007).
O. W. Webster. Living polymerization methods. Science (Wash. D.C.). 251:887–893 (1991).
E. Franta, and P. F. Rempp. The block copolymer bag of tricks. Chemtech. 26:24–28 (1996).
W. Van Camp, V. Germonpre, L. Mespouille, P. Dubois, E. J. Goethals, and F. E. Du Prez. New poly(acrylic acid) containing segmented copolymer structures by combination of “click” chemistry and atom transfer radical polymerization. React. Functl. Polym. 67:1168–1180 (2007).
J. A. Opsteen, and J. C. M. Van Hest. Modular synthesis of ABC type block copolymers by “click” chemistry. J. Polym. Sci., Part A: Polym. Chem. 45:2913–2924 (2007).
D. Quemener, T. P. Davis, C. Barner-Kowollik, and M. H. Stenzel. RAFT and click chemistry: a versatile approach to well-defined block copolymers. Chem. Commun. 48:5051–5053 (2006).
S. R. S. Ting, A. M. Granville, D. Quemener, T. P. Davis, M. H. Stenzel, and C. Barner-Kowollik. RAFT chemistry and Huisgen 1,3-dipolar cycloaddition: a route to block copolymers of vinyl acetate and 6-O-methacryloyl mannose. Aust. J. Chem. 60:405–409 (2007).
W. Agut, D. Taton, and S. Lecommandoux. A versatile synthetic approach to polypeptide based rod-coil block copolymers by click chemistry. Macromolecules. 40:5653–5661 (2007).
G. Deng, D. Ma, and Z. Xu. Synthesis of ABC-type miktoarm star polymers by “click” chemistry, ATRP and ROP. Eur. Polym. J. 43:1179–1187 (2007).
O. Altintas, G. Hizal, and U. Tunca. ABC-type hetero-arm star terpolymers through “Click” chemistry. J. Polym. Sci., Part A: Polym. Chem. 44:5699–5707 (2006).
S. Fleischmann, H. Komber, D. Appelhans, and B. I. Voit. Synthesis of functionalized NMP initiators for click chemistry: a versatile method for the preparation of functionalized polymers and block copolymers. Macromol. Chem. Phys. 208:1050–1060 (2007).
K. Kataoka, A. Harada, and Y. Nagasaki. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv. Drug Delivery. Rev. 47:113–131 (2001).
A. Mitra, A. Nan, B. R. Line, and H. Ghandehari. Nanocarriers for nuclear imaging and radiotherapy of cancer. Curr. Pharm. Des. 12:4729–4749 (2006).
V. P. Torchilin. Micellar nanocarriers: pharmaceutical perspectives. Pharm. Res. 24:1–16 (2007).
F. Henselwood, and G. Liu. Water-soluble nanospheres of poly(2-cinnamoylethyl methacrylate)-block-poly(acrylic acid). Macromolecules. 30:488–493 (1997).
B. Loppinet, R. Sigel, A. Larsen, G. Fytas, D. Vlassopoulos, and G. Liu. Structure and dynamics in dense suspensions of micellar nanocolloids. Langmuir. 16:6480–6484 (2000).
R. K. O’Reilly, M. J. Joralemon, C. J. Hawker, and K. L. Wooley. Preparation of orthogonally-functionalized core click cross-linked nanoparticles. New J. Chem. 31:718–724 (2007).
R. K. O’Reilly, M. J. Joralemon, K. L. Wooley, and C. J. Hawker. Functionalization of micelles and shell cross-linked nanoparticles using click chemistry. Chem. Mater. 17:5976–5988 (2005).
F. Kratz, K. Abu Ajaj, and A. Warnecke. Anticancer carrier-linked prodrugs in clinical trials. Expert. Opin. Investig. Drugs. 16:1037–1058 (2007).
R. Duncan. Designing polymer conjugates as lysosomotropic nanomedicines. Biochem. Soc. Trans. 35:56–60 (2007).
K. S. Albain, C. P. Belani, P. Bonomi, K. J. O’Byrne, J. H. Schiller, and M. Socinski. PIONEER: a phase III randomized trial of paclitaxel poliglumex versus paclitaxel in chemotherapy-naive women with advanced-stage non-small-cell lung cancer and performance status of 2. Clin. Lung. Cancer. 7:417–419 (2006).
S. A. Shaffer, C. Baker-Lee, J. Kennedy, M. S. Lai, P. de Vries, K. Buhler, and J. W. Singer. In vitro and in vivo metabolism of paclitaxel poliglumex: identification of metabolites and active proteases. Cancer Chemother. Pharmacol. 59:537–548 (2007).
M. J. Roberts, M. D. Bentley, and J. M. Harris. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 54:459–476 (2002).
R. B. Greenwald, Y. H. Choe, J. McGuire, and C. D. Conover. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 55:217–250 (2003).
A. Nathan, S. Zalipsky, S. I. Ertel, S. N. Agathos, M. L. Yarmush, and J. Kohn. Copolymers of lysine and polyethylene glycol: a new family of functionalized drug carriers. Bioconjug. Chem. 4:54–62 (1993).
M. Pechar, K. Ulbrich, V. Subr, L. W. Seymour, and E. H. Schacht. Poly(ethylene glycol) multiblock copolymer as a carrier of anti-cancer drug doxorubicin. Bioconjug. Chem. 11:131–139 (2000).
J. Cheng, K. T. Khin, G. S. Jensen, A. Liu, and M. E. Davis. Synthesis of linear, beta-cyclodextrin-based polymers and their camptothecin conjugates. Bioconjug. Chem. 14:1007–1017 (2003).
Wang, A. Dong, H. Tang, E. A. Van Kirk, P. A. Johnson, W. J. Murdoch, M. Radosz, and Y. Shen. Synthesis of degradable functional poly(ethylene glycol) analogs as versatile drug delivery carriers. Macromol. Biosci. 7:1187–1198 (2007).
X. M. Liu, A. Thakur, and D. Wang. Efficient synthesis of linear multifunctional poly(ethylene glycol) by copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition. Biomacromolecules. 8:2653–2658 (2007).
S. Svenson, and D. A. Tomalia. Dendrimers in biomedical applications–reflections on the field. Adv. Drug Delivery Rev. 57:2106–2129 (2005).
L. Y. Qiu, and Y. H. Bae. Polymer architecture and drug delivery. Pharm. Res. 23:1–30 (2006).
A. Gopin, S. Ebner, B. Attali, and D. Shabat. Enzymatic activation of second-generation dendritic prodrugs: conjugation of self-immolative dendrimers with poly(ethylene glycol) via click chemistry. Bioconjugate Chem. 17:1432–1440 (2006).
Y. Qiu, and K. Park. Environment-sensitive hydrogels for drug delivery. Adv. Drug Delivery Rev. 53:321–339 (2001).
J. K. Tessmar, and A. M. Gopferich. Matrices and scaffolds for protein delivery in tissue engineering. Adv. Drug Delivery Rev. 59:274–291 (2007).
V. Crescenzi, L. Cornelio, C. Di Meo, S. Nardecchia, and R. Lamanna. Novel hydrogels via click chemistry: synthesis and potential biomedical applications. Biomacromolecules. 8:1844–1850 (2007).
M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury, and C. J. Hawker. Synthesis of well-defined hydrogel networks using click chemistry. Chem. Commun. 2006:2774–2776 (2006).
D. A. Ossipov, and J. Hilborn. Poly(vinyl alcohol)-based hydrogels formed by “click chemistry.”. Macromolecules. 39:1709–1718 (2006).
B. Sieczkowska, M. Millaruelo, M. Messerschmidt, and B. Voit. New photolabile functional polymers for patterning onto gold obtained by click chemistry. Macromolecules. 40:2361–2370 (2007).
B. Voit, S. Fleischmann, H. Komber, A. Scheel, and K. Stumpe. Cycloaddition reactions and dendritic polymer architectures-a perfect match. Macromol. Symp. 254:16–24 (2007).
J. Panyam, and V. Labhasetwar. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Delivery Rev. 55:329–347 (2003).
E. Katz, and I. Willner. Integrated nanoparticle-biomolecule hybrid systems: synthesis, properties, and applications. Angew. Chem., Int. Ed. Engl. 43:6042–6108 (2004).
P. Couvreur, G. Barratt, E. Fattal, P. Legrand, and C. Vauthier. Nanocapsule technology: a review. Crit. Rev. Ther. Drug Carrier Syst. 19:99–134 (2002).
J. L. West, and N. J. Halas. Engineered nanomaterials for biophotonics applications: improving, sensing, imaging, and therapeutics. Annu. Rev. Biomed. Eng. 5:285–292 (2003).
P. W. Faulk, and M. G. Taylor. Immunocolloid method for the electron microscope. Immunochem. 8:1081–1083 (1971).
B.-S. Tan, X.-H. Cao, and Z.-H. Mo. Gold nanoparticles preparation and their application in DNA detection. Chongqing Daxue Xuebao, Ziran Kexueban. 26:58–62 (2003).
X. Huang, I. H. E.-Sayed, and M. A. E.-Sayed. The use of surface enhanced absorption, scattering and catalytic properties of gold nanoparticles in some bio-and biomedical applications. Proc. SPIE-Int. Soc. Opt. Eng. 5929 (2005).
G. Han, P. Ghosh, M. De, and V. M. Rotello. Drug and gene delivery using gold nanoparticles. NanoBiotech. 3:40–45 (2007).
J. L. Brennan, N. S. Hatzakis, R. T. Tshikhudo, N. Dirvianskyte, V. Razumas, S. Patkar, J. Vind, A. Svendsen, R. J. M. Nolte, A. E. Rowan, and M. Brust. Bionanoconjugation via click chemistry: the creation of functional hybrids of lipases and gold nanoparticles. Bioconjugate Chem. 17:1373–1375 (2006).
D. A. Fleming, C. J. Thode, and M. E. Williams. Triazole cycloaddition as a general route for functionalization of Au nanoparticles. Chem. Mater. 18:2327–2334 (2006).
A. Ito, M. Shinkai, H. Honda, and T. Kobayashi. Medical application of functionalized magnetic nanoparticles. J. Biosci. Bioeng. 100:1–11 (2005).
A. H. Lu, E. L. Salabas, and F. Schüth. Magnetic nanoparticles: synthesis, protection, functionalization, and application. Angew. Chem., Int. Ed. Engl. 46:1222–1244 (2007).
P.-C. Lin, S.-H. Ueng, S.-C. Yu, M.-D. Jan, A. K. Adak, C.-C. Yu, and C.-C. Lin. Surface modification of magnetic nanoparticle via Cu(I)-catalyzed alkyne-azide [2+3] cycloaddition. Org. Lett. 9:2131–2134 (2007).
M. A. White, J. A. Johnson, J. T. Koberstein, and N. J. Turro. Toward the syntheses of universal ligands for metal oxide surfaces: controlling surface functionality through click chemistry. J. Am. Chem. Soc. 128:11356–11357 (2006).
A. D. Bangham, D. H. Heard, R. Flemans, and G. V. Seaman. An apparatus for microelectrophoresis of small particles. Nature (London). 182:642–644 (1958).
T. M. Allen, and P. R. Cullis. Drug delivery systems: entering the mainstream. Science (Washington, D.C.). 303:1818–1822 (2004).
M. L. Immordino, F. Dosio, and L. Cattel. Stealth lipsomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 1:297–315 (2006).
X. Guo, and F. C. Szoka Jr. Chemical approaches to triggerable lipid vesicles for drug and gene delivery. Acc. Chem. Res. 36:335–341 (2003).
L. Nobs, F. Buchegger, R. Gurny, and E. Allemann. Current methods for attaching targeting ligands to liposomes and nanoparticles. J. Pharm. Sci. 93:1980–1992 (2004).
O. Ishida, K. Maruyama, H. Tanahashi, M. Iwatsuru, K. Sasaki, M. Eriguchi, and H. Yanagie. Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm. Res. 18:1042–1048 (2001).
J. T. Derksen, and G. L. Scherphof. An improved method for the covalent coupling of proteins to liposomes. Biochim. Biophys. Acta. 814:151–155 (1985).
L. Bourel-Bonnet, E. I. Pecheur, C. Grandjean, A. Blanpain, T. Baust, O. Melnyk, B. Hoflack, and H. Gras-Masse. Anchorage of synthetic peptides onto liposomes via hydrazone and alpha-oxo hydrazone bonds. Preliminary functional investigations. Bioconjugate Chem. 16:450–457 (2005).
S. F. Hassane, B. Frisch, and F. Schuber. Targeted liposomes: convenient coupling of ligands to preformed vesicles using “click chemistry”. Bioconjugate Chem. 17:849–854 (2006).
M. J. Copland, M. A. Baird, T. Rades, J. L. McKenzie, B. Becker, F. Reck, P. C. Tyler, and N. M. Davies. Liposomal delivery of antigen to human dendritic cells. Vaccine. 21:883–890 (2003).
S. Gal, I. Pinchuk, and D. Lichtenberg. Peroxidation of liposomal palmitoyllinoleoylphosphatidylcholine (PLPC), effects of surface charge on the oxidizability and on the potency of antioxidants. Chem. Phys. Lipids. 126:95–110 (2003).
S. Cavalli, A. R. Tipton, M. Overhand, and A. Kros. The chemical modification of liposome surfaces via a copper-mediated [3+2] azide-alkyne cycloaddition monitored by a colorimetric assay. Chem. Commun. 2006:3193–3195 (2006).
A. H. Dent. Optimizing bioconjugation processes. Pharm. Tech. Eur. 19:39–40, 43–46 (2007).
R. B. Silverman. Drug Discovery, Design, and Development. The Organic Chemistry of Drug Design and Drug Action, 2nd edn. Elsevier Scientific, San Diego, CA, 2004.
K. A. Stephenson, J. Zubieta, S. R. Banerjee, M. K. Levadala, L. Taggart, L. Ryan, N. McFarlane, D. R. Boreham, K. P. Maresca, J. W. Babich, and J. F. Valliant. A new strategy for the preparation of peptide-targeted radiopharmaceuticals based on an Fmoc-lysine-derived single amino acid chelate (SAAC). Automated solid-phase synthesis, NMR characterization, and in vitro screening of fMLF(SAAC)G and fMLF[(SAAC-Re(CO)3)+]G. Bioconjugate Chem. 15:128–136 (2004).
S. Liu, S. D. Edwards, and J. A. Barrett. 99mTc labeling of highly potent small peptides. Bioconjugate Chem. 8:621–636 (1997).
P. Bouziotis, M. Fani, S. C. Archimandritis, G. Loundos, M. Paravatou, R. Bicknell, A. L. Harris, S. Xanthopoulos, N. Stratis, and A. D. Varvarigou. Samarium-153 and technetium-99m-labeled monoclonal antibodies in angiogenesis for tumor visualization and inhibition. Anticancer Res. 23:2167–2171 (2003).
M. P. White, A. Mann, D. M. Cross, and G. V. Heller. Evaluation of technetium-99m red blood cell labeling efficiency in adults receiving chemotherapy and the clinical impact on pediatric oncology patients. J. Nucl. Med. Tech. 26:265–268 (1998).
R. Alberto, R. Schibli, A. Egli, A. P. Schubiger, U. Abram, and T. A. Kaden. A novel organometallic aqua complex of technetium for the labeling of biomolecules: synthesis of [99mTc(OH2)3(CO)3] + from [99mTcO4]- in aqueous solution and its reaction with a bifunctional ligand. J. Am. Chem. Soc. 120:7987–7988 (1998).
T. L. Mindt, H. Struthers, L. Brans, T. Anguelov, C. Schweinsberg, V. Maes, D. Tourwe, and R. Schibli. “Click to chelate”: synthesis and installation of metal chelates into biomolecules in a single step. J. Am. Chem. Soc. 128:15096–15097 (2006).
W. Chen, and G. Georgiou. Cell-surface display of heterologous proteins: from high-throughput screening to environmental applications. Biotechnol. Bioeng. 79:496–503 (2002).
J. A. Link, and D. A. Tirrell. Cell surface labeling of Escherichia coli via copper(I)-catalyzed [3 + 2] cycloaddition. J. Am. Chem. Soc. 125:11164–11165 (2003).
K. L. Kiick, E. Saxon, D. A. Tirrell, and C. R. Bertozzi. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. U.S.A. 99:19–24 (2002).
A. Basle, G. Rummel, P. Storici, J. P. Rosenbusch, and T. Schirmer. Crystal structure of osmoporin OmpC from E. coli at 2.0 ANG. J. Mol. Bio. 362:933–942 (2006).
Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless, and M. G. Finn. Bioconjugation by copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 125:3192–3193 (2003).
Q. Wang, E. Kaltgrad, T. Lin, J. E. Johnson, and M. G. Finn. Natural supramolecular building blocks: wild-type cowpea mosaic virus. Chem. Bio. 9:805–811 (2002).
Q. Wang, T. Lin, L. Tang, J. E. Johnson, and M. G. Finn. Icosahedral virus particles as addressable nanoscale building blocks. Angew. Chem., Int. Ed. Engl. 41:459–462 (2002).
Q. Wang, K. S. Raja, K. D. Janda, T. Lin, and M. G. Finn. Blue fluorescent antibodies as reporters of steric accessibility in virus conjugates. Bioconjugate Chem. 14:38–43 (2003).
M. H. Caruthers. Gene synthesis machines: DNA chemistry and its uses. Science (Washington, D.C.). 230:281–285 (1985).
H. K. Tewary, and P. L. Iversen. Qualitative and quantitative measurements of oligonucleotides in gene therapy: part II in vivo models. J. Pharm. Biomed. Anal. 15:1127–1135 (1997).
A. M. Gewirtz. Antisense oligonucleotide therapeutics for human leukemia. Curr. Opin. Hemat. 5:59–71 (1998).
M. Schena, D. Shalon, R. W. Davis, and P. O. Brown. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science (Wash. D.C.). 270:467–470 (1995).
T. S. Seo, Z. Li, H. Ruparel, and J. Ju. Click chemistry to construct fluorescent oligonucleotides for DNA sequencing. J. Org. Chem. 68:609–612 (2003).
F. Seela, V. R. Sirivolu, and P. Chittepu. Modification of DNA with octadiynyl side chains: synthesis, base pairing, and formation of fluorescent coumarin dye conjugates of four nucleobases by the alkyne-azide “click reaction”. Bioconjugate Chem. 19:211–224 (2008).
G. M. Cooper. The Cell Surface. The Cell: A Molecular Approach, 2nd edn. Sinauer, Sunderland, MA, 2000.
D. B. Werz, and P. H. Seeberger. Carbohydrates as the next frontier in pharmaceutical research. Chem. Eur. J. 11:3194–3026 (2005).
T. Liebert, C. Hänsch, and T. Heinze. Macromol. Rapid Commun. 27:208–213 (2006).
J. Hafrén, W. Zou, and A. Córdova. Macromol. Rapid Commun. 27:1362–1366 (2006).
C. Glaser. Beiträge zur kenntniss des acetenylbenzols. Ber. Dtsch. Chem. Ges. 2:422–424 (1869).
F. Straus. Acetylene bonding. Justus Liebig Ann. Chem. 342:190–265 (1905).
G. Eglinton and A. R. Galbraith. Cyclic diynes. Chem. Ind. (London). 1956:737–738 (1956).
P. Siemsen, R. C. Livingston, and F. Diederich. Acetylenic coupling: a powerful tool in molecular construction. Angew. Chem., Int. Ed. Engl. 39:2632–2657 (2000).
M. D. Cameron, and G. E. Bennett. Use of amines in the glaser coupling reaction. J. Org. Chem. 22:557–558 (1957).
F. Bohlmann, H. Schonowsky, E. Inhoffen, and G. Grau. Polyacetylenic compounds. LII. The mechanism of oxidative dimerization of acetylene compounds. Chem. Ber. 97:794–800 (1964).
E. H. Ryu, and Y. Zhao. Efficient synthesis of water-soluble calixarenes using click chemistry. Org. Lett. 7:1035–1037 (2005).
T. Wang, and Z. Guo. Copper in medicine: homeostasis, chelation therapy and antitumor drug design. Curr. Med. Chem. 13:525–537 (2006).
K. D. Held, C. F. Sylvester, K. L. Hopcia, and J. E. Biaglow. Role of Fenton chemistry in thiol-induced toxicity and apoptosis. Radiat. Res. 145:542–553 (1996).
J. E. Macdonald, J. A. Kelly, and J. G. C. Veinot. Iron/iron oxide nanoparticle sequestration of catalytic metal impurities from aqueous media and organic reaction products. Langmuir. 23:9543–9545 (2007).
Acknowledgements
We acknowledge financial supports from NIH Grant AR053325 and the College of Pharmacy, University of Nebraska Medical Center (teaching assistantship to CDH).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hein, C.D., Liu, XM. & Wang, D. Click Chemistry, A Powerful Tool for Pharmaceutical Sciences. Pharm Res 25, 2216–2230 (2008). https://doi.org/10.1007/s11095-008-9616-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9616-1