
Abstract
Rice yield is most sensitive to salinity stress imposed during the panicle initiation (PI) stage. In this study, we have focused on physiological and transcriptional responses of four rice genotypes exposed to salinity stress during PI. The genotypes selected included a pair of indicas (IR63731 and IR29) and a pair of japonica (Agami and M103) rice subspecies with contrasting salt tolerance. Physiological characterization showed that tolerant genotypes maintained a much lower shoot Na+ concentration relative to sensitive genotypes under salinity stress. Global gene expression analysis revealed a strikingly large number of genes which are induced by salinity stress in sensitive genotypes, IR29 and M103 relative to tolerant lines. We found 19 probe sets to be commonly induced in all four genotypes. We found several salinity modulated, ion homeostasis related genes from our analysis. We also studied the expression of SKC1, a cation transporter reported by others as a major source of variation in salt tolerance in rice. The transcript abundance of SKC1 did not change in response to salinity stress at PI stage in the shoot tissue of all four genotypes. However, we found the transcript abundance of SKC1 to be significantly higher in tolerant japonica Agami relative to sensitive japonica M103 under control and stressed conditions during PI stage.
Similar content being viewed by others

Introduction
Rice is one of the most economically important crops of the world. Rice yields can be reduced by up to 50% when grown under moderate (6 dS m−1) salinity levels (Zeng et al. 2002). Soil salinity is a serious constraint to rice cultivation under irrigated agriculture in countries like Pakistan and elsewhere due to poor quality of water (Abdullah and Ahmad 1982). Rice is the major crop of many coastal regions which are prone to sea water ingress during high tide and of rainfed systems which experience increasing salinity in the root zone due to a rising water table. Despite significant yield losses under saline conditions, rice is potentially a good candidate for cultivation in areas prone to coastal flooding due to its ability to tolerate standing water in the field for long periods. In California, salinity is an increasing concern for rice cultivation due to irrigation practices that involve use of recirculating water systems and the requirement of water holding after pesticide application during early growth stages (Scardaci et al. 1996).
The susceptibility of rice to salinity stress varies with growth stage. Rice is relatively salt-tolerant at germination, and in some cases is not affected significantly by up to 16.3 dS m−1 of salinity (Heenan et al. 1988; Khan et al. 1997). Rice becomes very sensitive at the young seedling stage, which impacts the stand density in salt-affected fields (Lutts et al. 1995). It is relatively less sensitive during late reproductive stage (grain ripening). From an agronomic perspective, sensitivity of yield components is of primary importance. Salinity significantly reduces the tiller number per plant, spikelet number per panicle, fertility, panicle length and primary branches per panicle (Heenan et al. 1988; Cui et al. 1995; Khatun et al. 1995; Zeng et al. 2002). Reductions in tiller number per plant and spikelet number per panicle were reported to be the major causes of yield loss in one cultivar of rice under salinity stress (Zeng and Shannon 2000). The number of spikelets per panicle was determined to be the most sensitive yield component. This component is determined at early reproductive stage, around panicle initiation (PI) (Hoshikawa 1989; Counce et al. 2000). The loss in spikelet number per plant was most significant when the stress was imposed before PI (3-leaf stage) or between PI and booting stage (∼16 days after PI) (Zeng et al. 2001). This loss of potential spikelets is attributed to degeneration of primary and secondary branches and flower primordia (Zeng et al. 2001). Considerable genetic variability for this yield parameter was reported based on evaluation of several diverse genotypes for salinity stress response (Zeng et al. 2002). Considerable variation for salt tolerance at critical stages in the cultivated gene pool has also been reported by other workers (Yeo and Flowers 1982; Moradi et al. 2003). This genetic variability can be utilized for improvement of salt tolerance by focusing on specific yield components. However, the underlying genes conferring tolerance during PI are at present unknown.
In the current study, we have focused on the salinity stress imposed during the PI stage considering the significant impact it has on rice yields and a lack of prior information on molecular components of the response to salinity stress at this critical stage. The genotypes selected for this study have contrasting physiological responses to salinity stress at PI based on several seasons of experimentation at the U.S. Salinity Laboratory, Riverside (Zeng et al. 2002). One of the most salt-tolerant genotypes identified by Zeng et al. (2002) was IR63731, an indica line developed from a cross between tolerant land race Nona Bokra and sensitive cultivar IR28. This genotype had the most favorable combination of salt tolerance at early vegetative and PI stages. This genotype possesses good yield potential under salinity stress due to high tillering ability and panicle weight (Zeng et al. 2002). Another genotype, Agami, was ranked high for its ability to yield high under salinity stress. Agami is a salt-tolerant japonica subspecies cultivar grown in Egypt. Several studies have reported the popular California japonica cultivar M103, to be very sensitive at all stages (Zeng et al. 2002, 2003). Therefore, for the current work we have used IR63731 and Agami as the tolerant genotypes representing indica and japonica subspecies and IR29 and M103 as the sensitive genotypes for each of the subspecies.
Previously, we focused on transcript accumulation responses of rice to salinity stress imposed during the early vegetative stage in two indica subspecies genotypes which differed in salt tolerance as manifested by higher tillering capacity and maintenance of favorable ion homeostasis (Walia et al. 2005). The sensitive genotype used for that study was IR29, a widely used sensitive standard which is susceptible at the vegetative and reproductive stages. IR29 was the sensitive parent of a recombinant inbred population developed at the International Rice Research Institute (IRRI) for mapping salt tolerance quantitative trait loci (QTL) in rice using Pokkali as the tolerant parent. A salt-tolerant recombinant inbred line (RIL) FL478 from this population was used as the tolerant genotype in our prior study (Walia et al. 2005).
The transcriptome of rice under salinity stress at PI has been largely uncharacterized. Considering the sensitivity of rice yield to salinity stress when imposed around PI, it is important to identify specific genes that are involved in the response to stress at this stage. Responsive genes are expected to include those with a role in salt adaptation and tolerance, and which respond to salt injury or constitute non-adaptive responses. The use of genotypes with well-characterized phenotypic responses to salinity stress at a particular growth stage can help in identifying potential candidate genes which may be important for heritable salt tolerance. Here we perform whole genome transcript profiling of genotypes from two major subspecies of cultivated rice chosen for their representative tolerance or sensitivity to salinity stress during the early reproductive stage, specifically at PI. By including indica genotype IR29 in the present study on PI, we now have mRNA expression level responses at both of the two most salt sensitive, yield-determining stages for this genotype (early vegetative as per Walia et al. 2005, and early reproductive in this study). Physiological characterization of the lines was also performed as part of the present study, and gene expression results were interpreted in the physiological context. We also incorporated information from known salt tolerance QTL in rice with the objective of correlating gene expression with gene location on the rice genome.
Material and methods
Plant materials for expression studies
Seeds of rice (Oryza sativa L.) genotypes IR29, IR63731-1-1-4-3-2 and Agami were obtained from G. B. Gregorio at IRRI of the Philippines and then propagated through quarantine at the George E. Brown, Jr. Salinity Laboratory, Riverside, CA, USA. Seed for M103 was supplied by California Cooperative Rice Research Foundation Inc., at Biggs, California. These genotypes were selected from the germplasm collections at the two sites based on their differences in salt tolerance in terms of agronomic and physiological performance. Extensive characterization of these lines was performed by Zeng et al. (2002, 2003) at George E. Brown, Jr. Salinity Laboratory, Riverside, CA, USA.
Plant culture and salinity treatments
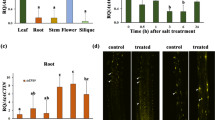
The experiment was conducted in the greenhouse at Riverside, California [33°58′24″ N latitude, 117°19′12″ W longitude] between May and July 2005. Plants were cultured in tanks (122 × 61 × 46 cm) filled with sand and irrigated with nutrient solution (Yoshida et al. 1976). The nutrient solution consisted of NH4NO3 (1.43 mM), NaH2PO4 · 2H2O (0.37 mM), K2SO4 (0.5 mM), CaCl2 (1.00 mM), and MgSO4 · 7H2O (1.6 mM). Nutrient solution pH was maintained between 5.0 and 6.5 by adding sulfuric acid as needed. Irrigation solutions were prepared in 1600 L reservoirs and pumped to provide irrigation to the sand tanks. Overflow irrigation was returned to the reservoirs through drainage by gravity. Each reservoir provided irrigation water to three sand tanks (replicates) three times daily for 30 min per irrigation cycle. Four rows of each genotype were planted per tank for control tanks. An extra row at each end of the tank was used as the border row. Each genotype was planted in separate tanks which were marked for salinization later in the experiment. The water level was maintained at the sand surface until emergence of the seedlings from the sand and then increased to 6–8 cm above the sand surface. Air temperature ranged from 30°C to 42°C during the day and 20–24°C during night. Relative humidity ranged from 40% to 80%. Light averaged 1104 μmol m−2 s−1 at noon. Weak plants were removed from the tanks. Plant growth stages were closely monitored. Rough estimates of the days to PI stage for each genotype were available from growing the genotypes for several years in the same greenhouse conditions and time of the year during previous experiments (Zeng et al. 2002). Salinity treatment was applied by adding NaCl and CaCl2 (5:1 molar concentration) in two steps over a period of 3 days to each genotype about 9–10 days before PI (Fig. 1). Electrical conductivities (ECw) of nutrient solutions were monitored using an EC meter daily (Hanna Instruments, Woonsockett, RI). The salinity in the treatment reservoir was allowed to stabilize for 5 days to a final electrical conductivity of 7.0 dS m−1. The non-saline control reservoir with Yoshida solution had an ECw of 1.1 dS m−1. Two plants were randomly selected each day for each genotype when the plants were approaching PI. The main culms were dissected under a microscope to observe the development of young panicles. The day when we observed any plant of a genotype with panicle about 0.5 mm long was defined as the PI date for that genotype under the treatment in which it was growing (control or salt stressed). Salinity delayed PI in the sensitive genotype M103, but not in other three lines.

Experimental design of salinity stress treatment. Salinity stress was applied 9–10 days before panicle initiation (PI). A final salinity level of 7 dS m−1 was reached by a two-step addition of NaCl and CaCl2. Conductivity in the control tanks is represented by a dotted line and the treated tank by a solid line. Diamonds represent the PI and ovals represent the harvest time points. The crown and growing point of the main shoot were harvested 4–5 days after reaching PI
Phenotypic characterization and gene expression studies
Plants were characterized for phenotypic responses to salinity stress on the day of harvest for RNA extraction. For phenotypic characterization the shoot tissue from six plants was harvested for ion analysis. Plants were washed with deionized water, dried in a forced air oven (70°C), and then ground into fine powder. Shoot Na+ and K+ concentrations were determined on nitric-perchloric acid digests by inductively coupled plasma optical emission spectrometry (ICP, Perkin-Elmer Co., Norwalk, CT, USA). Transpiration rate of the youngest fully expanded leaf was measured between 10.00 h and 12.00 h one day before harvesting the tissue for RNA using a LI-COR 6400 Photosynthesis System (LI-COR Biosciences, Lincoln, NE). The following conditions for leaf gas measurements were used: photosynthetic photon flux density, 1200 μmol m−2; chamber CO2 concentration, 380 μmol CO2 mol−1; leaf temperature, 27°C; and chamber vapor concentration, 20 mmol H2O mol−1.
The plants were harvested 4 or 5 days after reaching PI. The main shoot was dissected for RNA extraction to obtain the growing point and crown tissue which was snap frozen. Approximately 12 plants were harvested per genotype per tank and tissue pooled to make each biological replicate for RNA extraction. Three biological replicates (one per array) were used for each treatment. Two plants from each tank were allowed to grow to maturity to verify that plants survived the imposed salinity stress.
RNA extraction and processing for GeneChip analysis
RNA samples were processed as recommended by Affymetrix, Inc. (Affymetrix GeneChip Expression Analysis Technical Manual, Affymetrix, Inc., Santa Clara, CA) at the Core Instrumentation Facility at the University of California, Riverside by Barbara Walter. Total RNA was initially isolated from frozen shoot tissue using TRIzol Reagent. The RNA was purified using an RNeasy spin column (Qiagen, Chatsworth, CA) and an on-column DNase treatment. Eluted total RNAs were quantified with a portion of the recovered total RNA and adjusted to a final concentration of 1 μg/μl. All RNA samples were quality assessed prior to beginning target preparation/processing steps by running out a small amount of each sample (typically 25–250 ng/well) onto a RNA Lab-On-A-Chip (Caliper Technologies Corp., Mountain View, CA) that was evaluated on an Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA). Single-stranded, then double-stranded cDNA was synthesized from the poly(A)+ mRNA present in the isolated total RNA (10 μg total RNA starting material each sample reaction) using the SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen Corp., Carlsbad, CA) and poly(T)-nucleotide primers that contained a sequence recognized by T7 RNA polymerase. A portion of the resulting ds cDNA was used as a template to generate biotin-tagged cRNA from an in vitro transcription reaction (IVT), using the Affymetrix GeneChip IVT Labeling Kit. Fifteen micrograms of the resulting biotin-tagged cRNA was fragmented to strands of 35–200 bases in length following prescribed protocols (Affymetrix GeneChip Expression Analysis Technical Manual). Subsequently, 10 μg of this fragmented target cRNA was hybridized at 45°C with rotation for 16 h (Affymetrix GeneChip Hybridization Oven 320) to probe sets present on an Affymetrix Rice Genome array. The GeneChip arrays were washed and then stained (SAPE, streptavidin-phycoerythrin) on an Affymetrix Fluidics Station 450 followed by scanning on a GeneChip Scanner 3000.
Rice Genome array
The Rice Genome array (Affymetrix, Santa Clara, CA) contains probe sets designed from ∼48,564 japonica and 1,260 indica sequences. The sequence information for this array was derived from NCBI UniGene Build #52 (http://www.ncbi.nlm.nih.gov/UniGene), GenBank mRNAs and 59,712 gene predictions from TIGR’s osa1 version 2.0. Gene models which had any indication of transposable elements were removed from the list of TIGR predicted genes. The array is believed to represent about 46,000 distinct rice genes. About 26,000 of these are 3′ anchored Unigene EST and mRNA clusters, including known rice full length cDNA clones and 19,431 are solely from TIGR gene predictions. To obtain annotations for the salt-regulated probe sets, we extracted the target sequence of identified probe sets from the sequence information file (.sif) for the rice genome array. The target sequence extends from the 5′ end of the 5′-most probe to the 3′ end of the 3′-most probe. The target sequences were then searched using BLASTn against the TIGR rice pseudomolecules, release 3 (http://www.tigr.org/tdb/e2k1/osa1) and the TIGR Arabidopsis database, version 5.
Statistical analysis of array data
The triplicated array data set was analyzed using GeneChip Operating Software (GCOS 1.2) and DChip (Li and Wong 2001; http://www.DChip.org) software. The scanned images were examined for any visible defects. Satisfactory image files were analyzed to generate raw data files saved as CEL files using the default settings of GCOS 1.2 from Affymetrix. We used a default target intensity value (TGT) setting of 500. The normalization factor value was set to 1 to extract data without normalization. Default parameter settings for the Rice Genome array were used. The scaling factor for the arrays ranged from 3.1 to 8.5. The detection calls (present, absent or marginal) for the probe sets were made by GCOS. Further analysis was done using DChip which incorporates a statistical model for expression array data at the probe level. The DChip program was set to import GCOS signals. The normalization of all arrays was performed using an invariant set approach. For calculating the expression index of probe sets we used the PM model and opted for truncating the low expression values to 10th percentile of the probe set signals called absent. The expression values were log2 transformed after calculating the expression index. DChip was used for comparative analysis of samples from salt stress and control treated plants for both genotypes. We identified the differentially expressed genes using the empirical criterion of more than 10-fold change (E-B > 1 or B-E > 1) and significant t-tests of P < 0.05 based on three biological replicates. The baseline array, which is a control sample, is denoted by B and salinity-treated experimental sample is denoted by E. The P-values assigned to differentially expressed genes were used as a ranking criterion for ordering of up-regulated and down-regulated genes. We used the DChip feature to assess the false discovery rate (FDR) in our data set using 250 permutations in most comparisons. Empirical FDR was below 15% when identifying differentially expressed genes. All microarray data from this work is available from NCBI GEO (http://www.ncbi.nlm.nih.gov/geo) under the series entry GSE4438.
Expression validation by semi-quantitative RT-PCR
Several key expression profiles obtained from chip hybridizations were further validated by semi-quantitative RT-PCR using first-strand cDNA. The japonica sequence of each gene was obtained from the TIGR rice database. Exonic sequences were used for the design of primers using Primer Express (Perkin-Elmer Applied Biosystems, Foster City, CA). The primer sequences for 18S rRNA (F-ATGATAACTCGACGGATCGC, R-CTTGGATGTGGTAGCCGTTT) were used as an internal control. The primer sequences for SKC1 were obtained from Walia et al. (2005). A cDNA first strand was synthesized using Taq-Man Reverse Transcription Reagents (Applied Biosystems, Forster City, CA; Ref: N808-0234) following the manufacturer’s instructions. Two micrograms of total RNA was converted into cDNA. Each cDNA was diluted 40 times and 5 μL of cDNA was used for three-step PCR. The number of PCR cycles was optimized (37 cycles) for the 5 genes selected for expression validated and 34 cycles for SKC1.
Results
A moderate level of salinity stress was imposed gradually nine or ten days before PI stage and plants were sampled for phenotypic characterization and expression profiling five days after PI (Fig. 1). All four genotypes used in this experiment were known to reach PI at different time points based on prior characterization of these genotypes over several growing seasons under comparable growing conditions and time of the year.
One confounding factor in experiment involving salinity stress is that salinity is known to delay PI in all genotypes. With this in mind we designed our experiment to harvest the control and salinity stressed tissue of each of the genotype at similar developmental stage i.e. 4–5 days after the plants reached PI. This was done because developmental equivalence when comparing control plants with stressed plants within each genotype is more relevant when the focus is on a specific sensitive stage such as PI.
Shoot ion uptake of salt-tolerant and sensitive lines
Shoot sodium ion analysis when assayed under salinity stress clearly distinguished tolerant from sensitive genotypes. Salinity stress increased the Na+ concentration in all genotypes significantly (Table 1). The increase was nearly 10-fold in all genotypes except IR63731. Sodium ion accumulation in the tolerant genotypes, IR63731 and Agami, was much lower than in the sensitive genotypes. Agami, the salt-tolerant japonica, maintained a lower Na+ concentration even under control conditions and had the lowest Na+ concentration under stressed conditions. Shoot K+ concentration decreased in Agami, while the concentration in other three genotypes did not change significantly under salinity stress. Since K+/Na+ ratio under salinity is considered a more important index of salinity tolerance than ion concentrations separately, we ranked the four genotypes by K+/Na+ ratio in descending order: Agami (4.9) ≈ IR63731 (4.7) > M103 (3.7) > IR29 (2.9). Under control conditions, indicas had a lower K+/Na+ ratio than japonica varieties. In addition to measuring Na+ and K+ levels in the shoot tissue, we also analyzed the chloride levels. Shoot chloride concentrations increased in all genotypes under salinity stress. Sensitive japonica, M103 however, maintained a much lower Cl− level (433 mmol kg−1 compared to 535 mmol kg−1 in Agami) than other genotypes.
To further characterize the physiological status of the four genotypes, we measured the transpiration rate of the youngest fully expanded leaf of the main shoot (Table 1). The transpiration rate decreased in all genotypes under stress. A noteworthy observation was the significantly lower rate of transpiration maintained by tolerant japonica Agami under control and stressed conditions relative to all other genotypes. Tolerance in Agami therefore seems to depend on an avoidance mechanism. We also observed the plants for visual damage symptoms of salinity stress. The sensitive genotypes, especially IR29, had necrosis on some leaves about 1/3 the length of the leaf from the tip roughly one week after the initiation of stress.
Expression responses of tolerant and sensitive genotypes to salinity stress
We used two japonica and indica rice genotypes with contrasting salt tolerance for global gene expression under salinity stress. The developmental stage targeted in this study was PI. Salinity stress was applied nine or 10 days before PI and the tissue (crown and growing point) was harvested 5 days after the genotypes reached PI stage (Fig. 1; see “Materials and methods”). To define differentially expressed genes we used the following criteria: (1) t-test P-value < 0.05, (2) ≥ 2-fold change, and (3) FDR below 15% (see Materials and methods). Using these criteria we identified 292 and 346 probe sets which were up- and down-regulated in salt-sensitive japonica M103 respectively (Fig. 2). The same selection criteria identified 54 up- and 54 down-regulated probe sets in the salt-tolerant japonica, Agami. A comparable number of probe sets (55) were differentially up-regulated in salt-tolerant indica IR63731 under salinity stress. A total of 35 probe sets were down-regulated in IR63731. The expression response of sensitive indica, IR29 was characterized by up-regulation of 589 probe sets. In contrast, only 57 probe sets were significantly down-regulated in response to salinity stress. Complete lists of probe sets identified as differentially expressed in each of the genotypes are provided as supplemental data (Datasheet 9–11). Five probe sets identified as differentially expressed from the microarray datasets were checked using semi-quantitative RT-PCR (Supplemental Fig. 1). The results agreed for four of the genes examined. The primers for the fifth gene, Os02g52390, did not amplify from the indica subspecies, since they were derived from japonica exonic sequence. Nineteen probe sets were significantly up-regulated in all genotypes in response to salinity stress. A selected list of probe sets which were commonly induced is presented in Table 2. Only one probe set, Os.17057.1.S1_at, with no hits to the database was down-regulated by salinity stress in all genotypes.

Overlap of up-regulated probe sets between different genotypes. (A) Overlap between the sensitive genotypes M103 and IR29. (B) Overlap between the tolerant genotypes. (C) Overlap among the japonica subspecies. (D) overlap among the indica subspecies. The up-regulated probe sets were identified using the combined criteria of a 2 or more fold change, P-value threshold of 0.05 and false discovery rate below 15%
We made several comparisons to identify genes which characterize the response of japonica and indica to salinity as well as genes which are expressed in tolerant and sensitive genotypes (Fig. 2). We identified 42 probe sets which were induced commonly between the two japonicas, Agami and M103. Similar comparison between the indica genotypes identified 41 probes sets as commonly induced. In an effort to search for transcripts which may be associated with salt tolerance of Agami, we identified 12 probe sets which were induced in Agami but not in M103. Two of these probe sets (Os.11975.2.A1_at and OsAffx.25546.1.S1_s_at) were induced in Agami but down-regulated in M103. Probe set Os.11975.2.A1_at had no sequence match to known genes. Probe set OsAffx.25546.1.S1_s_at represented a germin-like gene (GPL4), Os03g44880.. We also identified several probe sets which were induced in IR63731 but not in sensitive indica, IR29. These included RD22, a jacalin lectin protein and nitrate reductase (NR1). The expression of RD22 is known to be induced in response to salt stress and dehydration (Yamaguchi-Shinozaki and Shinozaki 1993).
Besides focusing on the genes which were differentially expressed in response to salinity stress within each of the genotypes, we also performed genotype–genotype comparisons under control conditions. This analysis was performed to identify genes associated with salt tolerance that may be inherently transcribed at a higher level in one genotype than in another. Interpretation of such genotypic comparisons using plants profiled under control conditions is done in light of the fact that the compared genotypes have similar developmental stage but not same chronological age. We found 539 probe sets which were significantly up-regulated in Agami relative to M103 under control conditions (Supplemental Data Sheet 13). A similar comparison yielded 686 probe sets expressed at a higher level in IR63731 than IR29 (Supplemental Data Sheet 14). We BLAST searched the target sequence of all the probe sets against the rice genomic sequence assembled at TIGR (http://www.tigr.org) to obtain the genomic position of the best BLAST hit gene model. We found several probe sets which were elevated in Agami relative to M103 to be tightly clustered. Clusters on chromosome 1, 5, 10 and 12 were particularly large (Fig. 3A). A dense 19-probe set cluster occurred on the long arm of chromosome 10. This cluster localized to a segment of 1.1 Mb and represented 15 rice gene loci. Another 11-probe set cluster on chromosome 1 extends from 11.07 Mb to 12.61 Mb (1.54 Mb in length). This region is of particular interest because two QTL for salt tolerance have been mapped to this region (Bonilla et al. 2002; Lin et al. 2004). The number of gene clusters obtained from the comparison of indica genotypes was relatively small and most were found on chromosome 1 (Fig. 3B). Two of the clusters on chromosome 1 were located in the region of a salt tolerance QTL Saltol. The first was an 8-probe set (six gene loci) cluster (9.03–9.91 Mb) and the second was a 14-probe set (eight gene loci) cluster extending from 11.19 Mb to 12.62 Mb. The positions of these two clusters may coincide with two separate QTL in the Saltol region reported by Lin et al. (2004) and Bonilla et al. (2002). Additionally, a dense 16-probe set, 537 kb long cluster was located on the distal end of chromosome 1.

Rice chromosomes with gene clusters based on genotypic comparisons. (A) Rice gene model clusters on chromosomes 1, 5, 10 and 12 obtained from genotypic comparison of M103 with Agami under control conditions. (B) Rice gene model clusters on chromosome 1, obtained by genotypic comparison of IR29 with IR63731 using unstressed samples. The chromosome displays only up-regulated rice genes in the tolerant lines under control conditions. The gene clusters with shaded area co-localize to previously identified as a salt tolerance QTL on chromosome 1
Genes involved in ion homeostasis
Maintenance of low Na+ and a favorable K+/Na+ ratio are desirable traits for rice growing under saline conditions and ultimately manifest as salt tolerance. Considering the importance of ion transporters in exclusion, redistribution, and compartmentalization of ions we searched for and identified several cation transport-related genes which significantly responded to the salinity treatment (Table 3). One such gene was a sodium transport protein (Os02g07830) induced (P < 0.02) in IR29 only. In contrast a sodium/calcium exchanger was down-regulated in M103. Another gene (Os12g25200), encoding for CLC-d chloride channel was induced in M103 and IR29 by 17-fold (P < 0.0001) and 7.3-fold (p < 0.00008), respectively. Several genes associated with transport of K+ were differentially regulated in response to salinity stress. Three of these genes belong to the KT-HAK-KUP family of transporters associated with high affinity K+ uptake from the roots (Santa-Maria et al. 1997). In M103, a transporter (Os08g36340) similar to HAK4 was up-regulated by stress, while HAK5 (Os07g01220) was down-regulated. Another cation channel gene (Os01g52070), with sequence similarity to KAT3 was down-regulated in M103. A potassium transporter, KUP1 (Os09g27580), was induced in IR29 by more than 5-fold (P < 0.002) under salinity stress. We did not detect any cation transporters significantly responding to salinity stress in the tolerant genotypes.
Since the shoot ion analysis indicated a strong Na+ exclusion phenotype in the tolerant lines, we further explored our data set for any association of cation transporters. On comparing the tolerant Agami with M103 under control and salinity stress conditions, we found a cation transporter (Os01g20160) to be expressing at a higher level in Agami by 2.6-fold (P < 0.01) and 2.8-fold (P < 0.003) compared to M103 under control and salt stressed conditions, respectively. On extending the genotypic comparison to indica genotypes, we found Os01g20160 transcript to be present in IR63731 at a higher level than in IR29 (1.98-fold, P < 0.01) under control conditions. The cation transporter gene 01g20160 is located in the vicinity of the Saltol region (Bonilla et al. 2002) and has a perfect sequence match to the SKC1 gene reported to be the tolerance conferring gene in the salt tolerance QTL studied by Ren et al. (2005). We pursued the expression of this gene using semi-quantitative RT-PCR in roots and shoot tissue of several rice genotypes which were part of the salinity stress experiment conducted during the vegetative stage study (Walia et al. 2005). The expression of 01g20160 in genotypes IR63731, Pokkali, FL478 and IR29 was analyzed in the root and shoot tissues for vegetative stage (V), and shoot tissue of IR63731 and IR29 at the PI stage (R) under control and stressed conditions (Fig. 4). The expression of Os01g20160 increased in response to salinity in the roots of tolerant lines IR63731, Pokkali and FL478 but not in salt-sensitive IR29. The mRNA accumulation of Os01g20160 in shoot tissue increased in response to salinity in all four genotypes analyzed during the vegetative stage.

Expression of SKC1 in different indica genotypes. Semi-quantitative RT-PCR was performed to study the expression of SCK1 in the root and shoot tissue of IR63731 and IR29 under control and stressed conditions during vegetative (V) and reproductive (R) stages. Expression was analyzed for roots and shoot tissue of tolerant lines Pokkali and FL478 during vegetative stage of growth. The curly bracket encloses a control and the corresponding salt stressed sample
Differential expression of auxin associated genes in rice under salinity stress
Phytohormones including auxin indole 3-acetic acid (IAA) play a central role in plant growth, and development. Auxin is also known to mediate responses to light and gravity. In our expression analysis, several auxin-related genes were differentially regulated by salinity stress, indicating that salinity stress is associated with auxin response in rice. Almost all of these auxin related genes were differentially expressed in sensitive genotypes M103 and IR29. Most striking was the response of M103 to salinity in this context. The expression of cytochrome P450 83B1 (CYP83B1) was significantly (P < 0.04) down-regulated under salt stress. The expression level of CYP83B1 is known to determine auxin levels in plants (Barlier et al. 2000). Other auxin related genes down-regulated in response to salinity include a putative axi1 and two auxin response factors. In contrast, a mitogen activated protein kinase cascade gene, NPK1 which is known to suppress early auxin response gene transcription is induced in response to salinity in IR29. An increased level of nonphototropic hypocotyl 3 (NPH3) transcript was observed in M103 under salt stress. A rice ortholog of NPH3 is involved in phototropic response of coleoptiles through lateral auxin translocation (Haga et al. 2004). Another gene involved in auxin transport, PIN1 was found to be down-regulated in Agami. Several of the auxin related genes and their response to salinity are listed in Table 3.
Effect of salinity stress on floral transition and development in rice
The transition from vegetative to reproductive stage, termed floral transition, is a dramatic phase change in plant development. The transition controlled by a complex genetic network that integrates environmental signals such as day length and temperature (Simpson et al. 1999). We found several rice genes which are associated with floral transition and morphological development to be responsive to salinity stress imposed at the PI stage. Centroradialis (CEN), a gene which plays a role in phase transition and panicle morphology, was induced in both the sensitive genotypes, M103 and IR29 in response to salinity. A zinc-finger protein indeterminant1 (ID1) linked to phase transition signaling in maize, was induced at a higher level (2.7-fold, P < 0.0001) in M103 compared to IR29 (1.5-fold, P < 0.02). In contrast, the flowering locus T (FT) (Os06g35940) gene was differentially regulated in M103 (down-regulated) compared to IR29 (up-regulated) under salinity stress. FT is known to be downstream of gigantea, which represses flowering in rice. A gigantea-like gene (Os01g08700) was down-regulated in both japonica genotypes M103 and Agami. Several other genes involved in phase transition and organ identity and size determination were differentially regulated in M103, IR29 and Agami but not in IR63731. On comparing the expression patterns of IR63731 with IR29, we found several genes including gigantea, FT, terminal flower 1 (TFL1), and embryonic flower 2 (EMF2) to be expressing at a higher level in IR63731 under control conditions. Some of these differentially regulated genes and their expression levels in different genotypes are detailed in Table 3.
Discussion
In this study, we applied global gene expression profiling analysis to study the transcriptional responses of four rice genotypes comprised of tolerant and sensitive indicas and japonicas. For these genotypes, each with well-characterized salinity tolerance from prior studies, we measured gene expression during PI, the most salt-sensitive stage of development, considered to be the major factor in yield loss in rice. The results from this study, which used a moderate, agronomically relevant salinity stress, demonstrate striking differences in expression responses between tolerant and sensitive genotypes. The genotypes were also characterized for their physiological responses to obtain insights which enabled us to correlate some of the phenotypic observations with expression results.
Under salinity stress two key features, one physiological and the other within the transcriptomes, clearly distinguished the tolerant genotypes from the sensitive genotypes. First is the ability of the tolerant lines IR63731 and Agami to exclude Na+ ion from the shoot tissue compared to sensitive lines. The second feature, observed at the gene expression level, is the strikingly small number of genes which responded to salinity stress in the tolerant genotypes. This is likely a manifestation of the low shoot Na+ trait in tolerant lines which experience significantly lower toxicity at the cellular level and therefore requires fewer adjustments of cell metabolism and architecture. Among the genes which were commonly induced by salinity in all genotypes were abiotic stress response related genes such as water stress induced (WSI76), low temperature induced protein (LTI30) and a drought-induced RNase S-like gene. The WSI76 encodes for galactinol synthase (Takahashi et al. 1994) and was responsive to low temperature and osmotic stress. Galactinol synthase is required for the committed step in raffinose family oligonucleotides (RFOs). It was also induced in the tolerant line FL478 when salinity was imposed at early vegetative stage, but not in IR29 (Walia et al. 2005). Besides the genes involved in response to dehydration, we also detected at least two genes involved in nitrate transport, which may be compensating for salinity induced nutritional deficiency. A nitrate transporter, NRT1 was induced in Mesembryanthemum crystallinum in leaf and root tissue under salt stress (Popova et al. 2003). A low-affinity nitrogen transporter and NRT2, both localizing to chromosome 1 of rice were induced in all rice genotypes in response to salinity stress (Table 2).
Ion homeostasis in rice under salinity stress
Our data indicated that the tolerant genotypes IR63731 and Agami maintained significantly lower Na+ concentration in the shoot tissue compared to sensitive lines under salinity stress. Sodium ion concentration in the plant is considered to be a good indicator of the ion toxicity levels to which the cells are exposed. The lower ion specific stress in tolerant genotypes is reflected in the relatively small number of genes which respond to salinity in tolerant lines when compared to sensitive IR29 and M103. We found several ion homeostasis related genes such as ion transporters which were responsive to salinity stress in the sensitive genotypes. One such gene encoding for Na+ transporter (Os02g07830) was induced by salinity stress in IR29. This gene has high sequence similarity to the Arabidopsis HKT1. The barley ortholog of Os02g07830 was up-regulated under salt stress in the shoot tissue of a barley genotype, Morex during early seedling stage (Walia et al. 2006, submitted). This barley genotype particularly and barley in general, is known to accumulate a high level of Na+ in the shoot tissue during salinity stress. Salt tolerance in barley is believed to be mainly achieved through efficient compartmentalization of toxic ions besides other mechanisms conferring tissue tolerance. High Na+ in the shoot tissue was also documented for rice genotype IR29 at vegetative and reproductive stage under salinity stress (Walia et al. 2005; Table 1). The expression of Os02g07830 did not change significantly in the other three genotypes examined. Based on these pieces of information, we can speculate that Os02g07830 may be involved in redistribution or compartmentalization of Na+ within the plant in genotypes which are relatively inefficient in Na+ exclusion from the plant.
Another rice gene recently proposed to confer salt tolerance through recirculation of Na+ ions is SKC1 (Ren et al. 2005). SKC1 was identified as gene underlying a rice QTL for maintaining high shoot K+ in the shoot under salt stress (Lin et al. 2004). The SKC1 gene also encodes a HKT-type transporter and functions as a Na+-selective transporter in rice. Rice locus, Os01g20160 previously annotated as a cation transporter corresponds to SKC1 based on sequence similarity. This locus has been reported to be induced in shoot tissue by salt stress in both the sensitive IR29 and tolerant FL478 genotypes under salinity stress imposed during early vegetative stage (Walia et al. 2005). However, the induction in salt-tolerant FL478 was significantly higher than in IR29 under stress. In contrast to these prior results with vegetative stage stress, we did not detect a significant up-regulation of SKC1 in the shoot tissue of any of the four rice genotypes at PI profiled under salt stress in this study. However, on making genotype–genotype comparisons between Agami-M103 and IR63731-IR29, we found SKC1 to be expressing at a higher level in Agami compared to M103 under both control and saline conditions. Interestingly, the shoot Na+ ion analysis indicated that Agami maintains lower Na+ concentration than M103 under both control and saline conditions (Table 1).
The expression of SKC1 was reported to be up-regulated by salt stress in root but not shoot tissue (Ren et al. 2005). In contrast, our results with indica genotypes at vegetative stage indicated an up-regulation of SKC1 by salt stress in the shoot (crown plus growing point tissues), an observational discrepancy that may be attributed to the very different methods of applying salt stress between the two studies. We pursued the expression analysis of SKC1 further by analyzing the shoot and root tissues of tolerant indicas Pokkali, IR63731 and FL478 and sensitive line IR29 (Fig. 4). Our results indicate that SKC1 is salt up-regulated in the shoots of all four indica genotypes under the conditions that we used. Under the more gradual and moderate stress conditions that we applied, SKC1 was up-regulated in the root tissues only in salt-tolerant lines, not in salt-sensitive IR29.
Auxin and response of rice to salinity stress
Salinity modulates the expression of several auxin-associated genes in the sensitive japonica genotype, M103. The expression level of the gene encoding CYP83B1 was down-regulated in M103 but not in other genotypes. A loss-of-function mutation in CYP83B1 resulted in accumulation of indole 3-actaldoxime (IAOx) which is redirected to auxin biosynthesis (Bak et al. 2001). Taken together, it is plausible that auxin homeostasis in M103 is disturbed in response to salinity by virtue of a reduction of CYP83b1 gene expression. Another gene involved in auxin signal transduction, axi1 was also down-regulated in M103 (Hayashi et al. 1992). Although the mechanisms of auxin signal transduction in plants are still largely obscure, there is considerable evidence linking auxin perception and various ion transporters located in plant cell membranes (Blatt and Thiel, 1993; Barbier-Brygoo, 1995). Anion channels have been indicated to be involved in auxin signaling (Thomine et al. 1997). More specifically, auxin stimulated Cl− efflux/influx has been of interest for several decades (Bentrup et al. 1973; Rubinstein, 1974; Babourina et al. 1998). Bentrup et al. (1973) reported that IAA significantly reduced the uptake of Cl− by cells of Petroselinum sativum, while Babourina et al. (1998) reported a stimulatory effect of auxin on Cl− uptake by oat coleoptiles. The modulation of auxin related genes in M103 during salt stress is attractive in context of the shoot Cl− concentration in M103. The sensitive genotype maintained a significantly lower Cl− concentration under salt stress compared to other genotypes (Table 1). Interestingly, a CLC-d chloride channel (Os12g25200) was induced in M103 by 17-fold (P < 0.0001) in response to salinity stress.
Three of the genes induced in IR29 in response to salinity were associated with auxin. The chloride channel (Os12g25200) was induced in IR29 by 7-fold (P < 0.00008). Also induced was a gene for an NPK1 kinase-like protein under salinity stress. Constitutive expression of a tobacco MAPKK NPK1 is reported to dampen auxin responsiveness of a reporter gene (Kovtun et al. 1998) and confer salt tolerance (Kovtun et al. 2000). Additionally, expression profiling of the salt-overly sensitive (sos2-1) mutant in comparison to wild type Arabidopsis revealed differential induction of auxin-related genes in the mutant under salt stress (Kamei et al. 2005). These reports and our data indicate a significant role of auxin in response of some rice genotypes to salinity stress.
Salinity stress and floral transition
In the current work we focused on shoot meristem tissue under salinity stress imposed during PI. We detected several genes associated with developmental transitions and flowering to be modulated by salinity stress. Salinity delays PI in rice (Zeng et al. 2002), and in our study this delay was generally greater for sensitive than tolerant genotypes. In this context we found the up-regulation of rice genes with sequence match to terminal flower 1 (TFL1)/centroradialis (CEN) in the sensitive genotypes to be interesting. Over expression of TFL1/CEN homologs of rice delayed phase transition and altered panicle morphology (Nakagawa et al. 2002). The delay in initiation of panicle in sensitive genotypes, especially M103 under salinity stress, was longer than tolerant genotypes. The expression of TFL1/CEN was not significantly changed in the tolerant lines under stress. Both TFL1 and CEN belong to the family of phosphatidyl-ethanolamine-binding proteins (PEBPs) (Bradley et al. 1996). Another gene which is involved in flowering transition is FT, which is also a member of the PEBP family. The role of FT is however antagonistic to TFL1, as FT induces floral transition. The expression level of FT was down-regulated in M103 as expected due to a delayed floral transition under salinity stress. Surprisingly, FT was up-regulated in IR29. Up-regulation of FT in IR29 was also observed during the vegetative stage (Walia et al. 2005). The expression of FT was not modulated by salinity stress in tolerant lines IR63731 and Agami during PI and in FL478 during vegetative stage. An up-stream member of the floral induction pathway to which FT belongs is gigantea (GI). Suppressed expression of the rice ortholog of gigantea (OsGI) through RNA-mediated interference (RNAi) resulted in a late flowering phenotype under short day conditions relative to wild-type plants (Hayama et al. 2003). The expression level of gigantea-like gene was significantly reduced in M103 and Agami under salinity stress (Table 3). None of these genes were differentially modulated in tolerant indica IR63731. In brief, the gene expression analysis identified genes which explain the observed delay in PI of M103 under salinity stress.
High salt tolerance has often been linked to genotypes which take relatively long to reach maturity. The genotypes selected for this experiment also differ in their time to reach maturity. Therefore it can be argued that the late maturing genotypes IR63731 and Agami are likely to be more salt tolerant merely as a consequence of higher biomass translating into higher yield. However, Zeng et al. (2002) reported a lack of correlation between the duration of reproductive growth period and salt tolerance based on seed yield. This study also included three of the four genotypes used in the current study, namely IR63731, Agami and M103 and employed a comparable level of salinity stress. Since difference in time to maturity for different genotypes can potentially lead to confounding effects on gene expression measured by microarrays, we decided to focus on the developmental stage just after PI was observed for each treatment regardless of the growth conditions (control or stressed) and genotype. This involved staggered harvesting of control and stressed plants for each of the genotype, hence normalizing for delay in PI resulting from salt stress. The effect of difference in development among genotypes used for transcriptome analysis can be avoided by using nearly isogenic lines which differ in salinity tolerance at PI but have similar developmental behavior. However such optimal lines focusing on tolerance at PI are presently not available.
Commonalties in the salinity stress response of IR29 at early vegetative versus reproductive stage
Sensitive genotype IR29 responded to salinity stress during PI by inducing a large number of genes. Up-regulation of large number of genes was also reported for IR29 under salinity stress during early vegetative stage relative to tolerant genotype FL478 (Walia et al. 2005). We explored the extent of commonalities in salinity induced responses of IR29 by comparing the induced genes during vegetative and reproductive stage. One of the most striking stress responses of IR29 during the vegetative stage experiment was the induction of genes involved in the flavonoid pathway. In the present study, we again observed that several of the flavonoid pathway related genes were induced in IR29 under salt stress, in this case at PI. Among the genes induced include those encoding for phenylalanine ammonia lyase, chalcone synthase, dihydroflavonol 4-reductase, and flavonone 3-hydroxylase. Therefore, up-regulation of the flavonoid pathway as a response to salinity stress appears to be a general characteristic of IR29. Interestingly, some of the genes involved in the flavonoid pathway were down-regulated in the sensitive japonica, M103. So the induction of flavonoid pathway in response to salinity stress is not a universal response of sensitive rice genotypes.
We further compared the IR29 data from the vegetative stage study with the current IR29 PI data. In general, we found that the genes responding to salinity stress at vegetative stage were strikingly different from those which responded when the stress was imposed during PI. A total of 524 probe sets were differentially expressed in response to salinity at reproductive stage but not during vegetative stage. We were especially interested in genes which mapped to regions of the rice genome associated with salt tolerance based on QTL studies. One such gene (Os01g24710) encoding for SALT protein was induced during the reproductive stage. This gene has a jacalin-like lectin domain and physically maps to the Saltol region on chromosome 1 (13.88 Mb). Interestingly, another gene (Os01g25280) also encoding a SALT-related protein was induced during the vegetative stage in IR29 (Walia et al. 2005). This gene also maps to the Saltol region (14.26 Mb) on chromosome 1. The salT gene in rice is responsive to salt stress and the phytohormones abscisic acid and jasmonic acid (JA) (Claes et al. 1990; Moons et al. 1997). We found some genes involved in JA biosynthesis, and JA responsive genes, to be induced by salinity stress in IR29 during reproductive stage but not during vegetative stage. These include two lipoxygenases (Os03g52860 and Os12g37260), a gene encoding for 12-oxo-phytodienoic acid reductase (OPR2) (Os06g11240) and a jasmonate induced protein (JIP). The induction of JA biosynthesis and responsive genes during salinity stress is interesting in context of the report that post-treatment of salt-stressed rice with JA dramatically improves the physiological activities otherwise inhibited by stress (Kang et al. 2005). Results from our laboratory have also indicated an ameliorating role of JA pretreatment in salt-stressed barley on shoot Na+ concentrations and photosynthetic performance (Walia et al. 2007).
Besides salT, we found several other key transcripts related to salinity stress response as differentially expressed during reproductive stage. A potassium uptake protein (Os09g27580) induced by salinity stress during the reproductive stage in IR29 was one such transcript. This gene localizes to a region on chromosome 9 (16.7 Mb) which is associated with a major QTL for K+ uptake in rice identified by Koyama et al. (2001). This region on chromosome 9 was more recently linked to a QTL controlling root Na+ concentration (Lin et al. 2004). Another gene Os12g39400 (Os.16453.1.S1_at) encoding for a salt-tolerance zinc finger protein (STZ/ZAT10) was up-regulated during reproductive stage under salinity stress. The STZ/ZAT10 is a transcriptional repressor which is believed to regulate the COR/RD gene expression (Lee et al. 2002). The expression of STZ/ZAT10 was not up-regulated in any other genotype under salt stress. Based on this comparison, we can state that very few genes are commonly induced in response to salinity stress in a given genotype when sampled at two different developmental stages.
References
Abdullah Z, Ahmed R (1982) Salt tolerance of Solanum tuberosum L. growing on saline soils amended with gypsum. Z Acker und Pflanzenbau 151:409–416
Babourina O, Shabala S, Newman I (1998) Auxin stimulates Cl− uptake by oat coleoptiles. Ann Bot 82:331–336
Bak S, Tax FE, Feldmann KA, Galbraith DW, Feyereisen R (2001) CYP83B1, a cytochrome P450 at the metabolic branch point in auxin and indole glucosinolate biosynthesis in Arabidopsis thaliana. Plant Cell 12:101–111
Barbier-Brygoo H (1995) Tracking auxin receptors using functional approaches. Crit Rev Plant Sci 14:1–25
Barlier I, Kowalczyk M, Marchant M, Ljung K, Bhalerao R, Bennett M, Sandberg G, Bellini C (2000) The SUR2 gene of Arabidopsis thaliana encodes the cytochrome P450 CYP83B1, a modulator of auxin homeostasis. Proc Natl Acad Sci USA 97:14819–14824
Bentrup FW, Pfruner H, Wagner G (1973) Evidence for differential action of indole-acetic acid upon ion fluxes in single cells of Petroselinum sativum. Planta 110:369–372
Blatt MR, Thiel G (1993) Hormonal control of ion channel gating. Annu Rev Plant Physiol Plant Mol Biol 44:543–567
Bonilla P, Dvorak J, Mackill D, Deal K, Gregorio G (2002) RFLP and SSLP mapping of salinity tolerance genes in chromosome 1 of rice (Oryza sativa L.) using recombinant inbred lines. Philippine J Agric Sci 85:68–76
Bradley D, Carpenter R, Copsey L, Vincent C, Rothstein S, Coen ES (1996) Control of inflorescence architecture in Antirrhinum. Nature 376:791–797
Claes B, Dekeyser R, Villarroel R, van den Bulcke M, van Montagu M, Caplan A (1990) Characterization of a rice gene showing organ-specific expression in response to salt stress and drought. Plant Cell 2:19–27
Counce PA, Keisling TC, Mitchell AJ (2000) A uniform, objective, and adaptive system for expressing rice development. Crop Sci 40:436–443
Cui H, Takeoka Y, Wada T (1995) Effect of sodium chloride on the panicle and spikelet morphogenesis in rice. Jpn J Crop Sci 64:593–600
Haga K, Takano M, Neumann R, Iino M (2004) The rice COLEOPTILE PHOTOTROPISM1 gene encoding an ortholog of Arabidopsis NPH3 is required for phototropism of coleoptiles and lateral translocation of auxin. Plant Cell 17:103–115
Hayama R, Yokoi S, Tamaki S, Yano M, Shimamoto K (2003) Adaptation of photoperiodic control pathways produces short-day flowering in rice. Nature 422:719–722
Hayashi H, Czaja I, Lubenow H, Schell J, Walden R (1992) Activation of a plant gene by T-DNA tagging: auxin-independent growth in vitro. Science 258:1350 1353
Heenan DP, Lewin LG, McCaffery DW (1988) Salinity tolerance in rice varieties at different growth stages. Aust J Exp Agr 28:343–349
Hoshikawa K (1989) Growing rice plant-An anatomical monograph. Nosan Gyoson Bunka Kyokai (Nobunkyo), Tokyo, Japan
Kamei A, Seki M, Umezawa T, Ishida J, Satou M, Akiyama K, Zhu J-K, Shinozaki K (2005) Analysis of gene expression profiles in Arabidopsis salt overly sensitive mutants sos2-1 and sos3-1. Plant Cell Environ 28:1267–1275
Kang D-J, Seo Y-J, Lee J-D, Ishii R, Kim KU, Shin DH, Park SK, Jang SW, Lee I-J (2005) Jasmonic acid differentially affects growth, ion uptake, and abscisic acid concentration in salt-tolerant and salt-sensitive rice cultivars. J Agron Crop Sci 191(4):273–282
Khan MSA, Hamid A, Karim MA (1997) Effect of sodium chloride on germination and seedling characters of different types of rice (Oryza sativa L.). J Agron Crop Sci 179:163–169
Khatun S, Rizzo CA, Flowers TJ (1995) Genotypic variation in the effect of salinity on fertility in rice. Plant Soil 173:239–250
Kovtun Y, Chiu WL, Tena G, Sheen J (2000) Functional analysis of oxidative stress-activated mitogen-activated protein kinase cascade in plants. Proc Natl Acad Sci USA 97:2940–2945
Kovtun Y, Chiu WL, Zeng W, Sheen J (1998) Suppression of auxin signal transduction by a MAPK cascade in higher plants. Nature 395:716–720
Koyama ML, Levesley A, Koebner RMD, Flowers TJ, Yeo AR (2001) Quantitative trait loci for component physiological traits determining salt tolerance in rice. Plant Physiol 125:406–422
Lee H, Guo Y, Ohta M, Xiong L, Stevenson B, Zhu JK (2002) LOS2, a genetic locus required for cold-responsive gene transcription encodes a bi-functional enolase. EMBO J 21:2692–2702
Li C, Wong WH (2001) Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA 98:31–36
Lin HX, Zhu MZ, Yano M, Gao JP, Liang ZW, Su WA, Hu XH, Ren ZH, Chao DY (2004) QTLs for Na+ and K+ uptake of the shoots and roots controlling rice salt tolerance. Theor Appl Genet 108:253–260
Lutts S, Kinet JM, Bouharmont J (1995) Changes in plant response to NaCl during development of rice (Oryza sativus L.) varieties differing in salinity resistance. J Exp Bot 46:1843–1852
Moons A, Prinsen E, Bauw G, Van Montagu M (1997) Antagonistic effects of abscisic acid and jasmonates on salt stress-inducible transcripts in rice roots. Plant Cell 9:2243–2259
Moradi F, Ismail AM, Gregorio G, Egdane J (2003) Salinity tolerance of rice during reproductive development and association with tolerance at seedling stage. Indian J Plant Physiol 8:105–116
Nakagawa M, Shimamoto K, Kyozuka J (2002) Overexpression of RCN1 and RCN2, rice TERMINAL FLOWER 1/CENTRORADIALIS homologs, confers delay of phase transition and altered panicle morphology in rice. Plant J 29:743–750
Popova OV, Dietz KJ, Golldack D (2003) Salt-dependent expression of a nitrate transporter and two amino acid transporter genes in Mesembryanthemum crystallinum. Plant Mol Biol 52:569–578
Ren ZH, Gao JP, Li LG, Cai XL, Wei H, Chao DY, Zhu MZ, Wang ZY, Luan S, Lin HX (2005) A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat Genet 37:1141–1146
Rubinstein B (1974) Osmotic shock inhibits auxin-stimulated acidification and growth. Plant Physiol 59(3):369–371
Santa-Maria GE, Rubio F, Dubcovsky J, Rodriguez-Navarro A (1997) The HAK1 gene of barley is a member of a large gene family and encodes a high-affinity potassium transporter. Plant Cell 9:2281–2289
Scardaci SC, Eke AU, Hill JE, Shannon MC, Rhoades JD (1996) Water and soil salinity studies on California rice. Rice Pub. 2. Coop Ext Univ California, Colusa
Simpson GG, Gendall AR, Dean C (1999) When to switch to flowering. Annu Rev Cell Dev Biol 99:519–550
Takahashi R, Joshee N, Kitagawa Y (1994) Induction of chilling resistance by water stress, and cDNA sequence analysis and expression of water stress-regulated genes in rice. Plant Mol Biol 26:339–352
Thomine S, Lelievre F, Boufflet M, Guern J, Barbier-Brygoo H (1997) Anion channel blockers interfere with auxin responses in dark-grown Arabidopsis hypocotyls. Plant Physiol 115:533–542
Walia H, Wilson C, Condamine P, Liu X, Ismail AM, Zeng L, Wanamaker SI, Mandal J, Xu J, Cui X, Close TJ (2005) Comparative transcriptional profiling of two contrasting rice genotypes under salinity stress during the vegetative growth stage. Plant Physiol 139:822–835
Walia H, Wilson C, Wahid A, Condamine P, Cui X, Close TJ (2006) Expression analysis of barley (Hordeum vulgare) during salinity stress. Func Intgr Genom 6(2):143–156
Walia H, Wilson C, Condamine P, Ismail AM, Close TJ (2007) Large-scale expression profiling and physiological characterization of jasmonic acid mediated adaptation of barley to salinity stress. Plant Cell Environ (in press)
Yamaguchi-Shinozaki K, Shinozaki K (1993) The plant hormone abscisic acid mediates the drought-induced expression but not the seed-specific expression of rd22, a gene responsive to dehydration stress in Arabidopsis thaliana. Plant J 238:17–25
Yeo AR, Flowers TJ (1982) Accumulation and localization of sodium ions within the shoots of rice (Oryza sativa) varieties differing in salinity resistance. Physiol Plant 56:343–348
Yoshida S, Forno DA, Cock JH, Gomez KA (1976) Laboratory manual for physiological studies of rice. 3rd edn. IRRI, Los Banos, Philippines, pp 61–66
Zeng L, Lesch SM, Grieve CM (2003) Rice growth and yield response to changes in water depth and salinity stress. Agric Water Manage 59(1):67–75
Zeng L, Shannon MC (2000) Salinity effects on the seedling growth and yield components of rice. Crop Sci 40(4):996–1003
Zeng L, Shannon MC, Lesch SM (2001) Timing of salinity stress affects rice growth and yield components. Agric Water Manage 48(3):191–206
Zeng L, Shannon MC, Grieve CM (2002) Evaluation of salt tolerance in rice genotypes by multiple agronomic parameters. Euphytica 127:235–245
Acknowledgements
We wish to thank Dr. Xuan Liu for photosynthesis measurments, Don Layfield (GEB Salinity Laboratory, USA) for ion analysis. We also wish to thank Drs. Timothy Kingan and Barbara Walter (Core Instrumentation Facility, University of California, Riverside) for generating GeneChip data. This work was supported by an International Rice Research Institute USAID Linkage Program grant (DPPC 2004-30LOA0704) and in part by NSF DBI-0321756, “Coupling Expressed Sequences and Bacterial Artificial Chromosome Resources to Access the Barley Genome” and USDA-NRI 02-35300-12548, “HarvEST: A Portable EST Database Viewer”.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below are the links to the electronic supplementary materials.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Walia, H., Wilson, C., Zeng, L. et al. Genome-wide transcriptional analysis of salinity stressed japonica and indica rice genotypes during panicle initiation stage. Plant Mol Biol 63, 609–623 (2007). https://doi.org/10.1007/s11103-006-9112-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-006-9112-0