
ABSTRACT
Purpose
The goal of this study was to test the hypothesis that by controlling intracellular uptake, organic cation transporter 1, Oct1 is a key determinant of the disposition and toxicity of cis-diammine(pyridine)chloroplatinum(II)(CDPCP) and oxaliplatin.
Methods
Pharmacokinetics, tissue accumulation and toxicity of CDPCP and oxaliplatin were compared between Oct1-/- and wild-type mice.
Results
After intravenous administration, hepatic and intestinal accumulation of CDPCP was 2.7-fold and 3.9-fold greater in Oct1 wild-type mice (p < 0.001). Deletion of Oct1 resulted in a significantly decreased clearance (0.444 ± 0.0391 ml/min*kg versus 0.649 ± 0.0807 ml/min*kg in wild-type mice, p < 0.05) and volume distribution (1.90 ± 0.161 L/kg versus 3.37 ± 0.196 L/kg in wild-type mice, p < 0.001). Moreover, Oct1 deletion resulted in more severe off-target toxicities in CDPCP-treated mice. Histologic examination of the liver and measurements of liver function indicated that the level of hepatic toxicity was mild and reversible, but was more apparent in the wild-type mice. In contrast, the effect of Oct1 on the pharmacokinetics and toxicity of oxaliplatin in the mice was minimal.
Conclusions
Our study suggests that Oct1 plays an important role in the pharmacokinetics, tissue distribution and toxicity of CDPCP, but not oxaliplatin.
Similar content being viewed by others


INTRODUCTION
Membrane transporters play important roles in drug disposition and response (1–5). Recently, organic cation transporter 1 (OCT1), encoded by SLC22A1, has been shown to affect the pharmacokinetics and pharmacodynamics of the anti-diabetic drug, metformin (6,7). In humans, OCT1 is primarily expressed in the liver (8–10), whereas in rodents, Oct1 is expressed in equal abundance in the liver, kidney, and small intestine (11,12). In general, the human OCT1 mediates intracellular uptake of a broad range of structurally diverse monovalent organic cations with molecular weights of less than 400 Daltons (13,14). Substrates of OCT1 include endogenous compounds, such as choline, creatinine, and monoamine neurotransmitters; clinically used drugs, such as metformin, oxaliplatin and cimetidine; and a variety of xenobiotics, such as tetraethylammonium (TEA, model cation), N-methylquinine, and 1-methyl-4-phenylpyridinium (MPP+) (13,15,16). In vivo, Oct1-/- mice display impaired liver uptake and intestinal excretion of a subset of organic cations, such as TEA, MPP+, and metformin (4,6,17), suggesting that the transporter Oct1 is important for the uptake of certain xenobiotics and drugs into various tissues, including the liver in the animal model.
Platinum-based chemotherapies have been widely used to treat solid tumors since the 1970s (18). Although cisplatin is effective against several solid tumors, its clinical use is limited because of its toxic effects as well as the intrinsic and acquired resistance of some tumors to this drug (19). To overcome limitations related to toxicities and resistance, other platinum agents have been developed (20). Oxaliplatin, approved in 2002 for the treatment of colorectal cancer in combination with 5-FU/leucovorin (21), exhibits a different pattern of sensitivity to that of cisplatin in the NCI60-cell human tumor panel, and in combination with 5-FU/leucovorin produces response rates twice that of 5-FU/leucovorin regimens alone in the treatment of colorectal cancer, against which cisplatin is inactive (20,22,23). Moreover, compared with cisplatin, oxaliplatin shows a more favorable toxicity profile.
Cis-diammine(pyridine)chloroplatinum(II) (CDPCP), belongs to a different series of platinum analogues from cisplatin, carboplatin and oxaliplatin. In particular, it is a cationic triamine complex that possesses desirable physical properties, such as high stability and solubility in aqueous media. Although CDPCP violates some of the classical structure-activity relationship, its anti-cancer activity was established in murine tumor models (24). However, CDPCP has never been tested in humans, possibly because this compound only forms monofunctional complexes with DNA, and due to its polarity, the drug was not expected to cross cell membrane readily (25). A recent study, however, showed that CDPCP can largely escape repair, yet inhibit transcription very effectively, and its adducts should persist longer than those of cisplatin yet produce a similar number of downstream consequences that might raise the therapeutic potential of CDPCP relative to cisplatin (25).
Recently, we showed that CDPCP is an excellent substrate of OCT1 (25). When overexpressed in transfected cells, OCT1 substantially increased the cellular uptake and cytotoxicity of CDPCP, suggesting that OCT1 facilitates intracellular accumulation of CDPCP, thereby sensitizing the cells to CDPCP cytotoxicity. Our in vitro studies also indicated that OCT1 facilitates the accumulation of oxaliplatin, though the effect was much smaller in comparison to CDPCP (15,25). Currently, there is an increased interest in further developing CDPCP as an anti-cancer drug regarding the high selectivity of CDPCP for cells expressing hOCT1 and/or hOCT2, which are broadly expressed in human liver, kidney and human colorectal cancer (8–10,13,15).
In this study, we determined the pharmacokinetics and toxic properties of CDPCP and oxaliplatin in mice and tested the hypothesis that Oct1 plays a major role in the pharmacokinetics and toxicities of these drugs using Oct1-/- mice. Specifically, we examined the role of Oct1 in the pharmacokinetics, tissue accumulation and toxicities of CDPCP and oxaliplatin in Oct1-/- and wild-type mice. Our results demonstrate that Oct1 plays an important role not only in the pharmacokinetics of CDPCP, but also in its toxicities. However, the role of Oct1 in the pharmacokinetics of oxaliplatin was proven to be minimal. Our study sets the stage for further preclinical and clinical studies of CDPCP.
MATERIALS AND METHODS
Drugs and Reagents
Oxaliplatin and disopyramide were purchased from Sigma (St. Louis, MO). ITS (Insulin-Transferrin-Selenium) was purchased from Invitrogen (Carlsbad, CA). CDPCP was synthesized in our laboratory as described below. Solutions of oxaliplatin (5 mM) and CDPCP (10 mM) were freshly prepared in saline or PBS. The stock solutions were stored frozen at −20°C and discarded after 1 month of preparation. The cell culture media DMEM H21 medium, William’s E, heat-inactivated fetal bovine serum (FBS), and L-glutamine were purchased from the Cell Culture Facility of the University of California, San Francisco (San Francisco, CA).
Synthesis of CDPCP
CDPCP was synthesized by a method adapted from Hollis et al. (24). Briefly, to a solution of cisplatin (900 mg, 3 mmol) in 15 mL of anhydrous DMF, AgNO3 (485 mg, 2.85 mmol) was added at room temperature, and the mixture was allowed to stir for 24 h. The resulting precipitate of AgCl was removed by filtration before the addition of pyridine (242 μL, 3 mmol) to the filtrate. After the solution was stirred for an additional day, the DMF was removed under vacuum, and the remaining oil was triturated with CH2Cl2 (100 mL), followed by trituration with diethyl ether (50 mL). The resulting solid was filtered and then recrystallized from hot methanol to obtain product cis-[Pt(NH3)2(Pyr)Cl](NO3) as a light yellow solid (yield 720 mg, 1.77 mmol, 59%). 1H NMR (400 MHz, D2O) δ8.90 (m, 1H, Ar), 8.45 (m, 2H, Ar), 7.31 (m, 2H, Ar), 4.72, 4.26 (bs, 3H, NH 3); ESI-MS 343.1 [M]+; Elemental Analysis: Found (%) C, 15.12; H, 2.50; N, 13.62. Calculate for C5H11ClN4O3Pt (%): C, 14.80; H, 2.73; N, 13.81.
Cell Lines and Transfection
Human embryonic kidney (HEK) 293 cells stably transfected with the full-length reference human OCT1 cDNA (HEK-hOCT1) and with the empty vector (HEK-MOCK) were established previously in our laboratory (6,15). HEK293 cells stably transfected with pcDNA5/FRT vector (Invitrogen) containing the full-length reference mouse Oct1 cDNA (HEK-mOct1) were established using Lipofectamine 2000 (Invitrogen) per manufacturer’s instructions. The stable clones were selected with 75 μg/ml hygromycin B.
Primary Mouse Hepatocytes
Primary hepatocytes were isolated from Oct1+/+ and Oct1-/- mice by the UCSF Liver Center using the standard collagenase method (26). For the CDPCP uptake assay, the isolated primary hepatocytes were seeded onto collagen-coated 6-well plates (Becton Dickenson, Bedford, MA) at a density of 0.8 × 106 cells/well in culture medium. After attachment (2–3 h), the cells were maintained in fresh medium for another 16–24 h followed by drug treatment as described below. For the uptake of oxaliplatin, the freshly isolated hepatocytes in suspension (1.5 × 105 cells/tube) instead of attached cells were used. During the incubation, the suspended cells were mixed several times by gentle vortex.
Cell Culture
The culture medium for stably transfected cells was DMEM H21 medium supplemented with 10% FBS, 100 units/ml penicillin and 100 μg/ml streptomycin and 60 μg/ml hygromycin B. The culture medium for primary hepatocytes was William’s E medium supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 10% fetal bovine serum (FBS), 0.1 μM dexamethasone, 2 mM L-glutamine, 1X ITS (100X ITS from Invitrogen). All cell lines were grown at 37°C in a humidified atmosphere with 5% CO2.
Cellular Accumulation of Platinum
The cellular accumulation of platinum was determined as described previously (15) with some modifications. Briefly, the cells were incubated in serum-free culture medium containing the indicated platinum compounds with or without a specific inhibitor at 37°C in 5% CO2 for 1 h unless specified. After incubation, cells were washed with ice-cold PBS three times. Then the cells were dissolved in 100 μl of 70% nitric acid at 65°C for at least 2.5 h. Distilled water containing 10 ppb of iridium (Sigma) and 0.1%Triton X-100 was added to the samples to dilute nitric acid to 7%. The platinum content was measured by inductively coupled plasma mass spectrometry (ICP-MS) in the Analytical Facility at the University of California at Santa Cruz (Santa Cruz, CA). Cell lysates from a set of identical cultures were used for BCA protein assay. Cellular platinum accumulation was normalized to the protein content.
Platinum-DNA Adduct Formation
The platinum content associated with genomic DNA was determined as described previously (15) with some modifications. Briefly, the cells were incubated in the serum-free culture medium containing platinum drugs with or without an OCT1 inhibitor at 37°C in 5% CO2 for 1 h unless specified. After incubation, cells were washed three times with ice-cold PBS. Genomic DNA was isolated from the cells using Wizard Genomic DNA Purification kit (Promega, Madison, WI) according to the manufacturer’s protocol. The DNA-bound platinum was determined by ICP-MS. The DNA content from the same DNA preparation was measured by absorption spectroscopy at 260 nm. Platinum-DNA adduct level was normalized to total DNA content.
RNA Isolation
Total RNA from primary hepatocytes was isolated using an RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer’s protocols. Total RNA from tissues was extracted using Trizol (Invitrogen). During the RNA purification procedure, DNase I was treated to digest residual genomic DNA. RNA was quantified spectrophotometrically at 260 nm. Extracted RNA was stored at −80°C until use.
Quantitative Real-Time PCR
Five μg of total RNA from each sample was reverse transcribed into cDNA using a superscript first-strand cDNA synthesis kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was carried out in a 96-well plate in a total volume of 10 μl reaction solution which includes a cDNA equivalent of 2 μg total RNA, specific probe and Taqman Universal Master Mix (Applied Biosystems, Foster City, CA). The thermal cycling conditions (ABI Prism 7700) were 95°C for 20 s followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. The amplification of glyceraldehydes-3-phosphate dehydrogenase (GAPDH) mRNA was used as an internal control.
Animals
Oct1-/- mice were generated as described elsewhere (6). The animals used in all experiments were in FVB background and were age-matched Oct1-/- and Oct1+/+ (wild-type) mice between 8 and 10 weeks of age. All animals were housed in a virus-free, temperature-controlled facility on a 12-h light-dark cycle. Mice were allowed standard mouse food and water ad libitum. All experiments on mice were approved by the Institutional Animal Care and Use Committee of University of California at San Francisco and carried out according to the “Principles of Laboratory Animal Care” (NIH publication #85-23, revised in 1985).
Tissue Accumulation Studies in Mice
Eight-week-old male Oct1+/+ and Oct1-/- mice were given platinum drugs in saline via tail vein injection. The animals were sacrificed at different time points after treatment. Blood samples were collected by heart puncture and transferred into heparinized BD vacutainer blood collection tubes. Blood was centrifuged for 30 min at 1000 x g, and the plasma was decanted and frozen at −80°C until analysis. Tissues (liver, kidney, heart, lung, brain, intestine, spleen and muscle) were collected as quickly as possible and snap frozen in liquid nitrogen. Five μl of plasma and about 10–30 mg of tissues were used for total platinum measurement by ICP-MS.
Pharmacokinetics Study in Mice
Eight-week-old male Oct1+/+ and Oct1-/- mice were given platinum drugs in saline via tail vein injection and placed in metabolic cages for 24 h. Blood samples (20 μl) were collected at different time points by tail vein bleeding into heparinized micro-hematocrit capillary tubes (Fisher, Pittsburg, PA). Blood was centrifuged for 5 min using Microhematocrit Centrifuge (Thermo Fisher Scientific Inc. Waltham, MA), and the plasma was decanted and frozen at −80°C until analysis. Urine was collected from tubes attached to the cages. Tissues (liver, kidney, intestine, muscle) were collected as quickly as possible upon the last time point and snap frozen in liquid nitrogen. Five μl of plasma, urine and about 10–30 mg of tissues were used for total platinum measurement by ICP-MS. The pharmacokinetics parameters were obtained by two-compartmental analysis using WinNonlin 4.0 (Pharsight Corporation, Mountain view, CA).
Toxicity Study in Mice
Eight-week-old male Oct1-/- and Oct1+/+ mice were randomized and treated with saline or platinum drugs via tail vein injection (CDPCP) or i.p. injection (oxaliplatin) once weekly for a total of 3 weeks (CDPCP) or once daily for a total of 5 days (oxaliplatin). The animals were closely monitored and sacrificed when they became moribund. Body weight was monitored and recorded. All surviving animals were sacrificed 24 h after the last treatment. On the day of the terminal sacrifice, the animals were anesthetized with isoflurane, and blood (500–1000 μl) was collected by heart puncture and centrifuged for 30 min at 1000 X g at 4°C. The serum was frozen at −80°C until analysis. Tissues (liver, kidney, intestine, and muscle) were collected and divided into two halves: one half of the tissues were fixed in 4% paraformaldehyde for histopathology examination, and the other half of the tissues were snap frozen in liquid nitrogen.
Hematological Evaluation
The whole blood (50–100 μl) was placed in K2EDTA coated tubes, and complete blood count (CBC) tests were conducted within 6 h by Mouse Pathology Core of UCSF Helen Diller Family Comprehensive Cancer Center.
Clinical Chemistry Evaluation
Alaninetransaminase (ALT), aspartatetransaminase (AST), alkaline phosphatase (ALKP), total protein, albumin, glucose, blood urea nitrogen (BUN), creatinine and electrolytes in serum were measured by the Clinical Laboratory of the San Francisco General Hospital.
Histopathological Examination
The tissues (liver, intestine and kidney) fixed in 4% paraformaldehyde were embedded in paraffin, cut into thin sections and mounted on glass slides. The tissue sections were stained with hematoxylin and eosin for microscopic examination.
Statistical Analysis
Unless specified, data are expressed as mean ± standard deviation (SD). Data were analyzed statistically using the unpaired Student’s t-test and Fisher exact test according to the sample size. Multiple comparisons were performed with Dunnett’s two-tailed test after one-way ANOVA. A p value of less than 0.05 was considered statistically significant.
RESULTS
Platinum Uptake in Cells Over-Expressing hOCT1 and mOct1 After Exposure to CDPCP and Oxaliplatin
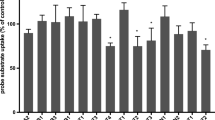
As shown in Fig. 1a, we observed that the uptake of platinum in HEK-mOct1 transfected cells exposed to CDPCP (10 μM) (1470 ± 66.6 pmol/mg protein/hr) was 53-fold greater than that in HEK-MOCK cells (27.4 ± 7.43 pmol/mg protein/hr) (p < 0.001). This effect was slightly greater than the effect of hOCT1 on platinum uptake after exposure to CDPCP (10 μM) (852 ± 135 pmol/mg protein/hr). The effect of hOCT1 and mOct1 on the cellular uptake of oxaliplatin was much smaller than on CDPCP uptake, and, again, mOct1, in comparison to hOCT1, promoted a greater uptake of platinum in cells exposed to oxaliplatin (p < 0.001; Fig. 1b).

Intracellular uptake of platinum after 1-h exposure to platinum drugs in cells over-expressing hOCT1 and mOct1. The cellular uptake of platinum in hOCT1-transfected (striped bar), mOct1-transfected (solid bar) cells and in the corresponding MOCK (white bar) cells after incubation with CDPCP (A) and oxaliplatin (B) were determined as described in Materials and Methods. Briefly, the cells were incubated in serum-free culture medium containing CDPCP (10 μM) or oxaliplatin (10 μM) at 37°C and 5% CO2 for 1 h. Total platinum concentration inside the cells was determined using ICP-MS. Total platinum concentration was normalized to protein content. Data are presented as the mean ± SD (n = 6). a: significantly greater than in MOCK cells (P < 0.001); b: significantly greater than in hOCT1-transfected cells (P < 0.001).
Intracellular Platinum Uptake and DNA Adduct Formation of Platinum Drugs in Primary Mouse Hepatocytes
To examine the role of Oct1 in the disposition of CDPCP in the liver, we compared the intracellular uptake and DNA adduct formation of CDPCP in hepatocytes derived from Oct1 wild-type mice with that in hepatocytes derived from Oct1-/- mice. The studies showed that the hepatocellular uptake of CDPCP (10 μM) after 1 hr exposure in hepatocytes from Oct1 wild-type mice (251 ± 94.6 pmol/mg protein/hr) was 2.9-fold of that in hepatocytes from Oct1 -/- mice (87.8 ± 7.61 pmol/mg protein/hr) (p < 0.01; Fig. 2a). Co-incubation of CDPCP with disopyramide (150 μM), an Oct1 inhibitor, resulted in a 4.5-fold decrease in the platinum uptake in hepatocytes from Oct1 wild-type mice (control versus disopyramide-treated, 251 ± 94.6 pmol/mg protein/hr versus 55.9 ± 1.41 pmol/mg protein/hr) (p < 0.01) with no effect in hepatocytes from Oct1-/- mice (control versus disopyramide-treated, 87.8 ± 7.61 pmol/mg protein/hr versus 95.8 ± 17.6 pmol/mg protein/hr) (p > 0.05; Fig. 2a). The platinum-DNA adduct level after 1 h exposure to CDPCP in hepatocytes from Oct1 wild-type mice (0.0857 ± 0.0232 pmol/μg DNA) was 2.4-fold greater than that in hepatocytes from Oct1-/- mice (0.0351 ± 0.00344 pmol/μg DNA) (p < 0.01; Fig. 2b). Co-incubation of CDPCP with disopyramide (150 μM) significantly decreased (3.7-fold; p < 0.01) platinum-DNA adduct formation in hepatocytes from Oct1 wild-type mice (control versus disopyramide treated, 0.0857 ± 0.0232 pmol/μg DNA versus 0.0233 ± 0.00847 pmol/μg DNA) (p < 0.01) with no effect in hepatocytes from Oct1-/- mice (control versus disopyramide-treated, 0.0351 ± 0.00344 pmol/μg DNA versus 0.0338 ± 0.00798 pmol/μg DNA) (p > 0.05; Fig. 2b). Collectively, data shown in Fig. 2a and b suggest that Oct1 mediates the uptake and binding to DNA of CDPCP in mouse hepatocytes.

Effect of Oct1 on the intracellular uptake of platinum in primary mouse hepatocytes after exposure to CDPCP and oxaliplatin. Intracellular platinum uptake (A) and platinum-DNA adduct formation (B) after 1 h exposure to 10 μM CDPCP in the absence (white bars) or presence (black bars) of an Oct1 inhibitor, disopyramide (150 μM) and intracellular platinum uptake after 30 min (C) and 120 min (D) exposure to 1 μM and 5 μM of oxaliplatin in hepatocytes from Oct1 wild-type (white bars) and Oct1-/- mice (black bars) were determined as described in Materials and Methods. Total platinum concentration inside the cells and DNA-bound platinum was determined using ICP-MS. Total platinum concentration was normalized to protein content, and DNA-bound platinum was normalized to DNA content. Data are presented as the mean ± SD (n = 3) and from a representative experiment. * P < 0.05, ** P < 0.005.
For oxaliplatin, the intracellular uptake rate of platinum increased proportionally with increasing concentrations of oxaliplatin in hepatocytes isolated from both Oct1+/+ and Oct1-/- mice (Fig. 2c and d). In all hepatocytes isolated from Oct1+/+ and Oct1-/- mice, the intracellular uptake rate of oxaliplatin was similar between 30 min exposure and 120 min exposure. Compared to hepatocytes isolated from Oct1-/-mice, hepatocytes isolated from Oct1+/+ mice had significantly higher platinum uptake rate after incubation with 1 μM and 5 μM of oxaliplatin for 30 min and 120 min. The hepatocellular uptake rate of oxaliplatin after 30 min exposure in hepatocytes from Oct1 wild-type mice (1 μM versus 5 μM, 1.54 ± 0.300 pmol/mg protein/hr versus 5.55 ± 0.384 pmol/mg protein/hr) was 1.8-fold and 1.4-fold of that in hepatocytes from Oct1 -/- mice (1 μM versus 5 μM, 0.855 ± 0.184 pmol/mg protein/hr versus 3.97 ± 0.500 pmol/mg protein/hr), respectively (p < 0.05; Fig. 2c). Similar results were obtained after 120 min of exposure (Fig. 2d). These data indicate that Oct1 is likely to play a role in facilitating the transport of oxaliplatin across hepatocytes. However, the effect of Oct1 on oxaliplatin uptake is less than its effect on CDPCP uptake.
Tissue Accumulation of Platinum After Treatment with Platinum Drugs in Oct1 Wild-Type and Oct1-/- Mice
To extend the in vitro cellular studies, we examined the role of Oct1 in the accumulation of platinum drugs in various tissues after dosing to Oct1 wild-type and Oct1-/- mice (Fig. 3). The tissue accumulation study of CDPCP (8 mg/kg) showed that total levels of platinum in the plasma 1 h after CDPCP treatment were significantly higher (1.3-fold) in Oct1-/- mice than in Oct1 wild-type mice (Fig. 3a, p < 0.001). Comparing the absolute platinum level in various tissues (Table 1 and Fig. 3a), we observed that the platinum accumulation levels in liver and intestine were significantly higher in Oct1 wild-type mice than in Oct1-/- mice (p < 0.01). However, the platinum accumulation levels in kidney, heart and skeletal muscle in Oct1 wild-type mice were significantly lower than in Oct1-/- mice (p < 0.05). When the absolute platinum level in the various tissues was normalized to the corresponding plasma concentration (Table 1 and Fig. 3b), we observed that platinum levels in the muscle and kidney were no longer significantly different between Oct1 wild-type and Oct1-/- mice (p > 0.05), while the normalization to plasma concentration had no effect on the platinum accumulation level in the heart (p < 0.01). The normalized platinum accumulation was 2.7-fold greater in the liver of Oct1 wild-type compared to the knockout mouse and 3.9-fold greater in the intestine of Oct1 wild-type compared to knockout mice. The highest platinum accumulation levels were found in liver and intestine in both Oct1 wild-type and Oct1-/- mice (Table 1). Taken together, this study suggests that the expression of Oct1 in liver and intestine contributes to the enhanced accumulation of CDPCP in these organs.

Effects of Oct1 on tissue accumulation of platinum drugs and plasma concentrations after intravenous dosing of CDPCP and oxaliplatin in mice. Absolute platinum accumulation levels (A, C), normalized platinum accumulation levels (B, D) in various tissues were measured 1 h post dosing of 8 mg/kg of CDPCP (A, B) and oxaliplatin (C, D), respectively in Oct1wild-type and Oct1-/- mice. Plasma concentrations of CDPCP (E), oxaliplatin (F) in Oct1wild-type and Oct1-/- mice were measured at 15, 30 min, 1, 2, 3, 4, 8, 10, 24, 48 hr after i.v. dosing of CDPCP (8 mg/kg) and oxaliplatin (8 mg/kg). The platinum levels in plasma and various organs were determined using ICP-MS. The unit for tissue accumulation of platinum is nmol/gram tissue, the unit for plasma is μM (A, C). Data are presented as the mean ± SD * P < 0.05; ** P < 0.001.
The effect of OCT1 on the tissue distribution of oxaliplatin was first examined 6 h after intravenous dosing of oxaliplatin (8 mg/kg). The absolute platinum levels and the platinum levels normalized to plasma platinum concentrations in various tissues including liver, kidney, intestine, spleen, bone marrow and muscle showed no significant difference between Oct1+/+ and Oct1-/- mice (p > 0.05). The effect of Oct1 on the tissue distribution of oxaliplatin was reexamined after a shorter treatment duration. After 1 h treatment with oxaliplatin (8 mg/kg), the plasma concentration of oxaliplatin in Oct1+/+ was slightly but significantly lower than that in Oct1-/- mice (Oct1+/+ versus Oct1-/-, 5.96 ± 0.54 μM versus 6.69 ± 0.62 μM; p < 0.05; Fig. 3c). Comparing the oxaliplatin accumulation in various tissues, we observed that the absolute oxaliplatin levels in intestine, kidney and heart in Oct1-/- mice were significantly higher than in Oct1+/+ mice (Fig. 3c; p < 0.05). However, after normalization with corresponding plasma concentrations, the differences in renal, cardiac, and intestinal accumulation between Oct1+/+ and Oct1-/- mice were not observed (Fig. 3d).
Platinum accumulation in various tissues was also measured at the end of pharmacokinetics studies, i.e. 48 h after dosing. The effects of Oct1 on the tissue accumulation of platinum drugs were similar to the data obtained at 1 h for both CDPCP and oxaliplatin, i.e. Oct1 enhanced the hepatic and intestinal accumulation of CDPCP, and exerted insignificant effect on the oxaliplatin accumulation.
Pharmacokinetics of Platinum Drugs in Oct1 Wild-Type and Oct1-/- Mice
The time-course of platinum in plasma following a single i.v. dose of CDPCP (8 mg/kg) to Oct1 wild-type and Oct1-/- mice is shown in Fig. 3e. Similar to the pharmacokinetics of AMD473 (picoplatin), a third-generation platinum agent, which is currently in Phase-III clinical trials for treatment of non-small-cell lung cancer (NSCLC) (27,28), the concentration versus time course of total platinum followed a two-compartment model in both Oct1 wild-type and Oct1-/- mice. The mean plasma concentrations of CDPCP in Oct1-/- mice were significantly higher than that in Oct1 wild-type mice at all time pointes (Fig. 3e). The mean pharmacokinetics parameters are shown in Table 2. Comparing the pharmacokinetics parameters of CDPCP in Oct1 wild-type mice with Oct1-/- mice, we observed that the plasma platinum exposure over 48 hrs (AUC0-48) in Oct1-/-mice was 1.69-fold of that in Oct1 wild-type mice (p < 0.01), and the volume of distribution of CDPCP in Oct1 wild-type mice was 1.77-fold of that in Oct1-/- mice (p < 0.01). Moreover, a significantly greater total clearance was observed in Oct1 wild-type mice in comparison to Oct1-/- mice (p < 0.05), although renal clearance showed no difference between Oct1+/+ mice and Oct1-/- mice. The renal clearance contributed to about 50% of the total clearance of platinum from plasma in both Oct1+/+ and Oct1-/- mice.
Consistent with the pharmacokinetics of CDPCP and the previous report of oxaliplatin pharmacokinetics in rats (29), the concentration versus time course of total platinum following i.v. administration of oxaliplatin (8 mg/kg) exhibited a biexponential decline in both Oct1 wild-type and Oct1-/- mice (Fig. 3f). The pharmacokinetics parameters were not significantly different between Oct1+/+ and Oct1-/- mice, as shown in Table 3. The clearance and volume of distribution of oxaliplatin in both Oct1+/+ and Oct1-/- mice were similar to that reported in BD2F1 mice (30).
Toxicity Study of CDPCP in Oct1 Wild-Type and Oct1-/- Mice
Based on the results of the dose-range finding toxicity study that CDPCP at 90 mg/kg and above resulted in significant changes in body weight, liver-to-body-weight ratio, liver function and hematology, we selected doses between 90 mg/kg and 120 mg/kg for use in evaluating the role of Oct1 in CDPCP toxicity. The role of Oct1 in toxicity of CDPCP was determined by comparing the toxicity profile of CDPCP between Oct1+/+ and Oct1-/- mice. The dose of CDPCP started with 120 mg/kg, which later was reduced to 90 mg/kg due to death or moribundity observed in Oct1-/- mice at 120 mg/kg.
CDPCP Resulted in Much Greater Body Weight Loss in Oct1-/- Mice
Compared with saline control group, CDPCP treatment caused body weight loss in both Oct1+/+ and Oct1-/- mice (Fig. 4a). However, the body weight loss was much greater in Oct1-/- mice than in Oct1+/+ mice. The greater decrease in body weight in Oct1-/- mice treated with CDPCP was consistent with the higher plasma platinum concentration in these mice (p < 0.005; Fig. 6a).

Effects of Oct1 on body weight loss and liver toxicities of CDPCP in mice. Oct1-/- andOct1+/+ mice were treated with saline or CDPCP via tail vein injection once weekly for a total 3 weeks (at least 9 animals per group). The mice were fasted for at least 16 h before sacrifice. Body weight (A) was monitored three times per week. Blood and various tissues were collected 24 hrs after the last dose; liver weight was measured. Liver to body weight ratios (B), total protein levels in serum (C), glucose levels in serum (D) and activities of liver enzymes (E), were compared among groups. Liver histopathology (F) was examined; the magnification is 40X. Data are presented as the mean ± SD (n ≥ 9). * P < 0.05; ** P < 0.001.
Hepatic Toxicity of CDPCP was Enhanced in Oct1+/+ Mice
The liver-to-body-weight ratio was significantly increased compared to saline control in both Oct1+/+ and Oct1-/- mice (Fig. 4b). However, the liver-to-body-weight ratio change in Oct1+/+ mice, which was 28.8% increase in CDPCP-treated mice compared with saline-treated mice, was more striking than that in Oct1-/- mice, which was 14.7% increase in CDPCP-treated mice compared with saline-treated mice. To exclude the confounding effect of less body weight decrease in Oct1+/+ mice on the liver-to-body-weight ratio, the actual liver weights between Oct1+/+ and Oct1-/- mice treated with CDPCP and saline were also compared. Our data showed that the actual liver weight was increased by CDPCP treatment in Oct1+/+ mice but not in Oct1-/- mice (CDPCP versus saline: 1.23 g versus 0.973 g in Oct1+/+ mice, p < 0.00001; 1.02 g versus 1.00 g in Oct1-/- mice, p > 0.05), indicating that CDPCP indeed resulted in hepatomegaly in Oct1+/+ mice. Moreover, CDPCP resulted in significant decrease in total serum protein levels and significant increase in glucose levels in Oct1+/+ mice, but not in Oct1-/- mice (Fig. 4c, d). However, the activities of ALT, AST and ALKP were not affected by CDPCP in either Oct1+/+ or Oct1-/- mice (Fig. 4e).
Histopathological examination of liver from Oct1-/- mice treated with either saline or CDPCP showed no clear signs of toxicity, while the liver from Oct1+/+ mice treated with CDPCP appeared to have enlarged and rounded hepatocytes, which are normally hexagonal (Fig. 4f). The irregular white spots, which were considered to be aqueous materials inside the hepatocytes pushed the nucleus aside, indicating CDPCP resulted in liver edema in Oct1+/+ mice. The hepatic lesion was more striking in the portal vein area than in other regions of the liver. Comparing the frequency and the severity of hepatic lesions, we observed that CDPCP caused more severe and frequent liver damage in Oct1+/+ mice in comparison to Oct1-/- mice, as indicated by mild to moderate swollen hepatocytes (Table 4). The differences did not reach statistical significance using Fisher exact test (p = 0.07), though trends were apparent. The greater hepatic toxicity in Oct1+/+ mice was consistent with the higher absolute hepatic platinum accumulation in these mice (Fig. 6b; p < 0.005). Further, at higher doses of CDPCP (120 mg/kg) used in our dose-finding studies, we observed significant increases in ALT in the wild-type, but not in the knockout, mice (data not shown). These data strongly suggest that Oct1 deletion protects animals from developing liver toxicity and that Oct1 plays a role not only in CDPCP disposition but also in CDPCP-associated organ toxicity.
Renal Toxicity of CDPCP was More Pronounced in Oct1-/- Mice
In both Oct1+/+ and Oct1-/- mice administered CDPCP, serum creatinine levels were not significantly different from mice administered saline (p > 0.05; Fig. 5a). Serum BUN levels were significantly reduced in CDPCP-treated Oct1+/+ mice compared with saline-treated Oct1+/+ mice (p < 0.05), while serum BUN was not affected by CDPCP in Oct1-/- mice (p > 0.05; Fig. 5a). CDPCP treatment increased renal mRNA levels of Kim-1, a more sensitive and quantitative renal biomarker of early tubular damage, by 19.3-fold over control value in Oct1+/+ mice. However, the elevation of renal Kim-1 expression in Oct1-/- mice following CDPCP treatment was much more striking than in Oct1+/+ mice; the fold difference over saline control was 117 (Fig. 5b).

Effect of Oct1 on renal toxicity of CDPCP in mice. Oct1-/- and Oct1+/+ mice were treated with saline or CDPCP via tail vein injection once weekly for a total 3 weeks (at least 9 animals per group). The mice were fasted for at least 16 h before sacrifice. Blood and various tissues were collected 24 hrs after the last dose. Creatinine and BUN in serum (A) were measured. Total RNA from kidney was reverse-transcribed and the synthesized cDNA was subject to qRT-PCR using Kim-1 (B) probe. The amplification of glyceraldehydes-3-phosphate dehydrogenase (GAPDH) mRNA was used as an internal control. Kidney histopathology (C) was examined; the magnification is 20X; arrows indicate kidney lesions. Data are presented as the mean ± SD. * P < 0.05; a: significantly greater than corresponding saline control groups (P < 0.01); b: significantly greater than Oct1+/+ mice (P < 0.05).
Histologic evaluation of kidneys from CDPCP-treated mice demonstrated tubular cast formation, which was revealed by eosinophilic amorphous material and pyknotic and karyorhectic debris (Fig. 5c.II) and degeneration of tubular cells, including tubular dilatation, tubular cell vacuolation, tubular cell detachment (Fig. 5c.III). The renal morphology change seemed to localize to the outer medulla (Fig. 5c.II and c.III). Consistent with the Kim-1 expression study, the kidney tubular lesions were most prominent in Oct1-/- mice treated with CDPCP compared with Oct1+/+ mice treated with CDPCP (Fig. 5c.I) and saline-treated control animals. The incidence and type of kidney lesion are listed in Table 4. We observed that CDPCP treatment resulted in a lower rate of kidney damage in Oct1+/+ mice than in Oct1-/- mice, and the rate difference was marginally significant (p = 0.07). The absolute renal platinum accumulation in Oct1-/- mice was remarkably greater than that in Oct1+/+ mice (Fig. 6c; p < 0.05). These data indicate that CDPCP is more toxic to the kidney in Oct1-/- mice.

Platinum concentrations in plasma and various tissues collected from the toxicity study. Oct1-/- and Oct1+/+ mice were treated with saline or CDPCP via tail vein injection once weekly for a total 3 weeks (at least 9 animals per group). The mice were fasted for at least 16 h before sacrifice. Blood (A), liver (B), kidney (C), and intestine (D) were harvested 24 hrs after the last dose. Platinum was measured by ICP-MS. Data are presented as the mean ± SD. * P < 0.05; ** P < 0.005.
Toxicities of CDPCP in Other Sites
The hematological toxicity of CDPCP was more pronounced in Oct1-/- mice, which was reflected by significant changes in white blood cell parameters, including neutrophil (%) and lymphocytes (%), and in platelet parameter, i.e. mean platelet volume (MPV), which were observed in CDPCP-treated Oct1-/- mice but not in Oct1+/+ mice (p < 0.05, data not shown). The higher plasma platinum concentration in Oct1-/- mice supported greater hematological toxicity of CDPCP in these mice (Fig. 6a; p < 0.005). Histological examination of intestine showed no signs of intestinal toxicity in either Oct1+/+ or Oct1-/- mice following CDPCP treatment, although significantly higher intestinal accumulation of CDPCP in Oct1+/+ mice was observed (Fig. 6d, p < 0.05).
Toxicity Study of Oxaliplatin in Oct1 Wild-Type and Oct1-/- Mice
To assess the role of Oct1 in the toxicities associated with oxaliplatin treatment, especially the hepatotoxicity, the Oct1+/+ and Oct1-/- mice (n = 5 per group) were treated with 6 mg/kg oxaliplatin or saline via i.p. injection once daily for a total of 5 days. Oxaliplatin caused more severe body weight loss in Oct1+/+ mice than in Oct1-/- mice; the relative body weight loss in Oct1+/+mice was 15.1%, whereas the relative body weight loss in Oct1-/- mice was 6.8%. Although there was a significantly greater hepatic accumulation of oxaliplatin in Oct1+/+ mice, no difference was observed in liver size or activities of three liver-associated enzymes: ALT, AST and ALKP between Oct1+/+ and Oct1-/- mice, respectively (data not shown). The liver histopathology examination showed no clear sign of liver damage associated with oxaliplatin treatment in either Oct1+/+ or Oct1-/- mice. The data suggest that the hepatic toxicity of oxaliplatin observed in humans was not observed under the conditions used in this study, and a clear role for Oct1 in oxaliplatin-induced hepatotoxicity could not be established. The effect of Oct1 deletion on body weight was striking and suggests that Oct1 plays a role in the overall toxicity of oxaliplatin in the mice. Since oxaliplatin was not hepatotoxic under the conditions used in this study, we could not assess the effect of Oct1 on hepatotoxicity of oxaliplatin. The dosing regimen or dosing route used in our study might not be appropriate to elicit the hepatic toxicity.
Oct1 Deletion Resulted in Enhanced Expression of Oct2 and Oct3 in Various Tissues
mRNA levels of Oct2, Oct3, Ent1, Mate1 and Ctr1 in various tissues were determined by quantitative RT-PCR. The expression levels of Oct2 in liver was increased by 2.6-fold in Oct1-/- mice compared with Oct1+/+ mice (Fig. 7a; p < 0.05). The increase of Oct2 expression in kidney and intestine of Oct1-/- mice was 1.8- and 2.5-fold compared with Oct1+/+ mice, respectively. However, these changes did not reach statistical significance due to the relatively large variation (Fig. 7b, c; p > 0.05). The expression of Oct3 in liver in Oct1-/- mice was 5.7-fold of that in Oct1+/+ mice (Fig. 7d, p < 0.001). There was no change in the expression levels of Ent1, Mate1 and Ctr1 in liver between Oct1+/+ mice and Oct1-/- mice.

Effects of Oct1 deletion on the expression of Oct2 and Oct3 in various tissues in mice. The expression of Oct2 in liver (A), kidney (B) and intestine (C) and the expression of various drug transporters in liver (D) were determined by quantitative RT-PCR. Various tissues were harvested from age-matched Oct1+/+ and Oct1-/- mice. Total RNA was extracted from these tissues and reverse-transcribed. The synthesized cDNA was subject to qRT-PCR using an Oct2, Ent1, Oct3, Mate1 and Ctr1 probes. The amplification of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used as an internal control. Data are presented as the mean ± SD (n = 3). * P < 0.05; ** P < 0.001.
DISCUSSION
Though shown to be a potent anti-tumor platinum compound, CDPCP was never tested clinically. Because of its positive charge, the drug may have been thought to have poor pharmacokinetic characteristics. In recent studies, OCT1 was shown to play a role in the cellular uptake and consequent cytotoxicity of CDPCP in cell lines over-expressing OCT1 (25). Accordingly, we hypothesized that OCT1 mediates the uptake of CDPCP in vivo in various tissues, particularly the intestine and liver, and that OCT1-mediated transmembrane flux may result in a favorable pharmacokinetic and toxicity profile for CDPCP. To test our hypothesis, we performed a comprehensive pharmacokinetic analysis using genetically modified Oct1-/- mice in this study. The results suggest that deletion of Oct1 not only decreased the uptake of cellular platinum but also reduced the level of platinum-DNA adduct formation in hepatocytes after CDPCP exposure (Fig. 2). These results are in agreement with those in cells transfected with human OCT1 (25), although the fold difference in platinum levels between hepatocytes harvested from Oct1+/+ versus Oct1-/-mice was much less than that between HEK293 cells over-expressing human OCT1 and control cells (25). These differences were not due to species differences between mOct1 and hOCT1, since our data suggest that CDPCP is a comparable or better substrate of mOct1 than of hOCT1 (Fig. 1). Drug- metabolizing enzymes and drug transporters normally are lost by liver cells in culture as a consequence of the adaptation of cells to the in vitro environment. Lecureu et al. reported that hepatocytes maintained in primary culture for several days were known to not only lose expression of albumin and cytochrome P-450 but also displayed low levels of OCT1 expression and activity (31). Studies from our laboratory also confirmed that the expression level of Oct1 in primary hepatocytes cultured from mouse liver was considerably lower than that in mouse liver (data not shown), suggesting that the isolation and culture procedures likely resulted in a reduced expression level of the Oct1 protein and therefore a reduction in the observed effect of Oct1 in the isolated hepatocytes.
The significantly higher hepatic and intestinal accumulation of CDPCP in Oct1+/+ mice was consistent with the abundance of Oct1 in liver and intestine of rodents (12,13). Normalization of tissue platinum levels to plasma concentrations of CDPCP excluded the effects of different plasma exposure on the uptake of CDPCP into the various tissues. Although there is strong Oct1 expression in the kidney of mice (12,13,32), it was notable that no differences were observed in the normalized levels of platinum in the kidneys of Oct1+/+ versus Oct1-/- mice. This may be explained by the redundant function of Oct1/2 in the rodent kidney, and the Oct2 strongly expressed in the kidney can completely compensate for the loss of Oct1. Previously, CDPCP was shown to be a substrate of OCT2 (25). It is notable that enhanced expression of Oct2 and Oct3 in the liver of Oct1-/- mice may have resulted in a reduction in the observed effect of Oct1 on CDPCP uptake in the liver (Fig. 7a and d).
Consistent with the high hepatic accumulation in Oct1+/+ mice, the hepatotoxicity of CDPCP was more prominent in these mice, as demonstrated by the increased liver-to-body-weight ratio, decreased total serum protein levels, increased serum glucose levels and histological changes at 90 mg/kg in the Oct1 wild-type mice. The low BUN in the wild-type mice treated with CDPCP is also consistent with hepatotoxicity in these mice. A low BUN usually has little significance, but its causes include liver problems, malnutrition (insufficient dietary protein), or excessive alcohol consumption. Although there was greater hepatic toxicity in Oct1+/+ mice, the level of toxicity was considered mild and reversible. If CDPCP is developed as an anti-cancer drug, hepatic toxicity may not be a concern.
Recently, use of oxaliplatin has been associated with development of hepatic lesions, which include sinusoidal alteration, portal hypertension, increase in transaminases, gamma glutamyltranspeptidase and alkaline phosphatase, and steatohepatitis (6,33). In our study, the role of Oct1 in the tissue accumulation and the pharmacokinetics of oxaliplatin was proven to be minimal, and oxaliplatin was not hepatotoxic under the conditions used in this study. However, total plasma platinum levels may not reflect platinum complexes available for DNA and protein binding, anti-tumor effects and toxicity of the oxaliplatin. The fact that oxaliplatin administration resulted in a greater reduction in body weight in the wild-type mice in comparison to the Oct1 knockout mice may suggest that Oct1 plays a role in the systemic toxicities of oxaliplatin.
In this study, we made an unexpected observation that Oct1 deletion markedly increased the renal toxicity of CDPCP in Oct1-/- mice, which was revealed by much greater Kim-1 mRNA expression level and histological changes of tubular cast formation and degeneration of tubular cells in Oct1-/- mice (Fig. 5b, c.II, and c.III). The differential localization of organic cation transporters Oct1 and Oct2 in the basolateral membrane of rodent kidney proximal tubules may account for the greater kidney toxicity in Oct1-/- mice. In particular, Oct1 was shown to be concentrated in the early proximal tubules in the renal cortex, whereas Oct2 was abundant in the late proximal tubules in the outer stripe of the outer medulla (34,35). In the report by Jonker et al. of the relative impact of Oct1 and Oct2 on the clearance of TEA (36), they observed that elimination of neither Oct1 nor Oct2 resulted in a decrease in renal TEA clearance, whereas elimination of both transporters eliminated TEA secretion. These data suggest that there is sufficient Oct1 transport capacity in the early renal proximal tubules to effectively clear the blood of substrate before it reaches the downstream Oct2-expressing cells of the late renal proximal tubules. CDPCP is an excellent substrate of both OCT1 and OCT2 (25). In Oct1-/- mice, CDPCP will be secreted by Oct2-expressing cells in late renal proximal tubules. The greater exposure of the late proximal tubule to CDPCP may contribute to the enhanced toxicity of CDPCP observed in the Oct1-/- mice. Further, Oct2 is likely to localize in the outer strip of the outer medulla and may produce a localized accumulation of CDPCP in this region. The outer medulla was the region most damaged by CDPCP (Fig. 5c.II and III).
The pharmacokinetics of CDPCP were consistent with the pharmacokinetics observed previously for oxaliplatin and picoplatin, suggesting that the drug has a pharmacokinetic profile similar to clinically used anti-cancer platinum agents. Deletion of Oct1 resulted in a reduction in the clearance and volume of distribution of CDPCP, which may be explained by the differences in the distribution of CDPCP to the tissues with high Oct1 expression, in particular, liver and intestine. The higher plasma platinum concentrations and AUC in Oct1-/- mice in comparison to Oct1+/+ mice can be attributed to the lower clearance and volume of distribution in Oct1-/- mice. Since the renal clearance of CDPCP was similar between Oct1+/+ and Oct1-/- mice, the lower total clearance of CDPCP in the Oct1-/- mice is likely due to a reduction in the uptake and subsequent covalent binding of CDPCP in the liver and intestine, organs that accumulate high levels of platinum.
Though other transporters may affect the disposition of CDPCP, this study clearly demonstrates that Oct1 plays a significant role in the pharmacokinetics of this drug. The lack of an important role of Oct1 in the pharmacokinetics of oxaliplatin may suggest a lower specificity of oxaliplatin for Oct1 and that other transporters (or diffusion) may contribute to its tissue distribution and pharmacokinetics. In this study, CDPCP exhibited excellent pharmacokinetic properties, comparable to other clinically used platinum analogs. Moreover, CDPCP appears to possess a favorable toxicity profile, and Oct1 plays an important role in determining the toxicity profile of CDPCP. Our study supported the concept that by targeting drug influx transporters, off-target toxicities, such as renal, hematological toxicities, can be spared. Whether CDPCP can be used for targeted therapy of various cancers is worthy of further investigation. Further, this study provides another example of a drug distinct from metformin for which OCT1 may play a profound role in its pharmacokinetics and toxicities (4,6,7).
Abbreviations
- ABC:
-
ATP Binding Cassette
- CDPCP:
-
cis-Diammine(pyridine)chloroplatinum(II)
- FBS:
-
fetal bovine serum
- GAPDH:
-
glyceraldehydes-3-phosphate dehydrogenase
- ICP-MS:
-
inductively coupled plasma mass spectrometry
- ITS:
-
insulin-transferrin-selenium
- Kim-1:
-
kidney injury molecule-1
- MPP+ :
-
N-methylquinine, and 1-methyl-4-phenylpyridinium
- OCTs:
-
organic cation transporters
- PK:
-
pharmacokinetics
- qRT-PCR:
-
quantitative real-time polymerase chain reaction
- SLC:
-
solute carrier
- TEA:
-
tetraethylammonium
REFERENCES
Maeda K, Ieiri I, Yasuda K, Fujino A, Fujiwara H, et al. Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin Pharmacol Ther. 2006;79:427–39.
Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug-drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34:1229–36.
Chung J-Y, Cho J-Y, Yu K-S, Kim J-R, Oh D-S, et al. Effect of OATP1B1 (SLCO1B1) variant alleles on the pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther. 2005;78:342–50.
Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, et al. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302:510–5.
Lin JH, Yamazaki M. Clinical relevance of P-glycoprotein in drug therapy. Drug Metab Rev. 2003;35:417–54.
Shu Y, Sheardown SA, Brown C, Owen RP, Zhang SZ, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–31.
Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83:273–80.
Zhang L, Dresser MJ, Gray AT, Yost SC, Terashita S, et al. Cloning and functional expression of a human liver organic cation transporter. Mol Pharmacol. 1997;51:913–21.
Gorboulev V, Ulzheimer JC, Akhoundova A, UlzheimerTeuber I, Karbach U, et al. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997;16:871–81.
Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–7.
Gruendemann D, Gorboulev V, Gambaryan S, Veyhl M, Koepsell H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature (London). 1994;372:549–52.
Schweifer N, Barlow DP. The Lx1 gene maps to mouse chromosome 17 and codes for a protein that is homologous to glucose and polyspecific transmembrane transporters. Mamm Genome. 1996;7:735–40.
Jonker JW, Schinkel AH. Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J Pharmacol Exp Ther. 2004;308:2–9.
Wright SH. Role of organic cation transporters in the renal handling of therapeutic agents and xenobiotics. Toxicol Appl Pharmacol. 2005;204:309–19.
Zhang SZ, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–57.
Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J Pharmacol Exp Ther. 2006;319:879–86.
Jonker JW, Wagenaar E, Mol C, Buitelaar M, Koepsell H, et al. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (Oct1 [Slc22a1]) gene. Mol Cell Biol. 2001;21:5471–7.
Wong E, Giandomenico CM. Current status of platinum-based antitumor drugs. Chem Rev. 1999;99:2451–66.
Weiss RB, Christian MC. New cisplatin analogs in development—a review. Drugs. 1993;46:360–77.
Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–84.
Ibrahim A, Hirschfeld S, Cohen MH, Griebel DJ, Williams GA, et al. FDA drug approval summaries: oxaliplatin. Oncologist. 2004;9:8–12.
Misset JL, Bleiberg H, Sutherland W, Bekradda M, Cvitkovic E. Oxaliplatin clinical activity: a review. Crit Rev Oncol Hematol. 2000;35:75–93.
Rixe O, Ortuzar W, Alvarez M, Parker R, Reed E, et al. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: Spectrum of activity in drug-resistant cell lines and in the cell lines of the national cancer institute’s anticancer drug screen panel. Biochem Pharmacol. 1996;52:1855–65.
Hollis LS, Amundsen AR, Stern EW. Chemical and biological properties of a new series of Cis-diammineplatinum(II) antitumor agents containing 3 nitrogen donors—Cis-[Pt(NH3)2(N-Donor)Cl]+. J Med Chem. 1989;32:128–36.
Lovejoy KS, Todd RC, Zhang SZ, McCormick MS, D’Aquino JA, et al. cis-diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: Uptake, structure, function, and prospects. Proc Natl Acad Sci USA. 2008;105:8902–7.
Moldeus P, Hogberg J, Orrenius S. Isolation and Use of Liver Cells. Fleischer S, Packer L, editors. Methods in Enzymology, Vol. Lii. Biomembranes. Part C: Biological Oxidations Microsomal, Cytochrome P-450, and Other Hemoprotein Systems. Xxii + 595p. Illus. Academic Press, Inc.: New York, N.Y., USA; London, England. Isbn 0-12-181952-3:60-71; 1978.
Raynaud FI, Boxall FE, Goddard PM, Valenti M, Jones M, et al. cis-aminedichloro(2-methylpyridine)platinum(II) (AMD473), a novel sterically hindered platinum complex: In vivo activity, toxicology, and pharmacokinetics in mice. Clin Cancer Res. 1997;3:2063–74.
More SS, Li S, Yee SW, Chen L, Xu Z, et al. Organic cation transporters modulate the uptake and cytotoxicity of picoplatin, a third-generation platinum analogue. Mol Cancer Ther. 2010;9:1058–69.
Luo FR, Wyrick SD, Chaney SG. Pharmacokinetics and biotransformations of oxaliplatin in comparison with ormaplatin following a single bolus intravenous injection in rats. Cancer Chemother Pharmacol. 1999;44:19–28.
Rice JR, Gerberich JL, Nowotnik DP, Howell SB. Preclinical efficacy and pharmacokinetics of AP5346, a novel diaminocyclohexane-platinum tumor-targeting drug delivery system. Clin Cancer Res. 2006;12:2248–54.
Lecureur V, Guillouzo A, Fardel O. Differential expression of the polyspecific drug transporter OCT1 in rat hepatocarcinoma cells. Cancer Letters. 1998;126:227–33.
Koepsell H. Polyspecific organic cation transporters: their functions and interactions with drugs. Trends Pharmacol Sci. 2004;25:375–81.
Rubbia-Brandt L, Audard V, Sartoretti P, Roth AD, Brezault C, et al. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann Oncol. 2004;460–466.
Sugawara-Yokoo M, Urakami Y, Koyama H, Fujikura K, Masuda S, et al. Differential localization of organic cation transporters rOCT1 and rOCT2 in the basolateral membrane of rat kidney proximal tubules. Histochem Cell Biol. 2000;114:175–80.
Urakami Y, Okuda M, Masuda S, Akazawa M, Saito H, et al. Distinct characteristics of organic cation transporters, OCT1 and OCT2, in the basolateral membrane of renal tubules. Pharm Res. 2001;18:1528–34.
Jonker JW, Wagenaar E, van Eijl S, Schinkel AH. Deficiency in the organic cation transporters 1 and 2 (Oct1/Oct2 [Slc22a1/Slc22a2]) in mice abolishes renal secretion of organic cations. Mol Cell Biol. 2003;23:7902–8.
ACKNOWLEDGMENTS
This study was supported by a grant from the National Institutes of Health, GM36780 and a Grant from the TRDRP grant 17RT-0126. This work was also made possible in part by core service provided by UCSF liver center (NIH P30 DK26743). Y.C. was supported by National Research Service Award T32 GM07546 from the National Institutes of Health. We acknowledge the services provided by Mouse Pathology Core of UCSF Helen Diller Family Comprehensive Cancer Center, the J David Gladstone Histology and Microscopy core and the Clinical Laboratory of the San Francisco General Hospital. We also thank Rob Franks at the Institute of Marine Sciences of University of California, Santa Cruz, for his great help in analyzing platinum using ICP-MS.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Li, S., Chen, Y., Zhang, S. et al. Role of Organic Cation Transporter 1, OCT1 in the Pharmacokinetics and Toxicity of cis-Diammine(pyridine)chloroplatinum(II) and Oxaliplatin in Mice. Pharm Res 28, 610–625 (2011). https://doi.org/10.1007/s11095-010-0312-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-010-0312-6