
Abstract
The story of hepatitis C virus (HCV) therapy is about to add one of its biggest chapters. From the serendipitous beginnings in the 1980s when investigators noted that interferon improves liver enzymes in non-A, non-B hepatitis, to the discovery and naming of the hepatitis virus, to the addition of ribavirin, to the pegylation of interferon, and now to the first direct-acting antivirals (DAA), the history of HCV is an intriguing one that continues to unfold. Along with the first DAAs, other important findings have helped explain long-observed differences between various ethnic groups, as well as new predictive information that can be gleaned from some of the observed adverse events.
Similar content being viewed by others

Introduction
Big changes are coming in the field of hepatitis C. In fact, the soon-to-arrive direct-acting antiviral (DAA) therapies will be the biggest advance in the field since the addition of ribavirin to interferon and the pegylation of interferon [1–5]. Addition of an oral antiviral to the pegylated interferon/ribavirin (PEG/RBV) backbone could nearly double the sustained virologic response (SVR) rate of genotype 1 disease. The first two DAAs will be the linear serine protease inhibitors, telaprevir (TVR) and boceprevir (BOC). These two compounds have more similarities than differences and will likely be widely used when available in 2011. Soon to follow are other compounds, and perhaps combinations of DAAs with different mechanisms of action.
The hepatitis C virus has genomic heterogeneity, and multiple genotypes and subtypes exist. Of the six predominant genotypes, hepatitis C virus (HCV) genotype 1 is the most common strain in the United States, Europe, and Japan, and is the most difficult to eradicate [6]. From dismal early results, with 6 months of interferon monotherapy curing fewer than 5% of patients, the addition of ribavirin and prolongation to 48 weeks increased this rate more than fivefold to 25% to 30%. [3] The pegylation of interferon led to overall improvement in efficacy to 40% [4, 5]. This combination has been the standard of care for most of the past decade. The 3000-patient, multicenter Individualized Dosing Efficacy versus Flat Dosing to Assess Optimal PegInterferon Therapy (IDEAL) trial was the largest study of treatment-naïve patients with HCV genotype 1 disease, and revealed that pegylated interferon-α2a and 2b, when combined with ribavirin, have comparable efficacy and adverse events [7].
The discovery of DAAs for the treatment of HIV set the stage for the development of similar classes of compounds for HCV [8]. The development of protease inhibitors was hindered by challenges related to binding inefficiencies and toxicity; the proof of concept turning point came with the Boehringer Ingelheim (Ingelheim am Rhein, Germany) compound 2061, which showed that over a 3-day course, viral declines of greater than 4 log could be achieved [9]. Although this compound did not see further development because of the risk of cardiac toxicity, it was catalytic in the development of other similar compounds.
In addition to new compounds, other changes are coming. Duration of therapy will be based on early viral responses. Baseline predictors of response have always been important in determining likelihood of response, but new genetic determinants will advance this field as well. Finally, the promise of an interferon-free regimen draws ever closer and will be the focus of many upcoming clinical trials in the next few years.
Protease Inhibitors
The serine protease NS3-NS4A is used by HCV for post-translational processing and viral replication. This was the first successful target of drug development in HCV. Protease inhibitors have been shown to be potent inhibitors of viral replication, but when used as monotherapy, resistance quickly develops [10]. Hence, the addition of this class of compound to an interferon/ribavirin-containing regimen is the current development paradigm. This development does not preclude the use of this class in future interferon-sparing regimens, as long as other compounds can cover emerging resistant strains.
Boceprevir
The first two protease inhibitors that have now completed phase 3 trials are boceprevir and telaprevir, both linear compounds taken orally three times per day and used for HCV genotype 1. Boceprevir (SCH-503034; BOC) in the phase 2b trial (HCV Serine Protease Inhibitor Therapy 1 [SPRINT 1]) of 595 patients was evaluated as triple therapy with peginterferon alfa 2b (PEG2b) and ribavirin (RBV) for 28–48 weeks. A unique lead-in strategy of 4 weeks of PEG2b/RBV was also evaluated followed by triple therapy for 24–44 weeks (discussed in detail below). This lead-in was found to lower viral breakthrough and was adopted for the phase 3 trials. The SVR rates in the phase 2b trial treating previously untreated patients ranged from 55% to 75% [11]. This study also assessed a low-dose ribavirin arm, which yielded a lower SVR than control, and a higher viral breakthrough and relapse. Hence, the importance of ribavirin dosing appears to be paramount even in the setting of a protease inhibitor. In this study, patients received either 24 or 44 weeks of BOC, and it appeared the longer duration was superior. However, the use of 24 weeks of BOC followed by another 24 weeks of only PEG/RBV was to be assessed in the phase 3 trial.
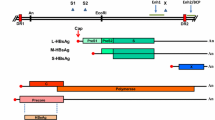
The phase 3 trial, called SPRINT 2 (Fig. 1), assessed about 1,100 patients in two separate cohorts, the second cohort being exclusively patients of African descent. In cohort 1, 68% of patients achieved SVR whereas 53% of black patients (cohort 2) achieved SVR with triple therapy using a 4-week lead in followed by 44 weeks of triple therapy. Using a response-guided treatment regimen allowed about half of the patients to be treated with a 4-week lead-in followed by 24 weeks for triple therapy with PEG/RBV/BOC. This led to SVR rates of 67% and 42% in non-blacks and blacks, compared to controls of 40% and 23% (P = 0.004 and P = 0.044), respectively [12]. From these data, it appears that BOC can be used for a 24-week period, and the total duration of PEG/RBV would be 28 weeks for patients who become viral negative by week 8 and remain so until week 24. Conversely, patients who become viral negative between weeks 8 and 24 would receive 24 weeks of BOC, but 48 weeks of PEG/RBV. This response-guided approach allows the minimization of treatment duration, but maximizes efficacy. The rate of anemia with BOC-containing regimens is almost double that of PEG/RBV, but rarely leads to treatment discontinuation.

Serine Protease Inhibitor Therapy (SPRINT) 2 and A New Direction in HCV Care: A Study of Treatment-naïve Hepatitis C Patients with Telaprevir (ADVANCE): design of the boceprevir (BOC) and telaprevir (TVR) phase 3 trials in treatment-naïve hepatitis C virus (HCV) genotype 1 patients. eRVR—extended rapid viral response; PR—pegylated interferon and ribavirin; TW8—treatment week 8
The nonresponder phase 3 trial of boceprevir is called the Retreatment with HCV Serine Protease Inhibitor Boceprevir and PEGINTRON/REBETOL 2 (RESPOND 2) trial. The phase 2 study that preceded this study did not use the ultimate dose of boceprevir with full-dose RBV, and led to SVR rates of only 14%. With appropriate dosing of boceprevir in the 404-patient RESPOND 2 trial, the SVR rates were 66% in the 48-week arm, and 59% in the response-guided arm, both significantly better than the control of 21% [12]. Although the study intended to exclude the so-called “null” responder patient, a large percentage of patients enrolled appear to fit that description. In fact, 25% of the patients achieved less than a 1-log viral decline at the end of the lead-in period, indicating that they had poor interferon response. Anemia occurred in up to 47% of patients on BOC-containing arms and in 20% of controls. Interestingly, discontinuations from anemia only occurred in 2% of patients, indicating that anemia may not be a treatment-limiting adverse event.
Lead-in Strategy
One of the unique features of the boceprevir development strategy was to use a 4-week lead-in of PEG/RBV prior to initiating boceprevir. The original premise was to lower viral load and theoretically decrease viral breakthrough and resistance. This premise was borne out in the SPRINT 1 trial. Additionally, other potential utilities of such a paradigm have come to light. Patients who achieve rapid viral response (RVR) at the end of the lead-in period are known to have high SVR rates to PEG/RBV, even with a shortened 24-week duration of therapy [13, 14]. Although RVR occurs in only 10% of US patients, it occurs in up to 26% of European patients [7, 15], leading to speculation that this lead-in paradigm could be used by health care plans to spare DAA therapy in this group, which has a high likelihood of viral clearance with current standard therapy. Conversely, patients who are minimally responsive to PEG/RBV in the lead-in have relatively low SVR rates in the mid-30% range. These patients may require more diligent monitoring for resistance and may benefit from more effective future therapies that increase response rates and minimize resistance. Finally, the lead-in tests the patient’s compliance and tolerance to PEG/RBV prior to committing to the increased adverse event profile associated with DAAs. Patients who cannot tolerate PEG/RBV should not be given a DAA.
Telaprevir
Telaprevir (VX-950; TVR) is another serine protease inhibitor that has completed its phase 3 trial (ADVANCE; A New Direction in HCV Care: A Study of Treatment-naïve Hepatitis C Patients with Telaprevir) in previously untreated genotype 1 patients, yielding a 75% SVR compared to a control of 44%. The response-guided therapy regimen of this study will be the preferred treatment paradigm, with over half of patients receiving 12 weeks of TVR and 24 weeks of PEG/RBV, and the others 48 weeks of PEG/RBV. Although this study also tested 8-week duration of TVR, it had a lower SVR of 69% and will likely not be the preferred regimen [16]. The two phase 2b studies, called Protease Inhibition for Viral Evaluation 1 and 2 (PROVE 1 and 2), were conducted in the United States and Europe, respectively. The SVR rates were 61% to 69%; several different arms were studied, including a short-duration therapy and a ribavirin-free arm. The trial design of the phase 3 study was different in that a response-guided approach was used. Patients who were viral-negative at weeks 4 and 12, the so-called extended RVR (eRVR), received a total duration of 12 weeks of TVR with 24 weeks of PEG/RBV. Patients who did not achieve eRVR received a total of 48 weeks of PEG/RBV. More than half of the patients were eligible for the shorter duration. This result was validated in the Illustrating the Effects of Combination Therapy with Telaprevir (ILLUMINATE) trial; this 540-patient trial did not have a control arm, but randomly assigned patients with eRVR to a total duration of therapy of 24 versus 48 weeks with 12 weeks of TVR. This trial revealed that the overall SVR in the study was 72%, but patients meeting eRVR did not benefit from a total duration of 48 weeks of PEG/RBV, with SVR rates of 92 and 88%, respectively, when eRVR was achieved [17].
The phase 2b trial assessing previously treated PEG/RBV patients, called the PROVE 3 study, tested several treatment durations. Based on the findings of that trial, it was decided that a total duration of 48 weeks of PEG/RBV with 12 weeks of TVR would be the paradigm for previously treated patients moving into the phase 3 trial. Hence, all arms of the phase 3 Re-treatment of Patients with Telaprevir-based Regimen to Optimize Outcomes (REALIZE) trial were 48 weeks in duration. One of the arms had the experimental 4-week lead-in of PEG/RBV preceding 12 weeks of triple therapy, followed by 32 weeks of PEG/RBV. The results of that arm were 66%, with 64% in the 48-week arm, and 17% in the control arm. In a pooled analysis of both study arms, relapsers had the highest SVR at 86% (24% control), followed by those who had a partial response to previous treatment at 57% (15% control), and finally, null responders at 31% (5% control) [18].
Similarities and Differences Between Boceprevir and Telaprevir
Based on the phase 2 data, it is clear that ribavirin plays a critical role in the treatment regimen of both protease inhibitors. Indeed, in the RBV-free or low-dose RBV arms of PROVE 2 and SPRINT 1, respectively, SVR rate was only 36% compared to controls of 46% and 38%. Hence, maintenance of optimal ribavirin dosing remains an important treatment goal with both regimens. This may be an issue given the higher rate of anemia seen in these studies. Anemia is a class effect of linear protease inhibitors, but is more common with BOC compared to TVR. Only 23% of patients in the SPRINT 1 trial maintained a hemoglobin above 11 g/dL, compared to 35% of patients in PROVE 1. In these two studies, 27% and 23% of patients had hemoglobin levels that fell below 9.5 g/dL with BOC- and TVR-based therapies, respectively [11, 19].
The major unique adverse event related to BOC appears to be dysgeusia (32%; 23% higher than control). About one third of patients experience dysgeusia, though it was not treatment limiting (Table 1). The adverse events that appear to be more common with TVR are rash and pruritus. Although 61% of patients had some degree of rash in the PROVE 1 study (20% higher than control), only a minority had to discontinue for this reason. Similarly, 48% had pruritus (25% higher than control) but it was not a treatment-limiting adverse event (Table 1). Discontinuation for rash occurs in 5% to 7% of patients, so it is likely not going to be a major limitation to treatment. With all of these adverse events, provider experience will be a major determinant of maintaining patients on treatment. Appropriate symptomatic treatment for these patient complaints will be a major part of the overall management, and will require diligence on the part of the health care provider team.
Treatment Regimen Differences
Based on the phase 3 study designs for BOC and TVR, it is likely that response-guided therapy will be the new paradigm for treatment-naïve patients. However, the paradigms are quite different. The BOC treatment will be a 4-week lead-in with PEG/RBV followed by 24 weeks of PEG/RBV/BOC and no further treatment for patients who became negative by week 8 and remain so until week 24. Patients who had detectable virus at any point between weeks 8 and 24, will stop BOC at week 28, but will continue PEG/RBV until week 48. Hence, all patients will have 24 weeks of BOC, and rapid responders will have total treatment duration of 28 weeks, whereas slow responders will have an extra 20-week extension of PEG/RBV. The treatment-experienced population will also likely have response-guided therapy with a lead-in and 36 weeks of PEG/RBV/BOC, with or without an additional 12 weeks of PEG/RBV for those patients who had detectable virus at week 8. Previously treated patients with detectable virus at week 12 will discontinue therapy for futility.
The TVR response-guided protocol for treatment-naïve patients will require viral negativity at weeks 4 and 12 for the total treatment duration to be 24 weeks, with TVR given over the first 12 weeks only. If patients do not have undetectable virus at both 4- and 12-week time points, they will require a total duration of 48 weeks of PEG/RBV. The paradigm for treatment-experienced patients will not be response-guided based on the design of the REALIZE trial. Patients will receive 12 weeks of TVR and 48 weeks of PEG/RBV.
In both the BOC and TVR phase 3 trials, the SVR in patients with a poor response to interferon, whether it was by historical assessment of 12-week lab values from referring doctors’ records, or lack of a 1-log decline at the end of the lead-in period, was very low, ranging from 29% to 34%. This population is perhaps not ideally served by either of these compounds, and further assessment of more potent compounds or more novel regimens is warranted. Until such time, it is likely that patients with null responses to interferon will be retreated with BOC and TVR, but retreatment should be done with great care, because viral breakthrough and relapse rates are high.
In distinction to this population, treatment-naïve patients, partial responders, and relapsers appear to have much better response rates and clearly are the prime groups to target for treatment. Patients with HIV coinfection, pre-transplant patients with low platelet counts, decompensated liver disease, or renal failure, and liver transplant recipients have yet to be studied. Although these patients represent very important subsets of the HCV population, treatment of these groups will have to await study results prior to any recommendations.
The study designs of BOC and TVR are quite different. The differences of these new treatment paradigms will require clinicians to apply different laboratory monitoring strategies based on the regimen being used. Although a learning curve is inevitable, the concept of individualization of therapy is beneficial for the patient and ultimately is designed to prevent over- and under-treatment. There are no head-to-head comparisons of these two compounds, but both BOC and TVR are being evaluated with both pegylated interferon alfa-2a and 2b to determine if the interferons can be used interchangeably with either drug.
Other Compounds in Development
No other HCV DAA compounds are in phase 3 development yet. Of the several other protease compounds, the Tibotec (Mechelen, Belgium) TMC 435 compound has shown the most promise based on daily dosing and a good safety profile. The polymerase inhibitors are more numerous and are beyond the scope of this article to review. They have been somewhat difficult to develop because of toxicity, but the hope is that in combination with a protease inhibitor, the lack of cross resistance and possible synergistic antiviral activity will allow for a more robust regimen when added to the PEG/RBV backbone. Along the same lines, the NS5A inhibitors are being evaluated as members of multi-compound cocktails. The Bristol-Myers Squibb (New York, NY) compound BMS790052 is currently being studied; if effective and safe, it will allow the further development of this interesting class of compound [20].
Other agents being evaluated (e.g., cyclophilin inhibitors, nitazoxanide, and silymarin) will likely not have major roles in this treatment arena. The need to find novel targets will drive further development, particularly to address the future BOC and TVR treatment failures, who will need a more complex regimen [20].
Other Important Developments
It has long been known that anemia is one of the major side effects of HCV therapy. What has recently been appreciated is the correlation with anemia and SVR. Of the 3,070 patients in the IDEAL trial, 28.6% (N = 865) developed anemia (hemoglobin <10 g/dL). About half of these patients received an erythropoiesis-stimulating agent (ESA). When anemia occurred within 8 weeks of treatment initiation, the use of ESA was associated with fewer patient discontinuations, and hence enhanced SVR (45.2% vs. 25.9%, P < 0.001). Similarly, in patients who had greater than a 3 g/dL decline in hemoglobin, SVR was more likely to occur (43.7% vs. 29.9%, P < 0.001) [21•]. The likely explanation is that these individuals have an adequate exposure to ribavirin. However, it has also been shown that genetic polymorphisms in inosine triphosphatase deficiency are protective against hemolytic anemia [22•]. Hence, some patients without anemia will also achieve sustained response. The association of anemia to SVR in the setting of DAAs has yet to be fully evaluated.
The identification of a single-nucleotide polymorphism (SNP), rs12979860 on chromosome 19, near the gene that encodes interleukin (IL)-28B was a major discovery that came out of the IDEAL trial. More than half of the 3,070 patients consented to genomic testing, and identification of this association with SVR has explained at least some of the genetic differences previously noted in interferon response rates. The favorable homozygous CC haplotype is associated with more than a twofold increase in SVR compared to other haplotypes, such as CT or TT. Interestingly, the prevalence of the CC haplotype is proportional to the SVR rates seen in various ethnicities: highest in Asians, lowest in African ethnicities. The favorable polymorphism leads to early viral decline and hence, higher SVR. In patients with the CC genotype, RVR was achieved more frequently. However, even when it was not achieved, SVR was more likely to occur in CC patients by a factor of two. As a baseline predictor of response, in multivariable regression analysis, IL-28B haplotype was the strongest forecaster of SVR (OR = 5.2; 4.1–6.7, 95% CI, P < 0.0001). Although RVR has a higher positive predictive value, it is an on-treatment variable, and not of utility prior to treatment. Patients with CC genotype have a high probability of response to the current standard of care therapy. The utility of DAAs in this population may be to shorten therapy to 6 months, perhaps even shorter. Data on IL-28B haplotypes were collected in some DAA trials, and further assessment on the utility in the setting of these new therapies has yet to be determined. The test is now clinically available through LabCorp (Burlington, NC) on a buccal swab or whole-blood specimen (test number 480630; IL28B polymorphism genotype by RT-PCR) [23•]. As more data emerge from the ongoing DAA trials, the utility of this test will be fully appreciated, and it is likely that it will be a part of the standard assessment of genotype 1 patients in particular.
Conclusions
The upcoming change in HCV genotype 1 therapy will be the most important advance in more than a decade. The first anticipated DAAs to be approved will be boceprevir and telaprevir. Both have shown efficacy over current standard therapy and appear to be effective in naïve and treatment-experienced patients. The complexity of these regimens and the added adverse events will require a new level of interest and diligence by health care providers, and understanding by patients who may have expected a simpler treatment regimen. However, the potential benefits are immense, and a much larger percentage of patients should be able to enjoy sustained viral clearance, altering the natural history of the disease in many. These big changes are likely only the beginning of many more changes to come, including other novel compounds and perhaps even all-oral therapies in some patients. Undoubtedly, 2011 will become a pivotal and historic year in the story of hepatitis C treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Hoofnagle JH, Mullen KD, Jones BD, et al.: Treatment of chronic non-A, non-B hepatitis with recombinant human alpha interferon. N Engl J Med 1986, 315:1575–1578.
Choo QL, Kuo G, Weiner AJ, et al.: Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244:359–362.
McHutchison JG, Gordon SC, Schiff ER, et al.: Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N Engl J Med 1998, 339:1485–1492.
Manns MP, McHutchison JG, Gordon SC, et al.: Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001, 358:958–965.
Fried MW, Shiffman ML, Reddy KR, et al.: Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002, 347:975–982.
Choo QL, Richman KH, Han JH, et al.: Genetic organization and diversity of the hepatitis C virus. Virol 1994, 68:4420–4426.
McHutchison JG, Lawitz EJ, Shiffman ML, et al.: Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med 2009, 361:580–593.
Billich S, Knoop MT, Hansen J, et al.: Synthetic peptides as substrates and inhibitors of human immune deficiency virus-1 protease. J Biol Chem 1988, 263:17905–17908.
Hinrichsen H, Benhamou Y, Wedemeyer H, et al.: Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 2004, 127:1347–1355.
Asselah T, Benhamou Y, Marcellin P: Protease and polymerase inhibitors for the treatment of hepatitis C. Liver Int 2009, 29(Suppl 1):57–67.
Kwo PY, Lawitz EJ, McCone J, et al.: Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT 1): an open-label, randomised, multicentre phase 2 trial. Lancet 2010, 376:705–716.
In pivotal phase III studies, Merck’s investigational medicine boceprevir helped majority of patients with chronic hepatitis C genotype 1 infection achieve sustained virologic response, the primary endpoint of the studies [news release]. Available at http://www.merck.com/newsroom/news-release-archive/research-and-development/2010_0804.html. Accessed September 2010.
Zeuzem S, Buti M, Ferenci P, et al.: Efficacy of 24 weeks treatment with peginterferon alfa-2b plus ribavirin in patients with chronic hepatitis C infected with genotype 1 and low pretreatment viremia. J Hepatol 2006, 44:97–103.
Jensen DM, Morgan TR, Marcellin P, et al.: Early identification of HCV genotype 1 patients responding to 24 weeks peginterferon alpha-2a (40 kd)/ribavirin therapy. Hepatology 2006, 43:954–960.
Ferenci P, Fried MW, Shiffman ML, et al.: Predicting sustained virological responses in chronic hepatitis C patients treated with peginterferon alfa-2a (40 KD)/ribavirin. J Hepatol 2005, 43:425–433.
Jacobson IM, McHutchison JG, Dusheiko GM, et al.: Telaprevir in combination with peginterferon and ribavirin in genotype 1 HCV treatment-naïve patients: final results of phase 3 ADVANCE study. Presented at the 62st Annual Liver Meeting of the American Association for the Study of Liver Diseases. Boston, MA; October 29–November 2, 2010.
Phase 3 ILLUMINATE study supports 24-week telaprevir-based therapy [news release]. Available at http://www.fiercebiotech.com/press-releases/phase-3-illuminate-study-supports-24-week-telaprevir-based-therapy. Accessed September 2010.
Telaprevir combination therapy boosts sustained response rates for previously treated hepatitis C patients [news release]. Available at http://www.hivandhepatitis.com/hep_c/news/2010/0910_2010_a.html. Accessed September 2010.
McHutchison JG, Everson GT, Gordon SC, et al.: Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009, 360:1827–1838.
Pockros P: Review: New direct-acting antivirals in the development for hepatitis C virus infection. Therapeutic Adv Gastroenterol 2010, 3:191–202.
• Sulkowski MS, Shiffman ML, Afdhal NH, et al.: HCV treatment-related anemia is associated with higher sustained virologic response rate. Gastroenterology 2010 (Epub ahead of print). This article provides a clinical description of the correlation of anemia and sustained virologic response. Anemia is not always a bad thing.
• Fellay J, Thompson AJ, Ge D, et al.: ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature 2010, 464:405–408. This important paper outlines a gene variant responsible for protecting against ribavirin-induced anemia.
• Thompson AJ, Muir AJ, Sulkowski MS, et al.: Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010, 139:120–129. This landmark clinical paper describes the power of the IL-28B polymorphism and its effect on interferon response between ethnicities. Much of the lower response in black patients is explained by this finding.
Disclosure
Conflicts of interest: F. Poordad has received research grants, fees and/or honoraria from Vertex, Schering Plough/Merck, Gilead, Abbott, Tibotec, Salix, Idenix, and Roche/Genentech.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Poordad, F. Big Changes Are Coming in Hepatitis C. Curr Gastroenterol Rep 13, 72–77 (2011). https://doi.org/10.1007/s11894-010-0153-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-010-0153-9