
Abstract
The term “gray-zone” lymphoma has been used to denote a group of lymphomas with overlapping histological, biological, and clinical features between various types of lymphomas. It has been used in the context of Hodgkin lymphomas (HL) and non-Hodgkin lymphomas (NHL), including classical HL (CHL), and primary mediastinal large B cell lymphoma, cases with overlapping features between nodular lymphocyte predominant Hodgkin lymphoma and T-cell/histiocyte-rich large B cell lymphoma, CHL, and Epstein–Barr-virus-positive lymphoproliferative disorders, and peripheral T cell lymphomas simulating CHL. A second group of gray-zone lymphomas includes B cell NHL with intermediate features between diffuse large B cell lymphoma and classical Burkitt lymphoma. In order to review controversial issues in gray-zone lymphomas, a joint Workshop of the European Association for Hematopathology and the Society for Hematopathology was held in Bordeaux, France, in September 2008. The panel members reviewed and discussed 145 submitted cases and reached consensus diagnoses. This Workshop summary is focused on the most controversial aspects of gray-zone lymphomas and describes the panel’s proposals regarding diagnostic criteria, terminology, and new prognostic and diagnostic parameters.
Similar content being viewed by others


Introduction
With application of modern immunophenotypic and molecular genetic techniques, most cases of lymphomas can be successfully diagnosed and classified as one of the currently recognized distinct disease entities. However, there are some cases which show overlapping or borderline morphological, biological, and clinical features between various types of lymphomas. The 2008 World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues [1] recognized this problem and introduced two new provisional categories of B cell lymphoma, unclassifiable, one with features intermediate between diffuse large B cell lymphoma (DLBCL) and Burkitt lymphoma (BL) [2] and the second with features intermediate between DLBCL and classical Hodgkin lymphoma (CHL) [3].
The first provisional category relates to the distinction of classical BL from DLBCL with morphology similar to BL and/or presence of an MYC translocation, with or without other translocations. Many of these cases occur in adults and morphologically resemble BL. Some of them were previously classified as “Burkitt-like” lymphoma, a term that has been deliberately abandoned [4]. Many lymphomas in this category present with translocations of both MYC and BCL2 and/or BCL6 (double or triple hit) and have a very aggressive clinical course [5]. The second provisional category recognizes cases that usually occur in the mediastinum and have features of both CHL and DLBCL, usually primary mediastinal large B cell lymphoma (PMBL). Although in most cases one or the other diagnosis can be made, there seems to be a true biological “gray-zone” between the two entities. Indeed gene expression profiling revealed striking similarities between CHL and PMBL [6, 7]. Although this category includes mainly lymphomas in young patients with mediastinal disease, similar cases have been reported in peripheral lymph nodes as a primary site in the adult population. The tumor cells in these cases exhibit an immunophenotype with transitional features between CHL and PMBL [8]. These lymphomas generally have a more aggressive clinical course and poorer outcome than either CHL or PMBL. A third group of gray-zone lymphoma, although not explicitly included as a provisional category in the 2008 WHO classification, deals with the relationship between nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) and T-cell/histiocyte-rich large B cell lymphoma (THRLBCL). Although both entities have distinctive clinical presentations and morphological features in the vast majority of cases, patients with NLPHL may show features of THRLBCL in initial or subsequent biopsies, which raises the question whether a diffuse variant of NLPHL might represent THRLBCL or whether NLPHL can progress to THRLBCL [9]. Because NLPHL and THRLBCL are both derived from follicle center B cells, problems in the differential diagnosis may occur. The immunophenotype of the tumor cells does not differ significantly; however, recognition of the small reactive B cell background and the growth pattern (nodular vs. diffuse) is of great help in the differential diagnosis. Nevertheless, there are cases of NLPHL with paucity of small B cells in the background and areas with a diffuse growth pattern and cases of THRLBCL with a nodular growth pattern of CD20+ tumor cells with few small reactive B cells, making distinction of the two entities difficult [10].
A fourth group of gray-zone lymphoma may be identified. This group is more heterogeneous than the others. Rather than having a basis in true biological overlap, it reflects diagnostic uncertainty. This group encompasses cases of EBV+ lymphoproliferations, including some cases of Hodgkin lymphoma and the newly recognized category of EBV-positive DLBCL of the elderly [11].
The European Association for Hematopathology (EAHP) and the Society of Hematology (SH) organized a workshop during the XIV Meeting of the EAHP held in Bordeaux, France in September 2008, in order to better characterize the pathologic features of gray-zone lymphomas, to clarify boundaries between these lymphomas and other well-defined lymphomas and highlight areas that require further study. One hundred and forty-five cases were received and reviewed by a panel of experienced hematopathologists. The cases submitted were initially categorized into four groups:
-
1.
Overlap between Burkitt lymphoma and diffuse large B cell lymphoma
-
2.
Primary mediastinal B cell lymphoma, mediastinal gray-zone lymphomas (MGZL), and cases with atypical immunophenotype
-
3.
Gray zone around nodular lymphocyte predominant Hodgkin lymphoma, T-cell/histiocyte-rich large B cell lymphoma, and classical Hodgkin lymphoma
-
4.
EBV+ lymphomas, lymphomas occurring in HIV+ individuals, and posttransplant lymphoproliferative disorders (PTLD)-related B cell lymphoproliferations.
The workshop forum provided a unique opportunity for open—and often lively—discussions between case submitters, the panel, and the other participants. The main topics of the discussions included ambiguities in the terminology and classification of different lymphomas under the umbrella of gray zone offering the opportunity to discuss the conclusions of the recent 2008 WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. The following report summarizes the discussions and conclusions emerging from the Workshop.
The overlap between Burkitt lymphoma and diffuse large B cell lymphoma (session 1)
The overlap between BL and DLBCL has been a matter of controversy among pathologists and clinicians, as well as between both groups. Separation of the two entities is considered relevant because treatment decisions are based on the different diagnoses, and prognostic impact is implied. Based on biological and clinical information gathered over the past few years, the subject was thoroughly re-evaluated for the fourth edition of the WHO classification, and some distinct decisions on classification were made [1]. Consequently, the possible overlap or gray zone between BL and DLBCL was chosen as one of the subjects of the EAHP workshop. In this section, we will review the background of these decisions, the newly set definitions, and the specific issues of differential diagnostic problems as were highlighted by the submitted cases.
Approach for classification in the WHO classification
For the fourth edition of the WHO classification, an attempt was made to retain the “clean entities.” Thus, BL and DLBCL were very narrowly defined based on morphology, immunophenotype, and genetic alterations in order to set very distinct biological entities with distinct clinical features. Terms like “Burkitt-like lymphoma” and “atypical BL” were eliminated since this terminology has led to much confusion in the past, mainly due to the fact that definitions were never clearly set and individual authors have used them in different ways. Inevitably, the choice implies that a larger number of cases will fall outside these narrow definitional boundaries and remain unclassifiable. To accommodate these cases, a novel category was established, “aggressive B cell lymphoma, unclassifiable with features intermediate between BL and DLBCL” [2]. This group should not be regarded as an entity but as a provisional sanctuary for differential diagnostic problems that await results from ongoing research to provide a sound scientific basis for future classification.
Burkitt lymphoma
Three clinical variants of BL have been recognized: the endemic form (also called African type), the sporadic or nonendemic form, and the immunodeficiency-related form (mostly HIV-associated). The endemic form is generally found in boys under the age of 10 years in the jaw or in the abdomen. This lymphoma is EBV positive in 100% of the cases. Recent investigations in Africa suggest that other epidemiologic pathogenetic cofactors might also play a role. The sporadic variant is observed everywhere in the world and affects boys between 5 and 15 years old. In the Western world, it is the most frequent lymphoma in children with 45%, in contrast to only 1–2% in adults. In most patients, the tumor presents in the ileocecal region, but other mainly extranodal (abdominal) localizations are observed. Extensive meningeal and bone marrow involvement can been seen. Patients may present with fully leukemic, fully “solid,” or combined patterns. The HIV-related form is very often predominantly nodal. EBV is present in 20–30% of all cases [12–14].
Typically, BL has a cohesive growth pattern with squared-off cell borders, and usually a starry-sky pattern is seen. The cells have a deeply basophilic cytoplasm with lipid vacuoles (Fig. 1a, f) The nuclei are medium-sized and are smaller than or similar in size to histiocyte nuclei. In well-preserved cases, the nuclei are round and contain a finely clumped chromatin and multiple medium-sized paracentral nucleoli. In the new definition of BL, some morphological variation is accepted to include a more pronounced range in nuclear size and presence of large central nucleoli, as were previously included in “atypical BL.” Much of this variation may be caused by differences in preservation and fixation, as well as by interobserver interpretation. Indeed, since gene expression studies suggest complete overlap, classical and atypical morphologies were lumped together in the new WHO classification [15, 16]. In spite of this morphologic diversity, the immunophenotype and genotype are very narrowly defined with high expression of CD20, surface IgM without IgD, and homogeneous expression of CD10, BCL6, CD77, CD38, and absence of BCL2. IRF4/MUM1 may be expressed in some cases but is generally weakly expressed (Fig. 1c–e, g, h). Expression of terminal deoxynucleotidyl transferase should be absent.

a Burkitt lymphoma. Note the typical morphology of BL cells and the starry-sky pattern. b DLBCL. The morphology of this case is consistent with a diagnosis of DLBCL. In case of an appropriate immunophenotype and genetic background, a diagnosis of (atypical) Burkitt lymphoma with plasmacytoid differentiation may be considered. The case illustrates the morphological overlap between BL and DLBCL. c–g Cytological features and immunophenotype of BL. Note the strong positivity of the tumor cells for CD10 (c) and BCL6 (d). The proliferation rate is almost 100% as demonstrated by MIB-1 staining (e). The cytological features are characteristic of BL (f). BCL2 is negative on tumor cells in the presence of positive T cells as internal control (g). h In contrast, note the strong BCL2 positivity, suspicious of dual translocation (double hit). i Session 1, interphase FISH showing MYC trisomy and possibly amplification (IGH green, MYC red). j Session 1, BCL6 dual-color break apart FISH showing a BCL6 break. k Session 1, complex karyotype with multiple structural alterations, amplifications, and deletions. l Session 1, simple karyotype, only showing t(8;14) and trisomy 12
The genetic basis of BL is defined by a translocation involving one of the immunoglobulin genes (IG) and MYC in the context of a very limited number of additional aberrations (simple karyotype; Fig. 1l). In 40% of the cases, no additional cytogenetic abnormalities are identified. This can be appreciated by conventional karyotyping but also by methods like comparative genomic hybridization (CGH) array [17]. In the majority of the cases, IGH is found as a translocation partner, whereas in approximately 15% of the cases IGL or IGK is found [18]. All MYC/8q24 breaks can be detected best using fluorescent in situ hybridization (FISH), which is applicable to formalin-fixed paraffin embedded (FFPE) tissue sections [19]. Classical karyotyping remains very helpful since the yield is high and the results give information on not only the translocation but also on the genetic complexity caused by other structural and/or numerical abnormalities [18]. Independent studies have shown that gene expression profiles are very characteristic for BL [15, 16].
In the workshop, four cases of BL were submitted that highlighted the morphological spectrum. Of this small series, IRF4/MUM1 was strongly positive in one case (submitted by R. Felgar), and aberrant CD4 expression was seen in an HIV-associated case (submitted by G. Fan).
Diffuse large B cell lymphoma with features of Burkitt lymphoma
In adult patients of Western countries, DLBCL represents almost 40% of all NHL, whereas in pediatric patients it accounts for up to 10–20% of all NHL. Various subtypes of DLBCL can be discerned, however, DLBCL, not otherwise specified still remains a mixed bag with a highly divergent clinical presentation, morphology, phenotype, and molecular background.
In less than 10% of the cases, differential problems with BL may be considered (Fig. 1b). These cases often show a cohesive growth pattern without any stromal reaction or admixture with reactive T cells, a high proliferation rate, numerous apoptotic bodies, and a starry-sky pattern [20]. However, in the absence of typical or atypical BL cellular morphology, a diagnosis of BL should not be considered, and cases with an unambiguous large cell morphology, “empty” vesicular nucleus, and multiple large nucleoli and/or abundant cytoplasm should rather be regarded as DLBCL, irrespective of the immunophenotype. Many of these cases do have a BL immunophenotype and may be negative for BCL2. Therefore, any tumor with a BL immunophenotype still requires the morphological and genetic context of BL in order to render this diagnosis.
In all DLBCL together, 8q24/MYC rearrangement is seen in 5–15% of the cases; however, in cases of DLBCL with features of BL, it increases up to 40% of the cases. Usually, such cases have a complex karyotype and many have a MYC breakpoint with non-IG partners (Fig. 1k). Such non-IG breakpoints are detected by an 8q24 segregation FISH assay but not by an 8q24–14q32 FISH fusion assay. DLBCL with a MYC breakpoint generally have a relatively poor outcome, and thus it has been advised to assess such breakpoints in all cases of DLBCL, irrespective of morphology [21, 22].
Several examples were submitted to the workshop, including a case with a highly complex karyotype with three copies of MYC, four copies of BCL2 and a BCL6 rearrangement, and a case with high-level amplification of MYC, submitted by N.L. Harris. In almost all submitted cases that were considered as DLBCL, CD10 was negative.
The biological relevance of separating BL from DLBCL, according to the current definitions, is supported by gene expression [15, 16] and array CGH studies [17] of MYC + DLBCL, DLBCL with a BL immunophenotype, and typical BL. Nevertheless, debatable cases still occur. Importantly, little solid information is available on the clinical relevance of this differential diagnosis. Accordingly, results from the MRC/NCRI/Ly10 clinical trial treating all BL and all highly proliferative DLBCL as BL suggest that the results in the DLBCL were not better than expected for treatment with rituximab, cyclophosphamide, adriamycin, vincristine, and prednisone (R-CHOP) [23].
The phenotypic spectrum of Burkitt lymphoma
BL has a characteristic immunophenotype, and the majority of the relevant markers for the differential diagnosis can be demonstrated on FFPE material (Fig. 1c–e, g, h). In contrast to the previous WHO classification, weak BCL2 expression is considered acceptable in BL. However, it should be realized that the normal counterpart cell of BL, the early germinal center B cell, does not express BCL2 and, therefore, another explanation should be found for this expression. For that reason, extensive ancillary studies are always indicated. Most importantly, a “double-hit” lymphoma (see below) should be actively excluded. Another explanation for (weak) BCL2 expression may be upregulation by EBV, and in EBV+ cases a diagnosis of BL would be fully acceptable in the presence of the correct genetic context. A prime example was submitted as a case with BL morphology with uniform positivity for CD10 and BCL6, but separate areas with a divergent immunophenotype of BCL2+/IRF4/MUM1− versus BCL2−/IRF4/MUM1+ (submitted by M. Ashton-Key). The diagnosis of BL was supported by the presence of the t(8;14), in the context of a simple karyotype (Fig. 1l). Similar considerations were present in a case submitted by G. Fan that lacked Bcl-6 expression but that was otherwise typical of BL. IRF4/MUM1 was found to be heterogeneous and noncontributory for classification.
Burkitt lymphoma without demonstrable MYC translocation
Currently, most MYC breakpoints are detected using FISH. In about 10% of all BL, including pediatric cases that otherwise fulfill all criteria, no 8q24/MYC breakpoint can be demonstrated using this technique [19, 24]. Of note, in-depth analysis may show MYC alterations with very small insertions/deletions or breakpoints that are located so far away from the gene that they are missed with standard FISH assays. So it is unknown how many cases of true BL really lack any MYC rearrangement or mutation. Copy number increase or amplification of MYC has been proposed as an alternative mechanism, but this phenomenon may be more indicative of a DLBCL than BL [25]. There is also one recent report suggesting that deregulated microRNAs may cause overexpression of MYC and can lead to a BL or a lymphoma-mimicking BL [26].
Two cases showing high level of MYC amplification were submitted to the workshop. The cases submitted by J.D. Nuckols (Fig. 1i) was diagnosed as DLBCL based on morphology and immunophenotype, whereas the case with several copies of MYC submitted by N.L. Harris showed a BL morphology and immunophenotype. Following the basic concept of the WHO classification of keeping well-set, clean entities, cases with MYC amplification, as well as those completely lacking 8q24/MYC alterations, should not be regarded as BL but rather be kept in the provisional category of B cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL, or alternatively they should be diagnosed as DLBCL, if the morphology is typical of this entity.
In conclusion, one likely has to accept rare cases of true BL that fulfill all epidemiological, clinical, histopathological, immunological, and molecular features of BL but, nevertheless, do not have a demonstrable MYC breakpoint.
“Double-hit” lymphomas
The “double-hit” lymphomas are characterized by a second translocation in addition to a t(8;14), t(8;22), or t(2;8). In general, the 8q24/MYC breakpoint is often a secondary alteration in these lymphomas. In the majority of the cases, a 18q21/BCL2 breakpoint can be found, mostly as a t(8;14) + t(14;18) karyotype, but sometimes as a complex translocation on a single chromosome t(8;14;18). Based on the molecular structure of the t(14;18), it can be assumed that this breakpoint has been the primary alteration caused by aberrant VDJ recombination in a precursor B cell. This event is followed by the 8q24/MYC translocation mediated by AID in the germinal center cell stage. Indeed, similar 8q24/MYC alterations are sometimes seen during follow-up in patients with follicular lymphoma leading to transformation to highly aggressive disease [5, 27–29]. Two such cases with a history of FL were submitted to the workshop. This type of “double-hit” lymphoma is characterized by a morphology that may completely mimic BL but with frequent BCL2 expression.
All together, six cases representing MYC/BCL2 double-hit cases were submitted to this workshop. These cases showed an immunophenotype with strong expression of CD10, BCL6, and BCL2. Analysis of IRF4/MUM1 was not helpful. Since both IG loci may be involved in the two translocations, IG expression may be absent in such cases [30].
Most importantly, the frequency of this diagnosis is steadily increasing with age and may increase to more than one third of all aggressive lymphomas with features of BL in patients over 60 years of age [18]. Therefore, the finding of a BCL2 protein-positive BL, especially in an elderly patient, should prompt a very careful search for a “double-hit” status (Fig. 1h). The clinical course of 18q21/BCL2 + 8q24/MYC double-hit lymphomas is extremely poor, and the disease responds rather poorly to both DLBCL-directed R-CHOP and BL-oriented “high-intensity–short-duration” treatment.
Much more rarely, 8q24/MYC alterations are seen in combination with 11q13/CCND1 and 3q27/BCL6 breakpoints. Examples of such cases were submitted by S. Dufresne and C. Keller (Fig. 1j). Even “triple-hit” cases of 18q21/BCL2+8q24/MYC/3q27/BCL6 breakpoints may occur, of which one case was submitted in the workshop by D. de Vinck. In contrast to 18q21/BCL2 and 11q13/CCND1 that can both be regarded as primary alterations, the sequence of events is less clear for 3q27/BCL6 and also little is known about the clinical consequences or prognosis of these patients. The immunophenotype (BCL2+, CD10+, BCL6+) seems to be rather similar to the cases with an additional 18q21/BCL2 alteration, as was supported by the three submitted cases.
B cell lymphoma, unclassifiable, with features intermediate between diffuse large B cell lymphoma and Burkitt lymphoma
The diagnosis of B cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL was coined for those cases that do not fit into the well-set category of BL but also show such features that would make it inappropriate to just lump them into the category of DLBCL. Many of the difficult differential diagnostic problems, as discussed during the workshop, indeed belonged to this category. Thus far, “double-hit” lymphomas represent the best-defined subgroup of this category. This group is an example of how increasing biological insight helps to define a distinctive disease entity and assists in recognizing its morphological spectrum and immunophenotype. For the other subgroups of this category, additional biological and clinical information will be needed to come to a better classification. From a practical point of view, a multidisciplinary discussion for treatment planning is mandatory in all these cases. Treatment decisions should not only depend on the classification by the pathologist but also on the clinical situation.
Mediastinal gray-zone lymphoma and other lymphomas with atypical immunophenotype (session 2)
This session was devoted to MGZL and other lymphomas with atypical immunophenotype that caused differential diagnostic problems, often between CHL and NHL. An emphasis was placed on MGZL given that, at the time of the workshop, the 2008 WHO classification was newly published with the new category of B cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL (herein denoted BCLu) [3]. In this section, we will briefly review primary mediastinal large B cell lymphoma (PMBL) and the concept of MGZL in order to provide the framework for the cases submitted that were related to this general topic. We will also discuss selected key diagnostic issues related to the other submitted cases of lymphomas with atypical phenotypic findings.
Primary mediastinal large B cell lymphoma
Primary mediastinal large B cell lymphoma was described in 1980 from a review of 184 adult NHLs. A group of 17 cases was identified that manifested as a mediastinal mass with diffuse aggressive histology [31]. Since this initial description, the features of typical PMBL have been well characterized [32–34].
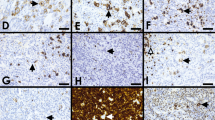
Often presenting in younger women, these lymphomas present as a rapidly enlarging mediastinal mass with symptoms related to compression of thoracic structures. Histologically, this lymphoma is composed of an infiltrate of large cells with round or lobulated nuclei. The cells often have abundant clear cytoplasm. There is background compartmentalizing sclerosis. Occasional multinucleated RS-like cells can be seen. While the architecture is usually diffuse, a minority of cases (30%) may show focal nodularity. Necrosis can be seen in 25% of cases, further mimicking CHL [35]. The immunophenotype is that of a mature B cell expressing CD20 and other pan B cell markers such as CD79a and PAX5 but lack surface immunoglobulin expression [36, 37]. The B-cell-associated transcription factors OCT-2 and BOB.1 are also strongly expressed [37]. CD30 is expressed in most cases, albeit with variable intensity [32, 35]. TRAF-1 and cREL are expressed in PMLBL, unlike other DLBCLs, and reflects activation of the nuclear factor kappa B pathway [38]. The cells are variably positive for germinal center markers BCL6 and CD10 and also express other markers such as CD23. The former two markers have been taken as evidence of germinal center derivation, and the latter, expressed in up to 85% of cases, has been taken as evidence of possible thymic B cell derivation [39, 40]. Although these characteristic pathologic features have been identified, none are entirely specific for PMBL. Thus, further proof that PMBL was indeed a specific type of aggressive large B cell lymphoma distinct from nodal DLBCL was needed. Application of molecular techniques confirmed that PMBL was a distinct entity. For example, it has been demonstrated that PMBL has characteristic genomic imbalances that include gains at chromosome 9p13.1–9p13.3 (approximately 70% of cases), 9p23-p24 (approximately 60% of cases), and 2p15-p16.1 (area of REL and BCL11A, approximately 60%), as well as losses [41–43]. Loss at chromosome 1p13.1-p13.2 has been reported in up to 43% of cases [44]. Gene expression profiling studies have also shown that PMBL has a distinct profile compared to diffuse large B cell lymphoma, and several highly expressed genes including MAL, CD23, FIG1, TARC, NFkB2, and PDL1/L2 have been identified [6, 7, 45, 46]. Indeed, molecular similarities were noted between PMBL and another lymphoma that characteristically occurs in the mediastinum CHL [6, 7]. Four cases of PMBL were submitted; two were from the mediastinum and two were from cervical lymph nodes, but it was unclear whether a mediastinal mass was present (Table 1). The features of the latter, however, were consistent with PMBL, including MAL expression. One case showed a nodular growth arising from the thymus (Fig 2a). The atypical cells showed a B cell immunophenotype but had some RS-like cells that expressed strong CD20, CD30, OCT-2, and BOB.1 but lacked EBER (this was also the case that lacked MAL expression). The second case was felt to be morphologically and phenotypically classic for PMBL with a diffuse histology and large cells expressing CD20, PAX5, and MAL (Fig 2b–d). One case added the twist of aberrant T cell marker expression (such as weak CD3) but lacked TRG@ rearrangement while showing IGH@ rearrangement (Fig. 3c–g).

a–d Session 2, case 38: A typical example of a primary mediastinal large B cell lymphoma arising from the thymus (a). Session 2, case 27 illustrates a primary mediastinal B cell lymphoma with large pleomorphic cells embedded within a background of sclerosis (b) that expresses CD20 (c) and CD30 (d) but lacks expression of CD15. e–i Session 2, case 1 shows a mediastinal “gray-zone” lymphoma demonstrating histologic features typical of primary mediastinal large B cell lymphoma (e) but with an atypical immunophenotype with expression of weak CD30 (f) and moderate to strong CD15 (g). The lack of CD20 among the large atypical cells (h) favors classical Hodgkin lymphoma whereas strong expression of PAX5 (i) favors a large B cell lymphoma

a, b Session 2, case 11 represents an example of a composite lymphoma and illustrates distinct areas resembling classical Hodgkin lymphoma and primary mediastinal B cell lymphoma. The classical Hodgkin-like areas exhibit large atypical cells with Reed-Sternberg cell morphology (a), whereas the primary mediastinal B cell lymphoma-like areas show sheets of large atypical cells embedded in a fibrotic stroma (b). c–i Session 2, case 20 shows an example of classical Hodgkin lymphoma with aberrant T cell marker expression. The large Reed-Sternberg-like cells (c) express multiple T-lineage markers including CD3 (d), CD5 (e), and CD4 (f), in addition to weak expression of the B cell transcription factor PAX5 (g)
Mediastinal gray-zone lymphoma
The similarities between PMBL and CHL had been recognized both from the clinical as well as histopathologic perspectives. Reports of sequential or composite CHL and NHL have been recognized [47, 48], and cases of mediastinal lymphoma with features of both a DLBCL and nodular sclerosis (NS) HL have also been reported [49]. Since these earlier reports, the occurrence of mediastinal lymphomas with features of both B cell NHL and CHL has been described and has been termed mediastinal gray-zone lymphoma [8, 50, 51]. In the largest series to date, 21 cases were characterized and two subtypes were identified [8]. Some cases had a morphology resembling CHL but had atypical features such as increased numbers of large mononuclear cells, lack of the typical mixed inflammatory background, and atypical immunophenotypic characteristics of the Hodgkin-like cells such as strong expression of CD20. The other subtype had morphology of PMBL with diffuse monomorphous large cells. Background compartmentalizing sclerosis was often present, and there was usually only a sparse inflammatory background. If a mixed inflammatory infiltrate was present, it was only a focal component. Occasional nodules or areas with RS-like cells or lacunar cells were present. The immunophenotype was atypical and had features suggestive of CHL. For example, CD20 was often only weakly expressed or absent in many cases, and CD15 was expressed at least focally in 60%. B cell transcription factors (PAX5, OCT-2, BOB.1) were usually expressed, albeit often weakly, in 13/14 cases in which all were evaluable. Finally, MAL was expressed in seven of nine cases [8].
Recognizing this phenomenon, the 2008 WHO classification included a chapter defining an “overlap” lymphoma: BCLu [3]. These lymphomas are present most commonly in younger males (age 20–40) with mediastinal involvement, although nodal sites may also be involved, with pathologic features described above. Although it is uncertain whether it is a related process, composite CHL and PMBL or sequential CHL and B cell lymphoma (in either order) is also recognized [3], although it is uncertain whether the biology underlying these composite or sequential lymphomas is related to the phenomenon of BCLu.
Seven cases of mediastinal gray-zone lymphoma were submitted (Table 1). Two cases represented mediastinal masses considered as examples of BCLu. The case presented by E. Hsi had the histology of a mediastinal large B cell lymphoma but had an atypical immunophenotype. Whereas the expression of strong CD45, CD79a, CD30, strong PAX5, and MAL might favor PMBL, lack of CD20 and B cell transcription factors OCT-2/BOB.1 with expression of CD15 is reminiscent of CHL (Fig 2e–j). The case presented by R. Gascoyne showed histology more in keeping with CHL but also showed hybrid phenotypic features (strong CD20+, CD45+, CD79a+, focal CD30+, CD15−, EBER+, MAL−, OCT-2+, BOB.1 weak+). An additional case illustrated how modern practice of needle biopsy may affect the pathologist’s ability to fully evaluate a case. In this case, a needle biopsy of a large mediastinal mass showed features compatible with NSHL; however, a repeat biopsy after only one cycle of chemotherapy showed mediastinal gray-zone features with sheets of mononuclear Hodgkin-like cells and sclerosis in a mixed inflammatory background. Immunophenotyping showed expression of CD30, OCT-2, BOB.1, CD20, and MAL. Additional variations of BCLu were seen in cases submitted by Drs. Alobeid, Chen, Imai, and Völker. The latter case was from an inguinal lymph node and added a further variation of not being from a mediastinal site. In summary, the 2008 WHO classification category BCLu was created to recognize the biologic overlap between some cases of B cell lymphoma and CHL, in particular the mediastinal gray-zone cases, and several illustrative cases were submitted that covered the morphologic and immunophenotypic spectrum. These include cases with morphology of CHL but immunophenotype more in keeping with PMBL and vice versa. The use of B cell transcription factor markers in the evaluation of these cases including PAX5, OCT-2, and BOB.1 is helpful and sometimes necessary, in addition to standard markers for CHL and DLBCL. Of interest, MAL expression is usually seen in such cases.
Composite PMBL and CHL
As noted previously [8], composite PMBL and CHL occurs, and this was illustrated in four cases submitted to this workshop (Table 1). The case presented by A. Dogan showed clear areas typical of either DLBCL or CHL and after treatment with R-CHOP, the patient relapsed with the CHL component (Fig 3a, b). Interestingly, in two cases with composite areas in a mediastinal tumor, one showed MAL expression in both components, whereas in the other case MAL expression was only seen in the mediastinal large B cell lymphoma component. The pathologic features in the submitted cases were consistent with what has been published. As one might expect from its relationship to PMBL, most cases expressed MAL protein. These cases help clarify diagnostic issues related to mediastinal lymphomas. Specifically, one should strive to separate PMBL from CHL, and this can be done in most cases using the presence of typical morphology, supported by characteristic immunophenotypic data. Morphologic variations such as presence of RS-like cells in PMBL or sheets of mononuclear RS variants in CHL are acceptable and should not prompt the use of the BCLu designation. Similarly, minor immunophenotypic variation such as strong CD20 expression in otherwise typical CHL should not prompt the use of the BCLu designation. An expanded immunophenotypic panel that includes use of markers such as CD23, PAX5, OCT-2, BOB.1, and MAL (if available) may help clarify how much overlap is present. The panel agreed that further work is needed to define better the boundaries between CHL and BCLu on one hand and to define acceptable phenotypic variation in CHL on the other hand. Finally, composite PMBL and cHL cases can be recognized when adequate sampling is present and reminds us of the problems that may be associated with the use of needle core biopsies for primary diagnosis. Importantly, composite cases containing distinct PMBL and CHL areas should be designated as composite lymphomas and not included in BCLu.
Classical Hodgkin lymphoma with immunophenotypic deviations
Another set of cases reviewed in session 2 had the theme of CHL with an unexpected immunophenotype. The case presented by E. Haralambieva was an axillary lymph node biopsy from a 38 years old with typical CHL morphology but strong CD20 expression in the RS cells. Other B-lineage markers such as CD79a and B cell transcription factors (PAX5, OCT-2) were negative. Another case illustrated a typical CHL case with MAL expression. MAL expression in CHL has been shown to be present in up to 20% of cases and associated with shorter survival [52]. Three cases were examples of CHL with aberrant T-cell-associated markers, a rare but reported phenomenon [53]. The case from U. Zanetto was particularly interesting, as in addition to the characteristic weak PAX5, CD15, and CD30, multiple T cell markers (CD2, CD3, CD4, and CD5) were also expressed in the RS cells (Fig 3c–i).
DLBCL with unusual immunophenotype
Seven cases could be grouped as DLBCL with unusual phenotype. Of these, three cases were cyclin D1 positive that lacked CCND1 rearrangement and did not appear to represent blastoid variants of mantle cell lymphoma. These rare cases have been the subject of recent study. Some cases may have additional copies of CCND1 by FISH analysis as a possible mechanism for overexpression, although cases of DLBCL with extra copies of the gene without protein overexpression do occur [54, 55]. Two cases with aberrant T-cell-associated antigen expression were also submitted along with two cases with unusual morphology and/or immunophenotype. The case from M. Kinney was felt to be related to an anaplastic variant of DLBCL occurring in a 34-year-old woman with laboratory findings of an underlying autoimmune process (antinuclear antibody). Anaplastic CD30+/EBER+ B cells were present in a sinusoidal pattern in a background of follicular hyperplasia and polytypic plasmacytosis and was reminiscent of a series of sinusoidal CD30+ large B cell lymphomas published previously [56].
T cell lymphomas with CD30 and/or CD15 expression
Four cases of T cell lymphomas were also submitted. One case was an ALK+ anaplastic large cell lymphoma that expressed Bcl-6, which has been reported in a substantial minority of cases [57]. Three cases of CD30+/ALK-negative anaplastic large cell lymphoma were submitted that were confirmed to have clonal T cell receptor gene rearrangement. Two were CD15+ and expressed other T-cell-associated markers such as CD4. Expression of CD15 in T cell lymphomas has been reported in rare cases and is a potential pitfall in misclassifying a case as cHL [58]. Lack of the typical mixed inflammatory background of CHL, presence of other atypical cells without features of Hodgkin cells, abnormal T cell immunophenotype (loss of pan T cell markers), and presence of a prominent T cell clone from whole-tumor DNA extracts are helpful diagnostic clues that one is not dealing with CHL. The case submitted by E. Remstein was representative of this problematic area. It had RS-like cells but lacked a typical mixed background of CHL and expressed CD4. Of note, all three of these cases expressed clusterin in the characteristic (albeit not entirely specific) “dot-like” pattern reported for ALCL, including ALK-negative ALCL [59, 60]. A minority of DLBCL, cases of cutaneous T cell lymphoma, and rare cases of peripheral T cell lymphomas or cHL may express clusterin [59–61].
The last three groups of cases with atypical immunophenotype illustrate and corroborate the general consensus that minor immunophenotypic variation (such as moderate/strong CD20 expression) in otherwise morphologically and immunophenotypically characteristic CHL should not prompt the use of BCLu. Furthermore, other immunophenotypic variation such as T cell marker expression can be seen in CHL. An additional twist showing potential problems in distinguishing T cell lymphoma and CHL was illustrated, as noted above, by submission of cases showing CD15+ T cell lymphoma.
Immunophenotypic changes in transformed B cell lymphomas
The final group of cases considered in this session was a group of heterogeneous lymphomas that appeared to occur as a transformation from prior lymphoma or represented a relapse event. These cases may have demonstrated a phenotypic “drift” from the original phenotype, not an uncommon occurrence. Two cases were diffuse large B cell lymphomas that expressed CD30 and had Hodgkin-like cells but also strong expression of other B cell markers such as CD79a. One case represented a DLBCL arising from a follicular lymphoma, again with unusual immunophenotypic findings such as CD30 positivity in the transformed areas, but a variety of other B cell markers including OCT-2 and PAX5 showed a range of expression in the large atypical RS-like cells. The case submitted by E. Jaffe was an example of DLBCL arising in splenic marginal zone lymphoma in which there was a history of SMZL with intervening CHL, followed by the submitted case with transforming SMZL. This last case pointed out the lack of well-defined criteria for diagnosing transformation of SMZL to DLBCL. Finally, we should keep in mind the possibility of immunophenotypic/morphologic changes occurring in the setting of relapse or transformation. The distinction between a second unrelated lymphoma vs. relapse/transformation will depend on molecular techniques to establish a clonal relationship and is not usually part of routine practice.
In summary, this session introduced the participants to evolving concepts in the area of mediastinal gray-zone lymphomas and the new WHO terminology. Furthermore, the submitted cases also illustrated a broad spectrum of cases in which unusual phenotypic variation could cause diagnostic problems between specific entities such as CD30+ sinusoidal large B cell lymphoma, cyclin D1+ DLBCL, or DLBCL with T cell marker expression. Weighing the evidence for lineage assignment and knowledge of such variability are required in order to arrive at the most appropriate diagnosis in these difficult cases.
Gray-zone around nodular lymphocyte predominant Hodgkin lymphoma, T-cell/histiocyte-rich large B cell lymphoma and classical Hodgkin lymphoma (session 3)
Gray zone between NLPHL and THRLBCL
NLPHL, a monoclonal B cell lymphoma, usually presents with a predominantly nodular or a nodular and diffuse growth pattern sometimes reminiscent of progressively transformed germinal centers. The infiltrate resides, in the nodular areas, within large meshworks of follicular dendritic cells. It is mainly composed of small B lymphocytes with varying numbers of intermingled histiocytes and epithelioid cells eventually forming small granulomas, and a proliferation of scattered large neoplastic B cells—the so-called Lymphocyte Predominant cells (LP cells; previously known as L&H cells). These blasts have scant to moderate amounts of pale cytoplasm and often folded or lobated nuclei and are sometimes very similar to, or even indistinguishable from, classical Hodgkin cells. Patients with NLPHL usually present in clinical stages I and II and have a long survival sometimes characterized by frequent relapses. Two cases submitted to the workshop represented classical examples of NLPHL (Fig 4a–d, submitted by X. Zhang) with a nodular growth pattern and scattered CD20+ CD79a+ epithelial membrane antigen (EMA)+ LP cells surrounded by reactive CD3+ CD57+ PD1+ T cells in a background rich in small B lymphocytes. In the 2008 WHO classification, THRLBCL is considered a variant of DLBCL. The reasoning behind this is that THRLBCL, in spite of a limited number of scattered, large neoplastic B cells, shows a diffuse growth pattern and usually has a prognosis similar to that of DLBCL. Morphological and immunophenotypical studies, however, have shown that the tumor cells in THRLBCL in many cases do resemble the LP cells of NLPHL. Because of the similarities in morphology and immunophenotype between NLPHL and THRLBCL, a biological relationship between these two entities has been discussed. In addition, there are cases of NLPHL with histological progression, in which the process becomes entirely diffuse, and the morphology is indistinguishable from de novo THRLBCL. The question, therefore, is whether these cases represent true progression to an aggressive B cell lymphoma or only “THRLBCL-like” areas in an otherwise typical case of NLPHL.

a–d Session 3, case 01: NLPHL. Lymph node with NLPHL with a characteristic nodular growth pattern (a). LP cells (a, insert) are strongly positive for CD20 and embedded in a background rich in small B cell lymphocytes (b). The LP cells are surrounded by reactive CD57+ (c) and PD1+ (d) T cells. e–g Session 3, case 03: NLPHL with diffuse areas, depletion of background B lymphocytes and IgD+. NLPHL with diffuse growth pattern infiltrating a lymph node (e). Note that the background infiltrate of small B lymphocytes is reduced (f); however, rosetting of PD1+ reactive T cells around the LP blasts is present (f, insert). The LP cells are strongly positive for IgD (g). The IgD+ LP cells are surrounding the nodules of reactive small B lymphocytes. h–i Session 3, case 09: NLPHL with THRLBCL-like areas. CD20 staining highlights the LP cells with areas of very reduced (h) or complete loss of CD20+ small B lymphocytes mimicking THRLBCL (i). j–k Session 3, case 18: NLPHL with CD15 positivity. Typical case of NLPHL with LP cells positive for CD20 (j) showing unequivocal strong expression of CD15 in the LP tumor cells (k). l–n Session 3, cases 07 and 27: NLPHL EBV+. CD20 staining highlights the typical characteristics of NLPHL (l) where the LP cells are EBV+ by EBER ISH (m). Case 27 showed in addition progression to a DLBCL. Note that the high-grade component (left), as well as the LP cells (right) are EBER+ (s)
Thus, one of the target issues addressed during the workshop was to define criteria for reliably distinguishing boundaries between NLPHL and THRLBCL. Although considered by some authors to represent a continuum ranging from an indolent (NLPHL) to an aggressive (THRLBCL) category, genetic evidence supports the concept of discrete entities [62] that infrequently can emerge from one another [51]. The WHO classification sharply defines NLPHL and THRLBCL based on characteristic features inferred by morphology and immunophenotype but also defines features that, obviously, are attributable to progression (the latter in a morphological, not necessarily clinical, sense). The spectrum of nodular and diffuse NLPHL, purely diffuse NLPHL, NLPHL rich in blasts, and NLPHL in possible transition to THRLBCL was presented and extensively discussed during the workshop, and some unique NLPHL cases associated with particular immunophenotypic and infection-associated features were presented.
NLPHL with diffuse areas, depletion of background B lymphocytes and IgD+
Purely diffuse NLPHL is exceedingly rare, and most cases diagnosed as such in the past would now be classified as THRLBCL. According to the recommendations of the 2008 WHO lymphoma classification, however, a single nodule of typical NLPHL, in an otherwise diffuse proliferation, is sufficient to exclude the diagnosis of de novo THRLBCL. In these cases, the term NLPHL with THRLBCL-like areas is recommended.
Altogether, seven cases submitted to the workshop were characterized by classical nodular areas of NLPHL but also by other features that might be attributed to progression. They either showed diffuse areas, with slight nodularity apparent only after immunohistochemical staining for B cell markers, or featured a moderate increase of blasts. The key diagnostic features of NLPHL, however, were present in all the cases. Although the characteristic background infiltrate of small B lymphocytes was reduced in some cases, predominantly in the diffuse areas, and the number of background T cells was higher, all these lymphomas featured typical morphological changes encountered in NLPHL, including CD21+ and/or CD23+ meshworks of follicular dendritic cells. Nevertheless, there were two remarkable immunohistochemical features recognized during the review of these cases (Fig 4e–g, submitted by C. Temmins). First, in all of the cases showing some features of morphological progression, IgD was expressed in the LP cells. Reactivity of the LP cells for IgD has been reported in roughly one third of NLPHL, and its expression was more commonly found in young males [63]. Accordingly, all seven cases of NLPHL with diffuse areas and IgD expression submitted to the workshop were young males aged between 4 and 40 years. In fact, the expression of IgD in the tumor cells is a useful argument against the diagnosis of de novo THRLBCL. Second, the expression of the PD1 antibody, a novel follicular T cell marker [64], turned out to be particularly helpful in delineating the presence of T cell rosetting around the LP blasts even in those cases that were B-cell-depleted or in which CD57 did not highlight the T cell rosetting. Furthermore, in contrast to de novo THRLBCL, which lack follicular T cell rosettes, all cases with borderline features between NLPHL and THRLBCL showed T cell rosettes, thus suggesting a biological relation with NLPHL.
NLPHL with T-cell/histiocyte-rich B-cell-like areas
Interestingly, two additional cases submitted to the workshop showed—sometimes discrete—areas entirely depleted of small background B cells, in which the tumor cells (with LP features or more reminiscent of peripheral B blasts or Hodgkin cells) were observed in a background of exclusively T cells (Fig 4h, i, submitted by S. Nam-Cha). According to the 2008 WHO lymphoma classification, these are the key defining features of NLPHL with THRLBCL-like areas. This morphological variant is seen more frequently in cases with a prominence of extranodular LP cells and in patients with recurrent disease. The 2008 WHO lymphoma classification recommends considering cases of NLPHL that progress to a diffuse T-cell-rich pattern as NLPHL, THRLBCL-like to distinguish them from primary or de novo THRLBCL. By introducing this term into the diagnostic algorithm of the NLPHL, Poppema et al. [65] challenged the classification of many published cases of NLPHL with “progression” to THRLBCL. In fact, some studies dealing with the accurate estimation of clinical risk inferred by the variant morphology in these patients have provided evidence that these tumors might be associated with higher clinical stages, but not necessarily with increased aggressiveness [10]. Accordingly, the two patients with THRLBCL-like areas in the workshop presented with stage III and IV disease and a history of multiple relapses. One case was treated as NLPHL and the other with R-CHOP. Nevertheless, both are alive and free of disease after several years of follow-up. Importantly, using the term NLPHL with THRLBCL-like areas in daily diagnostic practice might prevent overtreatment of these patients. The use of this terminology, on the other hand, does not exclude, by itself, the existence of true THRLBCL cases evolving from (classical) NLPHL. During the diagnostic workup of the cases, the panel found the use of the PD1 antibody particularly useful in differentiating NLPHL with extensive THRLBCL-like areas from THRLBCL because a large fraction of the background T cells in NLPHL with THRLBCL-like areas expressed PD1, while this was not the case in THRLBCL.
In this regard, the case presented by T. Molina especially highlighted the diagnostic challenges encountered when dealing with the THRLBCL–NLPHL interface. In this case, the patient had an original diagnosis of THRLBCL rendered on a needle biopsy of a retroperitoneal lymph node. After aggressive treatment, the patient achieved complete remission. The disease recurred after 5 years with enlarged lymph nodes that, on histological evaluation, revealed hyperplastic germinal centers surrounded, and in part colonized by, tumor cells reminiscent of LP cells. A diagnosis of NLPHL was made. The case was submitted as an example of coexisting THRLBCL and NLPHL, nevertheless raising the important issue of whether the first biopsy was representative (NLPHL with diffuse areas?) and whether or not a diagnosis of THRLBCL should be made in a needle biopsy. Unfortunately, in this case, there was no tissue available to perform a PD1 staining. Furthermore, the diagnosis of NLPHL in the second biopsy most probably represents very early morphological changes of the disease.
NLPHL with CD15 positivity
Characteristically, LP cells are positive for CD20, CD79a, CD75, BCL6, and CD45 in nearly all cases and the EMA is expressed in more than 50% of the cases. In contrast to the Hodgkin and Reed-Sternberg cells of CHL, OCT-2, PU.1, and BOB.1 are coexpressed in LP cells, and CD15 and CD30 are usually negative. Interestingly, two cases showed unequivocal expression of CD15 in the LP tumor cells, in addition to expression of B cell markers (Fig 4j, k, submitted by D. Norris). CD15 expression is regarded as one of the hallmark immunophenotypic features of classical Hodgkin lymphoma. It is worth mentioning that both cases were also IgD+, thus again suggesting that the expression of this antigen might be valuable in recognizing particular NLPHL subtypes. The practical consequence of the finding is that the expression of a rather promiscuous antigen like CD15 does not necessarily preclude the diagnosis of NLPHL. Conversely, and as shown in session 2, CD15 expression does not by itself point to the diagnosis of classical HL.
NLPHL EBV+
According to the 2008 WHO lymphoma classification, one of the defining characteristics of NLPHL, in contrast to CHL, is the absence of EBV infection in the LP cells. Nevertheless, in three NLPHL cases submitted to the workshop, EBER in situ hybridization (ISH) unequivocally demonstrated the presence of EBV genomes in the tumor cells (Fig 4l, m, submitted by Garcia-Cosio). This finding challenges the view that EBV can occasionally be seen in some small bystander B lymphocytes but is consistently absent in the tumor (LP) cells of NLPHL [65, 66]. More interestingly, one of the cases presented by S. Rodriguez also featured the transition of EBV-associated NLPHL to EBV-associated diffuse large B cell lymphoma (Fig 4n). It is currently unknown whether EBV is involved directly in transformation of NLPHL into DLBCL in some cases. One interesting feature of all three cases was that they occurred in patients over the age of 50 years, thus suggesting the possibility of impaired immune surveillance as a potential factor in lymphomagenesis as in EBV-associated DLBCL of the elderly.
Gray zone between THRLBCL and CHL
According to the criteria of the 2008 WHO classification, THRLBCL is a DLBCL characterized by a limited number of scattered, large, atypical B cells embedded in a background of abundant T cells and histiocytes. The large tumor cells express B cell markers and BCL6, with variable expression of BCL2 and EMA and are, as a rule, CD15 and CD30 negative. An important defining morphological feature is the absence of T cell rosettes around the tumor cells and the absence of remnants of B follicles or clusters of small B lymphocytes. Only one case was submitted to the workshop representing a classical example of de novo THRLBCL.
A recently recognized morphological variant of THRLBCL predominantly infiltrates the spleen with multifocal or micronodular involvement of the white pulp (Fig 5a, b) [67]. Morphologically, the cellular composition in the spleen-scattered large neoplastic B cells in a background of reactive T cells is similar to that seen in THRLBCL of the lymph nodes. Three cases were submitted to the workshop representing this splenic micronodular variant of THRLBCL. The case presented by L. de Leval (Fig 5a–d), in addition to demonstrate the typical features of the tumor, showed areas with progression to DLBCL forming clusters and sheets of large neoplastic B cells (Fig 5d). The tumor cells displayed a characteristic phenotype (CD20+, BCL6+, CD30−, CD15−), and the background lymphocytes were CD3+, CD5+, CD57 negative, with some PD1-positive intermingled T cells. T cell rosettes around the large neoplastic B cells were absent or only rarely found (Fig 5c) [64].

a–d Session 3, case 10: THRLBCL of the spleen with micronodular involvement. Micronodular involvement of the white pulp of the spleen characterized by scattered large neoplastic B cells in a background of reactive T cells (a). CD20 expression highlights the abnormal infiltrating nodules compared to the normal white pulp (b). PD1-positive T cells are scattered and T cell rosettes around the large neoplastic B cells are absent (c). THRLBCL shows areas of progression to DLBCL (d). e–j Session 3, case 17: B cell lymphoma unclassifiable with features intermediate between DLBCL and CHL. Lymph node with a diffuse infiltration (e) and numerous large cells with typical RS cell morphology (f). The RS-like cells are negative for CD15 (f, insert), strongly and uniformly positive for CD20 (g), CD79a (h), Oct 2 (i), and EBER ISH (j). k–n Session 3, case 20: B cell lymphoma unclassifiable with features intermediate between DLBCL and CHL. Lymph node with vaguely nodular infiltration reminiscent of NLPHL but with numerous large cells with typical RS cell morphology (k). The RS cells are strongly and uniformly positive for CD20 (l), Oct 2 (m), and EBER ISH (n), in addition to CD15 and CD30 positivity
Although THRLBCL is a well-defined subtype of DLBCL, morphologic similarities with and immunohistochemical transitions to CHL have raised the possibility of a relationship or a gray zone between these two entities [68]. Lim et al. [69] described a subtype of THRLBCL with features similar to classical Hodgkin lymphoma, where the tumor cells exhibited the characteristic morphology of classical Reed-Sternberg cells—large, pleomorphic multinucleated cells containing prominent eosinophilic or amphophilic nucleoli and abundant cytoplasm. However, the background was unusual for CHL in that eosinophils, neutrophils, and plasma cells were absent. Immunophenotypically, unlike CHL, the neoplastic cells showed strong—and particularly uniform—expression of CD20 with coexpression of CD79a in 50% of the cases. Expression of CD30 was frequently observed; however, CD15 was consistently negative. EBER in situ hybridization was found to be positive in 50% of the cases. Previous studies have shown that expression of CD20 and absence of CD15 reactivity are associated with a poorer prognosis in CHL, suggesting that these cases may be distinct from CHL with a typical phenotype [70].
The 2008 WHO lymphoma classification sharply defines CHL and THRLBCL based on characteristic morphological and immunophenotypical features. Accordingly, cases previously classified as THRLBCL [69] with strong and uniform expression of CD20 and Reed-Sternberg-like morphology, EBV positivity, and a rich background of reactive T lymphocytes should no longer be classified as THRLBCL but as EBV-positive DLBCL. Nevertheless, the 2008 WHO classification also recognizes that there are cases of B cell lymphomas, unclassifiable, with overlapping clinical, morphological, and/or immunophenotypic features between CHL and DLBCL in lymph nodes as the primary site [3]. As with the mediastinal gray-zone cases, one may encounter cases with RS cells, expression of CD45, a preserved B cell program (OCT-2+, BOB.1+, PU.1+, PAX5 strongly expressed, unlike the weak positivity found in CHL), strong expression of CD20 and/or CD79a, and aberrant expression of CD30 and CD15. Strong and uniform expression of CD20 (with or without other B cell markers) in an otherwise typical Hodgkin lymphoma setting is not enough to place a case into the borderline category. On the other hand, cases with CD20 reactivity and absence of CD15 should be diagnosed as gray-zone lymphoma.
During the workshop, the significance of strong, uniform expression of CD20 and a preserved B cell program in cases morphologically resembling CHL was extensively discussed. Five cases were submitted to the workshop highlighting problems in distinguishing between CHL and THRLBCL. All five patients were young males aged between 19 and 44 years with nodal presentations. In all cases, there were morphological features of CHL but also strong expression of B cell markers and a partially or completely preserved B cell program. Three of the cases were EBV positive and two were EBV negative. An example of the complexity of some of these cases was presented by A. Ramsay (Fig 5e–j). The case corresponded to a 39-year-old male presenting with lymphadenopathy, splenomegaly, and pancytopenia. The lymph node featured numerous large cells with typical RS cell morphology. The immunophenotype showed that the RS cells were strongly and uniformly positive for CD20, CD79a, CD30, Bcl-6, and the transcription factor OCT2 and negative for CD15 and BOB.1. EBER in situ hybridization was positive. The possibility of an EBV+ DLBCL of the elderly in a young patient was raised. However, the panel felt that this case rather fulfilled the criteria of a B cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL, based on morphology (CHL) and immunophenotype (strong CD20+, CD15−). Even more challenging was the case presented by B. Nelson (Fig 5k–n), of a 44-year-old man with localized lymphadenopathy. The lymph node showed CHL morphology; however, the immunophenotype was unusual in that the RS cells were strongly and uniformly positive for CD20, PAX5, and OCT-2 and partially positive for BOB.1 and CD45; however, CD30 and CD15 were positive. EBER ISH was also positive. Although the morphology and the expression of CD30 and CD15 suggested a diagnosis of CHL in this case, the unusual, uniform, and strong CD20 and PAX5 expression with a preserved B cell program (OCT-2, BOB.1+/−) makes the diagnosis of CHL difficult to render. After extensive discussions, the panel arrived at the conclusion that cases with features such as the two cases presented should be diagnosed as B cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL, until data from more studies are available, revealing whether these cases represent true biologically borderline cases or whether they are related to one specific entity. Indeed, the question remains how these patients should be treated. Keeping these cases separate will allow further study with the hope that these issues can be resolved as new data become available.
EBV-positive lymphomas, lymphomas occurring in HIV+ individuals, and PTLD-related B cell lymphoproliferations (session 4)
This session, which included the highest number of cases, turned out to be quite heterogeneous. In fact, the submitted cases corresponded to either recently described entities or cases which showed deviations from the prototypic descriptions quoted in the 2008 WHO classification [1]. Thus, they did not strictly fulfill the concept of gray zone applied to the other sessions of the workshop but focused attention on differential diagnostic problems or addressed issues related to the pathobiology of these lymphomas. Notably, most of these cases showed association with EBV or other viral agents. Due to their heterogeneity, the cases were subdivided into eight main groups.
EBV+ DLBCL of the elderly
EBV+ DLBCL of the elderly (EBV+ DLBCL-E) is a provisional entity in the recent WHO classification. It is defined as an EBV+ clonal B cell lymphoid proliferation that occurs in a patient older than 50 years and without any known immunodeficiency or prior lymphoma. The workshop panel included eight cases in this category. All occurred in HIV-negative individuals with no previous history of autoimmune disease and/or immunodeficiency. The median age was 74.5 years (range 60–84), and the male/female ratio was 1.66/1. Contrary to what has been reported in the literature concerning the prevalent extranodal location of EBV+ DLBCL-E [71, 72], seven of eight cases presented with lymphadenopathy, while the remaining one had a small-bowel tumor. The clinical course was known in four patients: three died within the first month after the diagnosis and one was in complete remission after 1-year follow-up. The latter had been treated with R-CHOP (eight cycles), intrathecal methotrexate, and autologous peripheral blood stem cell transplantation. Although based on a small series, these figures are in keeping with the concept that EBV+ DLBCL-E has a much worse prognosis than DLBCL, not otherwise specified [71, 73–75].
Morphologically, all but one case were characterized by a diffuse proliferation of clearly atypical transformed large cells/immunoblasts associated with scattered Hodgkin/Reed-Sternberg (HRS)-like elements and variable amounts of inflammatory cells. Three out of eight cases showed areas of necrosis. The case presented by C.H. Dunphy had a more polymorphic population with numerous HRS-like elements, raising the differential diagnosis of ALCL or CHL of the lymphocyte depleted type (LD; see below). Immunophenotypically, the neoplastic cells showed positivity for at least one B cell marker including CD20 (five of eight), PAX5 (six of eight), and CD79a (eight of eight). OCT-2 was detected in seven of eight cases, BOB.1 in six, BCL6 in four, and IRF4/MUM1 in all. The latter finding underlines that the submitted cases had either activated germinal center (GC) or activated post-GC phenotype (as the one presented by J. van der Walt) [76]. In seven of eight cases, the neoplastic cells were largely positive for CD30 but negative for CD15. The remaining case expressed both CD15 and CD30 and showed EBV positivity by EBER ISH in most if not all neoplastic cells. In this case, the panel retained the diagnosis of EBV+ DLBCL-E because of the absence of obvious HRS cells and the positivity of the neoplastic cells for CD79a, BOB.1, OCT-2, and PAX5. Notably, CD15 positivity seldom occurs among DLBCLs [77] and represents an unprecedented finding in the EBV+ DLBCL-E setting [71, 73]. EBV infection of the neoplastic cells was shown by LMP-1 immunochemistry and EBER ISH in seven of eight and eight of eight cases, respectively. EBV-encoded nuclear antigen 2 (EBNA2) was recorded in only one instance; this figure is definitely lower than the one reported by Oyama et al. in their series (22%) [71]. IGVH@ gene analysis was performed in seven cases; five showed a monoclonal B cell population, one a polyclonal profile, and one was not conclusive due to DNA degradation.
One case, presented by J. Burke, merits specific attention because of its complexity (Fig 6a–f). It occurred in a 65-year-old male who presented with generalized lymphadenopathy. A lymph node biopsy showed complete effacement due to a lymphoid population consisting of large atypical cells, often with HRS-like morphology. The cells were uniformly positive for CD20, CD79a, PAX5, BOB.1, OCT-2, CD30, and LMP1 but negative for CD15, CD45, and T cell markers. No clonal IGVH@ gene rearrangement was detected by polymerase chain reaction (PCR). This case obviously raised the possibility of LDCHL, its phenotype allowing the straightforward exclusion of ALCL (see above). After extensive discussion, the panel decided to regard it as EBV+ DLBCL-E because of the strong expression of B cell markers and CD15 negativity, which is also in keeping with the criteria recently published by Asano et al. [73]. Although limited in number, the present cases somewhat differ from those originally reported in Japan [75] and should prompt further studies in order to determine whether EBV+ DLBCL-E is a distinct entity in Western populations. In a small series from the Midwestern USA, the overall rate of EBV positivity in otherwise typical DLBCL in older adults was less than 1.1% [78].

a-f Session 4, case 03: EBV+ DLBCL of the elderly. The neoplastic population—provided with some HRS-like features—is admixed with a rich reactive T cell component (a) and expresses CD30 (b), CD20 (c), PAX5/BSAP (d), OCT-2 (e), and LMP1 (f). g–i Session 4, case 09: HIV-associated plasmablastic lymphoma. The neoplasm infiltrates the lachrymal gland (g; cytological details in the inset) and shows EBV integration (h) and Ig kappa monoclonality (i) at ISH. j–n Session 4, case 29: Extracavitary PEL in the absence of immunodeficiency. Lymphomatous cells vary in size and shape (from immunoblastic to plasmablastic; j), are negative for CD45 (k), express IRF4 (l), and turn positive for HHV8 LANA (m) and EBV (n). o–r Session 4, case 14: Monomorphic PTLD. The surgical specimen (colon) is characterized by multiple ulcers (o). The tumor consists of large cells with plasmablastic features (p) and does express LMP1 (q) and EBNA2 (r)
In conclusion, DLBCL-E should be distinguished from infectious mononucleosis-type proliferations (IM) and CHL. Although IM may display some polymorphism with HRS-like cells and foci of necrosis, it lacks overt cytological atypia and consists of a mixture of polyclonal activated B and T cells. On the other hand, DLBCL-E differs from CHL because of regular CD79a expression, frequent CD20 positivity, CD15 negativity, usually strong PAX5 staining, common detection of the transcription factors BOB.1 and OCT-2, and possible BCL6 expression. Finally, the positivity of a DLBCL for CD30 should encourage the search for EBV by EBERs ISH or LMP-1 immunohistochemistry, although the two features are not invariably associated.
HIV-associated lymphomas
The WHO classification states that these tumors are predominantly represented by aggressive B cell lymphomas. In a proportion of cases, they correspond to the initial manifestation of the acquired immunodeficiency syndrome. These disorders are heterogeneous and include lymphoid neoplasms seen in immunocompetent individuals as well as those more commonly encountered in the setting of HIV infection. The commonest HIV-associated lymphomas are: BL, DLBCL (frequently involving the central nervous system), primary effusion lymphoma (PEL), and plasmablastic lymphoma. CHL shows increased prevalence in HIV-positive patients. Peripheral T cell lymphomas are indeed rare.
Five cases were included in this category. Two of them represented morphologic examples of plasmablastic lymphoma. This entity—that in the previous edition of the WHO classification had been strictly related to the occurrence of HIV infection and location in the oral cavity—can actually develop in patients with other immunodeficiency states as well as at other extranodal sites. Interestingly, both cases showed some peculiar findings. In particular, the case presented by R.L. Christensen (Fig 6g–i)—that had the typical phenotypic profile (CD20−, PAX5−, BCL6−/+, IRF4/MUM1+, CD138+, and EBER+)—was diagnosed in the lacrimal gland and characterized by mesenteric and retroperitoneal lymphadenopathy, ascites, and bone marrow (BM) infiltration and carried the t(8;14) with MYC rearrangement. Involvement of lymph nodes is a rare event in this setting, but even rarer is the occurrence of t(8;14), so far recorded in only two cases in the literature [79, 80]. The other plasmablastic lymphoma revealed an aberrant phenotype. In keeping with a plasma cell, it was CD20−, PAX5−, BCL6−, IRF4/MUM1+, CD38+, and EBV+. However, it expressed CGET1+ and PRDM1/Blimp1+ while lacking CD138−, a profile intermediate between a GC B cell (GCB) and a plasma cell. CGET1 is a highly restricted marker for a subset of GCB-derived lymphoma [81], while the lack of CD138 in the presence of Blimp1 [82, 83] suggests late GC/early plasma cell differentiation. Notably, such a phenotype was not associated with t(8;14) as reported by Chuah et al. in a plasmablastic lymphoma aberrantly expressing CD10 [79].
Of the remaining three cases, one represented an ALK-negative ALCL associated with DLBCL, as supported by the phenotypic profiles of the two neoplastic components, both EBV+. To the best of our knowledge, such a composite lymphoma has never been observed in the HIV setting. Another case, a patient with a long history of HIV infection, represented a B cell lymphoma unclassifiable with features intermediate between DLBCL and BL. In particular, the tumor (corresponding to an ileocecal mass) consisted of two distinct populations. Of these, one showed cytological features consistent with BL, while the other was composed of large cells, partly multinucleated, with a wide rim of clear cytoplasm, resembling DLBCL primary arising in the mediastinum. Notably, both components shared the same BL-like phenotype (CD10+, BCL6+, BCL2−, and Ki-67 = 100%) and were negative for EBV and t(8;14). The last case, observed in a 33-year-old HIV-positive individual, fulfilled the clinical, morphologic, and phenotypic criteria for the diagnosis of PEL. Notably, besides bilateral pleural effusion and ascites, it showed lymphadenopathy and a mass involving the mesentery, large bowel wall, rectum, and left psoas, as observed in PEL. However, it was HHV8 LANA negative, a fact that contradicts the definition of PEL itself. Although two HHV8-negative PELs are quoted in the literature [84, 85], the panel decided to classify this case as HIV-related DLBCL. Ideally, molecular studies to exclude the presence of HHV8 genome could be done in such cases to exclude aberrant loss of HHV8-LANA.
PEL and DLBCL with plasmablastic morphology in immunocompetent patients
Three cases were included in this group. In particular, PEL is a large B cell neoplasm that presents as serous effusions in the absence of detectable tumor masses, is universally associated with HHV8 infection, and most often occurs in the setting of immunodeficiency. Rarely, HHV8+ tumors indistinguishable from PEL present as solid masses: they are termed “extracavitary PEL.” Two cases were consistent with the diagnosis of extracavitary PEL and notably occurred in immunocompetent hosts, a finding rarely encountered in elderly individuals from areas with high prevalence for HHV8 infection like the Mediterranean [1]. The case presented by C. Bacon (Fig. 6j–n) was a 15-year-old HIV-negative African boy who developed large cervical nodes, a mediastinal mass, splenomegaly, and abdominal lymphadenopathy. Morphologically, the tumor showed a cytological spectrum from immunoblastic to anaplastic with some Reed-Sternberg (RS)-like elements that were CD79a+ (weak), IRF4/MUM1+, EMA+, HHV8 LANA+, HHV8 vIL-6+, EBERs+, CD45−, CD20−, PAX5−, CD138−, CD38−, CD30−, BCL6−, T cell, cytotoxic and myeloid markers−, Ki-67 = 90%, and clonal rearrangement by IGK and DH-JH primers. Comparative genomic hybridization revealed gains and losses of regions containing PEL-associated genes [86]. The case presented by F. Camacho occurred in a 77-year-old Spanish male with several chronic diseases. The tumor presented as an obstructive jejunal mass with plasmablastic morphology. The tumor cells were CD45+, EMA+, CD30+, IRF4/MUM1+, CD138+, CD38+, CD20−, CD79a-, CD10−, BCL6−, BCL2−, and HHV8 LANA+. The neoplastic population was EBER−, a feature that—as mentioned above—can be seen in old immunocompetent patients from the Mediterranean area. The third case was seen in an 84-year-old female with a history of Wolff-Parkinson White syndrome. She had a 12-cm mass, involving the right psoas muscle. All clinical and laboratory tests, including the search for HIV and BM biopsy, were unremarkable. On microscopic examination, the tumor showed plasmablastic morphology and had a CD38+, CD138+, CD79a+, PAX5+, IRF4/MUM1+, CD20−, CD30−, CD56−, ALK−, and BCL6− phenotype. ISH displayed cytoplasmic kappa and negativity for EBERs. The panel classified it as DLBCL with plasmablastic features because of EBV negativity that excludes DLBCL-E and strong expression of PAX5, a feature inconsistent with a diagnosis of plasmablastic lymphoma, as stated in the WHO classification.
PTLD
This category included five cases, which were classified as polymorphic (one) and monomorphic (four). The case presented by F. Facchetti was a 46-year-old male, who developed a polymorphic PTLD 20 years following renal transplantation. The patient presented with fever, diffuse lymphadenopathy, lung nodules, and pleural and pericardial effusions. Microscopic examination showed scattered large cells with HRS-like morphology that revealed strong CD20 positivity, variable CD30 expression, and negativity for both CD15 and CD138, in a background of numerous reactive T lymphocytes. The blastic elements were EBV positive (EBERs ISH+, LMP1+, and EBNA2 partly+). The process had features intermediate between THRLBCL and CHL: similar cases, nowadays included among polymorphic PTLD, have in the past possibly been referred to as “Hodgkin-like” PTLD [1]. Notably, the administration of Foscarnet produced a dramatic improvement of the clinical conditions with reduction of EBV viremia from 26,100 to 1,500 after 1 week of treatment. The diagnosis of CHL-type PTLD was excluded because of the strong and homogeneous expression of CD20 with variable positivity for CD30, CD15−, and EBNA2+ and response to antiviral treatment.
Of the monomorphic cases, two were EBV+ and two EBV−. EBV+ monomorphic PTLD usually have a late GC/post-GC phenotype (CD10−, BCL6±, IRF4/MUM1+, CD138variably+), while the EBV− ones are more likely to have a GC-type phenotype (CD10±, BCL6+, IRF4/MUM1−, CD138−). However, all four cases revealed a post-GC profile. The first of these cases—presented by N. Rooney (Fig. 6o–r)—occurred in a 47-year-old woman who underwent kidney transplantation. The patient developed bleeding per rectum caused by multiple ulcers in the colon in the absence of previous inflammatory bowel disease. PTLD with DLBCL morphology was diagnosed on a colon biopsy. Despite the reduction of tacrolimus and the administration of rituximab, the patient’s general conditions worsened and a subtotal colectomy was required. Interestingly, rituximab had almost abolished CD20 expression when compared to the endoscopic biopsy but had no impact on the lymphoid cells that maintained their EBV positivity and high Ki-67 reactivity. Notably, different clonal IGVH@ rearrangements were detected in three different blocks from the large bowel. The other EBV+ monomorphic PTLD presented in the graft of a 17-year-old male who—1 year prior—had undergone kidney transplantation for end-stage renal disease secondary to reflux nephropathy. It was diagnosed by the contributors and panelists as monomorphic PTLD, DLBCL type with HL-like features. This interpretation is consistent with the statement that “in monomorphic PTLD, some cells may be bizarre and even multinucleated and even resemble RS-like cells” [1]. The phenotype also supported this interpretation (CD79a+, CD20+/−, PAX5+strong, CD138+, IRF4/MUM1+, CD30−/+, and CD15−). Notably, endothelial cells were affected by CMV infection. Of the two EBV-PTLD, one occurred in the stomach of a 58-year-old male who had received a kidney transplant 22 years previously. The interest of this case resided in the phenotypic characteristics of the neoplastic cells that morphologically fulfilled the criteria for the diagnosis of DLBCL. In particular, they showed negativity for B cell markers (CD20, CD79a, CD138, and PAX5) but carried mature B cell clonality genotype. The last case, also EBV negative, occurred in a female 10 years after a heart transplant performed when aged one for Kawasaki disease. She developed multiple polypoid masses in the terminal ileum with intussusceptions that led to partial small-bowel resection. In spite of CHOP chemotherapy that turned out totally ineffective, the tumor relapsed several times in the bowel and lung. Microscopically, it showed immunoblastic morphology with plasmablastic features. This was supported by the molecular profile that showed CD45−/+, CD20+variable, CD79a+, PAX5−, IRF4/MUM1+, BLIMP1+/−, CD56−, IgA/k+, and LANA−. IGVH@ genes were clonally rearranged.
LYG and related conditions
This group of cases was to some extent related to the previous and following categories. LYG is an angiocentric and angiodestructive lymphoproliferative disease involving extranodal sites, composed of EBV+ B cells admixed with reactive T lymphocytes, which usually predominate. The lesion has a spectrum of histological grades and clinical aggressiveness, which is related to the proportion of large B cells. Three of the submitted cases fulfilled such criteria. They occurred in patients in the adult life (aged 34, 37, and 39, respectively) with a male/female ratio of 2:1. Morphologically, all cases were grade 3 and showed angiocentricity and angioinvasiveness by large B cells (CD20+, CD30+, LMP1+, EBERs+) that caused extensive necrosis and were admixed with variable amounts of reactive T lymphocytes. When performed, molecular analysis revealed clonal IGVH@ gene rearrangement. Interestingly, one case—submitted by G. Rassidakis (Fig. 7a–d)—occurred in the skin in the recipient of a kidney transplant. Solid organ transplantation is regarded as one of the LYG predisposing conditions along with Wiskott-Aldrich syndrome, HIV infection, and X-linked lymphoproliferative syndrome [1]. The other two cases showed lung involvement, as commonly seen in LYG and originally described by Liebow et al. [87]. In one of them, the disease also affected lymph nodes, a finding that is rare in this setting [1]. Finally, the third case was recorded in a patient with a history of Wegener granulomatosis (WG) that lasted 10 months and had been treated with methotrexate, i.e., an immunosuppressive agent implicated in the development of lymphoproliferative disorders [1]. Although originally described as a variant of WG [87], LYG does represent a distinct disease and so far no transformation of WG into LYG has been reported in the literature. Thus, in the absence of any morphological evidence, it remains a matter of speculation whether or not the original clinical diagnosis of WG was correct.

a–d Session 4, case 24: LYG grade 3. The neoplastic growth shows angiocentricity and angioinvasiveness (a). At immunohistochemistry, lymphomatous cells express CD20 (b) and partly CD30 (b, inset) and are admixed with abundant CD3+ T lymphocytes (c). ISH reveals EBV integration (arrowed; d). e–h Session 4, case 16: EBV+ LPD mimicking DLBCL, leg type. The tumor infiltrates the deep dermis and subcutaneous tissue (e) and consists of large cells with plasmablastic features (f) that display high Ki-67 rate (g) and EBV integration at ISH (h). i–l Session 4, case 23: DLBCL associated with chronic inflammation (PAL). The lymphomatous population, showing immunoblastic/plasmablastic features (i), expresses CD20 (j), CD79a (k), and EBNA2 (l). m–p Session 4, case 32: HHV6+ THRLBCL. Scattered neoplastic cells (m) are comprised within a reactive population consisting of small lymphocytes and histiocytes (m). At immunohistochemistry, they express CD20 (n), are surrounded by CD3+ T lymphocytes (n, inset) and turn positive for the virus-associated antigen HIND (o). Electron microscopy shows viral particles consistent with HHV6 (p)
The last case included in this group shared the age (36 years), disease spread, and morphological features with the other cases described above. However, it turned out to be EBV negative, contradicting one of the criteria for the diagnosis of LYG. Therefore, the case was diagnosed as angiocentric DLBCL, HHV8 negative by the panel. A literature search revealed only one similar case in a patient with a 3-year-history of methotrexate treatment for rheumatoid arthritis [88].
EBV+ lymphoproliferative disorders in the course of immunosuppressive hematological malignancies
Three cases were included in this group, which corresponded to EBV-driven lymphoproliferative disorders. The underlying hematological diseases were represented by NK cell large granular lymphocyte disorder (LGL), refractory anemia with excess of blast (RAEB)-2, and chronic lymphocytic leukemia (CLL), respectively. The latter occurred in a 76-year-old male who had been treated with fludarabine, a drug which is well known for its immunosuppressive action [89, 90], as well as for the risk of inducing EBV+ lymphoproliferative disorders in patients treated for indolent B cell lymphomas [91]. In this case, it remained unsettled whether or not the abdominal large B cell tumor (expressing CD20, CD79a, CD30 (partly), BCL2, and LMP-1) was clonally related to the underlying CLL (CD20+, CD79a+, CD5+, CD23−, EBV−), since the only molecular analysis performed consisted in EBERs ISH. In particular, a review of the literature showed that an EBV+ large B cell component (so-called Richter’s syndrome) may develop within the CLL setting as either transformation of the existing tumor or a de novo process [92–95]. The focal positivity of large B cells for CD5 slightly favored a clonal relationship. The patient with LGL was a 79-year-old female who did not receive any therapy for her disorder. Suddenly, she developed a skin lesion on her right leg, composed of large cells with plasmablastic features that were CD20+ (in part), CD79a+, CD30+ (a few), CD138+ (most), and Ki-67+ (70%). FISH displayed BCL2 and BCL6 abnormal translocation patterns as well as p53 gene deletion. The panel diagnosed this case—submitted by M Twohig (Fig. 7e–h)—as EBV+ lymphoproliferative disorder mimicking DLBCL, leg type [1]. Finally, the third case occurred in a 59-year-old male with RAEB-2 and high EBV IgG and IgA titers that developed generalized lymphadenopathy. A laterocervical lymph node biopsy was consistent with THRLBCL. The neoplastic cells (CD20+, CD79a+/−, BCL6−, IRF4/MUM1+, CD30+/−, EBERs+, LMP-1+/−, and CD15−) were embedded within a reactive T cell component. PCR revealed an IGVH@ gene monoclonal band in a strong polyclonal background. Since RAEB-2 had apparently remained untreated, the panel presumed that the myelodysplastic syndrome was sufficient to cause immunosuppression, as LGL in the previous case. Therefore, a final diagnosis of immunodeficiency-related lymphoproliferative disorder was made. The disease had a fatal outcome, since various courses of chemotherapy with rituximab remained ineffective.
Chronic inflammation-associated DLBCL
In the WHO classification, DLBCL associated with chronic inflammation is a subtype of DLBCL occurring in the context of long-standing chronic inflammation and showing association with EBV. Most cases involve body cavities or narrow spaces. Pyothorax-associated lymphoma (PAL) is a prototypic form and develops in the pleural cavity of patients with long-standing pyothorax. Other settings of long-standing chronic suppuration/inflammation, which can favor the onset of the tumor, are chronic osteomyelitis, metallic implant, or chronic skin ulcer. There is probably a role for chronic inflammation at the local site in the proliferation of EBV-transformed B cells by enabling them to escape from the host immune surveillance (through IL-10, IL-6, and IL-6 receptor). Although most cases have been recorded in Japan, DLBCL associated with chronic inflammation can be encountered also in Western Countries [96, 97]. A typical example of PAL was presented by P. Gaulard (Fig. 7i–l). It occurred in a 69-year-old male who had undergone artificial pneumothorax 35 years prior. The tumor presented as a huge mass that involved the anterior chest wall and was associated with weight loss and pleural effusion. It consisted of immunoblasts with features of plasmablastic/plasmacellular differentiation, frequent apoptotic bodies, and numerous mitotic figures. These expressed CD20, CD79a, CD138, CD30 (heterogeneously), and IRF4/MUM1. They were monotypic kappa with immunohistochemistry and monoclonal by PCR. Typically, they were EBERs+ and expressed latency III pattern as found in about 60% of PAL cases.
Other virus-associated lymphoproliferations
This group included the instructive case from a 21-year-old female who had undergone right tonsillectomy when she was 6 years old. She presented with malaise of several weeks duration, bilateral cervical lymphadenopathy, hepatomegaly, atypical lymphocytosis in the peripheral blood (7.6 × 109/L), and left necrotising tonsillitis, refractory to antibiotics and dexamethazone. Monospot was positive, as was serology for EBV IgG and IgM. A left tonsillectomy was performed for air obstruction. Monospot was positive, as was serology for EBV IgG and IgM. Morphologically, there was a diffuse effacement of the normal tonsil structure due to a lymphoid population with blastic morphology and high mitotic index that expressed CD20, CD79a, IRF4/MUM1, EBERs, LMP-1, CD30 (in part), and CD45 and was negative for CD10, BCL6, CD138, ALK1, and HHV8. Ig light chains revealed a polytypic pattern. Admixed with the blasts were numerous cytotoxic T lymphocytes (CD3+, CD8 > CD4, and granzyme B+). The patient recovered 2 months later. This case was diagnosed by both the contributors and panel as large B cell proliferation in acute EBV infection (infectious mononucleosis). This case underlines the risk of misinterpreting such pattern as DLBCL, if the clinical and laboratory data are unknown and a limited panel of reagents is applied [98–101]. Another remarkable case—submitted by P. Adam (Fig. 7m–p)—was a 74-year-old male who underwent a laterocervical lymph node biopsy. The morphologic and immunohistochemical findings were consistent with THRLBCL. However, viral inclusion bodies were seen both in the tumor cells and reactive elements. Antibodies against early and late antigens as well as electron microscopy showed that such inclusions corresponded to HHV6 infection. This case expands the spectrum of herpesvirus infections associated with malignant lymphomas. HHV6 has been associated only with angioimmunoblastic T cell lymphoma [102, 103]. There is, however, no clear evidence for a causative role.
Conclusions
The workshop jointly organized by the EAHP and the SH in 2008 in Bordeaux focused on the difficult topic of gray-zone lymphomas. In the 2008 WHO classification, these borderline categories were added mainly to delineate the borderlands between BL and DLBCL and DLBCL and HL. As emerged from the discussions during the workshop—among the panel members, the auditorium, and the authors of the WHO classification—several issues were considered important for the proper use of these borderline categories. The main two issues were that all information—cytomorphologic, biologic, and clinical—should be considered to arrive at an accurate diagnosis, and second, whenever possible, distinct entities should be diagnosed. It was agreed, however, that this was not possible in all cases, thus justifying the creation of borderline diagnoses (unclassifiable), looked upon as practical categories awaiting for more data to become available and not as discrete entities.
Session 1 was devoted to the possible overlap between BL and DLBCL. Specifically, this overlap is caused by similarities in morphology, phenotype, and genetic aberrations. During the session, it became clear that the “double-hit” cases (a combination of a MYC breakpoint with mostly BCL2 breakpoints and other recurrent chromosomal breakpoints), often with distinct morphological features of BL should fall in a novel category of “B cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL.” In fact, there was some discussion whether or not B cell lymphomas, irrespective of morphology, should be moved to this novel category, but at the end it was felt that this change would be premature. The second most difficult category that remains in the gray zone consists of lymphomas with morphological features of both DLBCL and BL, but with a phenotype that is consistent with BL, in particular since up to 40% of such cases may have a MYC breakpoint. Many of such cases might fall in this novel category; however, it was felt that cases with a typical morphology of DLBCL should be kept in the DLBCL category, in spite of the fact that DLBCL patients with a MYC breakpoint will have a relatively poor prognosis.
Session 2 was devoted to mediastinal gray-zone lymphoma and other lymphomas with atypical immunophenotype that caused differential diagnostic problems, often between CHL and NHL. During the discussion, it was stressed that deviation of a single marker or criterion, e.g., strong and uniform CD20 expression in otherwise typical HL, was not sufficient for placing a case in the category of “B cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL,” but that a hybrid phenotype not being in line with current morphologic and/or biologic concepts would well be. Examples of those features could be a significant mononuclear large B cell component not compatible with HL or uniformly strong expression of pan B cell markers along with the expression of one or more of the transcription factors BOB.1, OCT2, and PU.1 in a mediastinal tumor with morphological features of CHL. This stresses the need of doing a complete phenotype (B cell markers and transcription markers) in cases with morphological features of CHL with strong and uniform CD20 expression.
Among the tumors with unusual phenotypes presented during this session were DLBCL with cyclin D1 overexpression and cases with aberrant expression of T-cell-associated markers. Also, in this group, several cases of T cell lymphomas with CD30 and/or CD15 expression including ALCL cases were shown.
Session 3 dealt with the differential diagnosis between NLPHL and THRLBCL, and the gray zones between CHL and DLBCL, mainly THRLBCL in lymph nodes. The main issue addressed during the workshop was to define criteria to reliably distinguish these entities. Within the first topic, it was considered important that NLPHL can manifest in various progression stages, being characterized by diffuse areas and by progressive loss of the typical small B cell background. While it was agreed that transition of NLPHL into THRLBCL might exist, the proposal of the WHO classification to name areas reminiscent of THRLBCL in an otherwise typical NLPHL background “THRLBCL-like areas” was generally considered a practical guideline. It implies that one single nodule of typical NLPHL in a THRLBCL background is sufficient to exclude de novo THRLBCL. Other important issues highlighted during the workshop were the reactivity of the LP cells for IgD in the progressed cases or the rare expression of CD15. In addition, three bona fide NLPHL showed the presence of EBV genomes in their nuclei, a feature considered unusual for NLPHL and rather characteristic of CHL.
Several cases submitted to the workshop exemplified the difficulties in accurately separating cases of CHL from THRLBCL due to overlapping clinical, morphological, and/or immunophenotypic features. In general, CHL is negative or weakly positive for B cell antigens and PAX5 and does show loss of transcription factors expression; however, deceptively similar cases may be placed in the THRLBCL category, when CD15 and/or CD30 are negative and CD20 and/or CD79a are strongly and uniformly positive. Similar to the mediastinal gray zone, cases with CHL and DLBCL boundary features also exist in lymph nodes. The cases presented during the workshop showed an amazing variety and overlapping immunohistochemical features, thus giving clear evidence for the need of a borderline category. In addition, the frequent association of these cases with EBV also discloses overlapping features with (THRBCL-like) EBV-associated DLBCL of the elderly in young patients.
Session 4 dealt with the heterogeneous lymphoma categories of EBV-positive lymphomas, lymphomas occurring in HIV+ individuals, and PTLD-related B cell lymphoproliferations. This session exemplify how meticulous diagnostic assessment of the cases is needed in arriving at the correct diagnosis. Clinical background information, and the exact knowledge of the sites involved, is absolutely essential in providing a correct diagnosis. EBV-associated lymphoproliferations such as posttransplant lymphoproliferative disorders, body-cavitary-based or extracavitary PEL or HIV-associated or lymphoproliferations following iatrogenic measurements cannot be diagnosed without proper clinical information. On the other hand, on purely morphological grounds, these cases might have been put into a different category. With respect to EBV association, the positivity of a DLBCL for CD30 should encourage performing EBER ISH or LMP-1 immunohistochemistry.
In the workshop, EBV+ DLBCL-E showed regular CD79a expression and frequent CD20 positivity, usually with strong PAX5, BOB.1, and OCT-2 staining, thus setting it apart from EBV-associated CHL. Among lymphomas occurring in HIV+ individuals, plasmablastic lymphomas and BL were the two categories represented. Two cases were consistent with the diagnosis of extracavitary PEL and, most interestingly, were diagnosed in immunocompetent hosts, a finding rarely encountered in elderly individuals from areas with high prevalence for HHV8 infection like the Mediterranean. In keeping with what has been stated above, the PTLD cases referred to the workshop often presented with hybrid morphological and immunophenotypic features intermediate between CHL and DLBCL; since two of the PTLD monomorphic DLBCL were EBV negative, clinical information is essential in arriving at the correct diagnosis.
Examples of lymphomatoid granulomatosis, EBV-associated lymphoproliferative disorders in the course of immunosuppressive hematological malignancies, cases of DLBCL associated with chronic inflammation, and other virus-associated lymphoproliferations completed the spectrum of diseases seen in session 4. For the latter category, two instructive cases were presented, both with an initial suspicion of malignant lymphoma but turning out to represent virus-associated reactive lymphoproliferations, one associated with EBV and one with HHV6. These difficult cases again underline the risk of misinterpreting such reactive disorders as DLBCL, if the clinical and laboratory data are unknown and/or a limited panel of antibodies is applied.
References
Swerdlow SH et al (2008) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. IARC, Lyon
Kluin PM et al (2008) B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma. In: Swerdlow SH et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, IARC, pp 265–266
Jaffe ES et al (2008) B-cell lymphoma, unclassifiable with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin Lymphoma. In: Swerdlow SH et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, IARC, pp 267–268
Macpherson N et al (1999) Small noncleaved, non-Burkitt’s (Burkitt-like) lymphoma: cytogenetics predict outcome and reflect clinical presentation. J Clin Oncol 17(5):1558–1567
Kanungo A et al (2006) Lymphoid neoplasms associated with concurrent t(14;18) and 8q24/c-MYC translocation generally have a poor prognosis. Mod Pathol 19(1):25–33
Rosenwald A et al (2003) Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 198(6):851–862
Savage KJ et al (2003) The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 102(12):3871–3879
Traverse-Glehen A et al (2005) Mediastinal gray zone lymphoma: the missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am J Surg Pathol 29(11):1411–1421
Fan Z et al (2003) Characterization of variant patterns of nodular lymphocyte predominant Hodgkin lymphoma with immunohistologic and clinical correlation. Am J Surg Pathol 27(10):1346–1356
Boudova L et al (2003) Nodular lymphocyte-predominant Hodgkin lymphoma with nodules resembling T-cell/histiocyte-rich B-cell lymphoma: differential diagnosis between nodular lymphocyte-predominant Hodgkin lymphoma and T-cell/histiocyte-rich B-cell lymphoma. Blood 102(10):3753–3758
Nakamura S, Jaffe ES, Swerdlow SH (2008) EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, IARC, pp 243–244
Burkitt D (1958) A sarcoma involving the jaws in African children. Br J Surg 46(197):218–223
Blum KA, Lozanski G, Byrd JC (2004) Adult Burkitt leukemia and lymphoma. Blood 104(10):3009–3020
Magrath I (1990) The pathogenesis of Burkitt’s lymphoma. Adv Cancer Res 55:133–270
Dave SS et al (2006) Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med 354(23):2431–2442
Hummel M et al (2006) A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med 354(23):2419–2430
Salaverria I et al (2008) Chromosomal alterations detected by comparative genomic hybridization in subgroups of gene expression-defined Burkitt’s lymphoma. Haematologica 93(9):1327–1334
Boerma EG et al (2009) Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: a historical review of cytogenetics in the light of today’s knowledge. Leukemia 23(2):225–234
Haralambieva E et al (2004) Interphase fluorescence in situ hybridization for detection of 8q24/MYC breakpoints on routine histologic sections: validation in Burkitt lymphomas from three geographic regions. Genes Chromosomes Cancer 40(1):10–18
Haralambieva E et al (2005) Clinical, immunophenotypic, and genetic analysis of adult lymphomas with morphologic features of Burkitt lymphoma. Am J Surg Pathol 29(8):1086–1094
Klapper W et al (2008) Structural aberrations affecting the MYC locus indicate a poor prognosis independent of clinical risk factors in diffuse large B-cell lymphomas treated within randomized trials of the German High-Grade Non-Hodgkin’s Lymphoma Study Group (DSHNHL). Leukemia 22(12):2226–2229
Savage KJ et al (2009) MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 114(17):3533–3537
Mead GM et al (2008) A prospective clinicopathologic study of dose-modified CODOX-M/IVAC in patients with sporadic Burkitt lymphoma defined using cytogenetic and immunophenotypic criteria (MRC/NCRI LY10 trial). Blood 112(6):2248–2260
Sevilla DW et al (2007) Clinicopathologic findings in high-grade B-cell lymphomas with typical Burkitt morphologic features but lacking the MYC translocation. Am J Clin Pathol 128(6):981–991
Mossafa H et al (2006) Non-Hodgkin’s lymphomas with Burkitt-like cells are associated with c-Myc amplification and poor prognosis. Leuk Lymphoma 47(9):1885–1893
Leucci E et al (2008) MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol 216(4):440–450
Johnson NA et al (2009) Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 114(11):2273–2279
Le Gouill S et al (2007) The clinical presentation and prognosis of diffuse large B-cell lymphoma with t(14;18) and 8q24/c-MYC rearrangement. Haematologica 92(10):1335–1342
Lin P, Medeiros LJ (2007) High-grade B-cell lymphoma/leukemia associated with t(14;18) and 8q24/MYC rearrangement: a neoplasm of germinal center immunophenotype with poor prognosis. Haematologica 92(10):1297–1301
Kramer MH et al (1991) De novo acute B-cell leukemia with translocation t(14;18): an entity with a poor prognosis. Leukemia 5(6):473–478
Lichtenstein AK et al (1980) Primary mediastinal lymphoma in adults. Am J Med 68(4):509–514
Higgins JP, Warnke RA (1999) CD30 expression is common in mediastinal large B-cell lymphoma. Am J Clin Pathol 112(2):241–247
Moller P et al (1986) Primary mediastinal clear cell lymphoma of B-cell type. Virchows Arch A Pathol Anat Histopathol 409(1):79–92
Paulli M et al (1999) Mediastinal B-cell lymphoma: a study of its histomorphologic spectrum based on 109 cases. Hum Pathol 30(2):178–187
Pileri SA et al (2003) Pathobiology of primary mediastinal B-cell lymphoma. Leuk Lymphoma 44(Suppl 3):S21–S26
Moller P et al (1987) Mediastinal lymphoma of clear cell type is a tumor corresponding to terminal steps of B cell differentiation. Blood 69(4):1087–1095
Pileri SA et al (2003) Primary mediastinal B-cell lymphoma: high frequency of BCL-6 mutations and consistent expression of the transcription factors OCT-2, BOB.1, and PU.1 in the absence of immunoglobulins. Am J Pathol 162(1):243–253
Rodig SJ et al (2007) Expression of TRAF1 and nuclear c-Rel distinguishes primary mediastinal large cell lymphoma from other types of diffuse large B-cell lymphoma. Am J Surg Pathol 31(1):106–112
Calaminici M et al (2004) CD23 expression in mediastinal large B-cell lymphomas. Histopathology 45(6):619–624
Salama ME et al (2009) The value of CD23 expression as an additional marker in distinguishing mediastinal (thymic) large B-cell lymphoma from Hodgkin lymphoma. Int J Surg Pathol (in press)
Bea S et al (2005) Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 106(9):3183–3190
Joos S et al (2000) Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res 60(3):549–552
Joos S et al (1996) Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood 87(4):1571–1578
Kimm LR et al (2007) Frequent occurrence of deletions in primary mediastinal B-cell lymphoma. Genes Chromosomes Cancer 46(12):1090–1097
Copie-Bergman C et al (1999) The MAL gene is expressed in primary mediastinal large B-cell lymphoma. Blood 94(10):3567–3575
Copie-Bergman C et al (2002) MAL expression in lymphoid cells: further evidence for MAL as a distinct molecular marker of primary mediastinal large B-cell lymphomas. Mod Pathol 15(11):1172–1180
Jaffe ES et al (1994) The interrelationship between Hodgkin’s disease and non-Hodgkin’s lymphomas. Ann Oncol 5(Suppl 1):7–11
Jaffe ES, Zarate-Osorno A, Medeiros LJ (1992) The interrelationship of Hodgkin’s disease and non-Hodgkin’s lymphomas—lessons learned from composite and sequential malignancies. Semin Diagn Pathol 9(4):297–303
Perrone T, Frizzera G, Rosai J (1986) Mediastinal diffuse large-cell lymphoma with sclerosis. A clinicopathologic study of 60 cases. Am J Surg Pathol 10(3):176–191
Poppema S et al (2005) Report: workshop on mediastinal grey zone lymphoma. Eur J Haematol Suppl 66:45–52
Rudiger T et al (1998) Workshop report on Hodgkin’s disease and related diseases (‘grey zone’ lymphoma). Ann Oncol 9(Suppl 5):S31–S38
Hsi ED et al (2006) MAL is expressed in a subset of Hodgkin lymphoma and identifies a population of patients with poor prognosis. Am J Clin Pathol 125(5):776–782
Seitz V et al (2000) Detection of clonal T-cell receptor gamma-chain gene rearrangements in Reed-Sternberg cells of classic Hodgkin disease. Blood 95(10):3020–3024
Rodriguez-Justo M et al (2008) Cyclin D1-positive diffuse large B-cell lymphoma. Histopathology 52(7):900–903
Ehinger M et al (2008) A subset of CD5- diffuse large B-cell lymphomas expresses nuclear cyclin D1 with aberrations at the CCND1 locus. Am J Clin Pathol 129(4):630–638
Lai R et al (2000) Sinusoidal CD30-positive large B-cell lymphoma: a morphologic mimic of anaplastic large cell lymphoma. Mod Pathol 13(3):223–228
Carbone A et al (1997) BCL-6 protein expression in human peripheral T-cell neoplasms is restricted to CD30+ anaplastic large-cell lymphomas. Blood 90(6):2445–2450
Barry TS et al (2003) Peripheral T-cell lymphomas expressing CD30 and CD15. Am J Surg Pathol 27(12):1513–1522
Saffer H et al (2002) Clusterin expression in malignant lymphomas: a survey of 266 cases. Mod Pathol 15(11):1221–1226
Wellmann A et al (2000) Detection of differentially expressed genes in lymphomas using cDNA arrays: identification of clusterin as a new diagnostic marker for anaplastic large-cell lymphomas. Blood 96(2):398–404
Chandra P et al (2009) Clusterin expression correlates with stage and presence of large cells in mycosis fungoides. Am J Clin Pathol 131(4):511–515
Franke S et al (2002) Comparative genomic hybridization pattern distinguishes T-cell/histiocyte-rich B-cell lymphoma from nodular lymphocyte predominance Hodgkin’s lymphoma. Am J Pathol 161(5):1861–1867
Prakash S et al (2006) IgD positive L&H cells identify a unique subset of nodular lymphocyte predominant Hodgkin lymphoma. Am J Surg Pathol 30(5):585–592
Nam-Cha SH et al (2008) PD-1, a follicular T-cell marker useful for recognizing nodular lymphocyte-predominant Hodgkin lymphoma. Am J Surg Pathol 32(8):1252–1257
Poppema S et al (2008) Nodular lymphocyte predominant Hodgkin lymphoma. In: Swerdlow SH et al (eds) B-cell lymphoma, unclassifiable with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin Lymphoma. Lyon, IARC, pp 323–325
Anagnostopoulos I et al (2000) European Task Force on Lymphoma project on lymphocyte predominance Hodgkin disease: histologic and immunohistologic analysis of submitted cases reveals 2 types of Hodgkin disease with a nodular growth pattern and abundant lymphocytes. Blood 96(5):1889–1899
Dogan A et al (2003) Micronodular T-cell/histiocyte-rich large B-cell lymphoma of the spleen: histology, immunophenotype, and differential diagnosis. Am J Surg Pathol 27(7):903–911
McBride JA et al (1996) T-cell-rich B large-cell lymphoma simulating lymphocyte-rich Hodgkin’s disease. Am J Surg Pathol 20(2):193–201
Lim MS et al (2002) T-cell/histiocyte-rich large B-cell lymphoma: a heterogeneous entity with derivation from germinal center B cells. Am J Surg Pathol 26(11):1458–1466
von Wasielewski R et al (1997) Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am J Pathol 151(4):1123–1130
Oyama T et al (2003) Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol 27(1):16–26
Shimoyama Y et al (2008) Age-related Epstein-Barr virus-associated B-cell lymphoproliferative disorders: special references to lymphomas surrounding this newly recognized clinicopathologic disease. Cancer Sci 99(6):1085–1091
Asano N et al (2009) Age-related Epstein-Barr virus (EBV)-associated B-cell lymphoproliferative disorders: comparison with EBV-positive classic Hodgkin lymphoma in elderly patients. Blood 113(12):2629–2636
Park S et al (2007) The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood 110(3):972–978
Wong HH, Wang J (2009) Epstein-Barr virus positive diffuse large B-cell lymphoma of the elderly. Leuk Lymphoma 50(3):335–340
Chang CC et al (2004) Immunohistochemical expression patterns of germinal center and activation B-cell markers correlate with prognosis in diffuse large B-cell lymphoma. Am J Surg Pathol 28(4):464–470
Garcia JF et al (2005) Large B-cell lymphoma with Hodgkin’s features. Histopathology 47(1):101–110
Gibson SE, Hsi ED (2009) Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol 40(5):653–661
Chuah KL et al (2009) Plasmablastic lymphoma affecting the lung and bone marrow with CD10 expression and t(8;14)(q24;q32) translocation. Int J Surg Pathol 17(2):163–166
Yotsumoto M et al (2009) CD20-negative CD138-positive leukemic large cell lymphoma with plasmablastic differentiation with an IgH/MYC translocation in an HIV-positive patient. Intern Med 48(7):559–562
Montes-Moreno S et al (2008) Gcet1 (centerin), a highly restricted marker for a subset of germinal center-derived lymphomas. Blood 111(1):351–358
Diehl SA et al (2008) STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol 180(7):4805–4815
Teruya-Feldstein J et al (2004) CD20-negative large-cell lymphoma with plasmablastic features: a clinically heterogenous spectrum in both HIV-positive and -negative patients. Ann Oncol 15(11):1673–1679
Jenkins C et al (2005) Human-immunodeficiency-virus-negative, human-herpes-virus-8-negative abdominal cavity primary effusion lymphoma. Clin Oncol (R Coll Radiol) 17(8):636–638
Tanaka S et al (2001) HHV8-negative primary effusion lymphoma of the peritoneal cavity presenting with a distinct immunohistochemical phenotype. Pathol Int 51(4):293–300
Ohshima K et al (2002) Chromosomal and comparative genomic analyses of HHV-8-negative primary effusion lymphoma in five HIV-negative Japanese patients. Leuk Lymphoma 43(3):595–601
Liebow AA, Carrington CR, Friedman PJ (1972) Lymphomatoid granulomatosis. Hum Pathol 3(4):457–558
Pfistershammer K et al (2009) Methotrexate-induced primary cutaneous diffuse large B-cell lymphoma with an ‘angiocentric’ histological morphology. Clin Exp Dermatol (in press)
Dodero A et al (2009) Haploidentical stem cell transplantation after a reduced-intensity conditioning regimen for the treatment of advanced hematologic malignancies: posttransplantation CD8-depleted donor lymphocyte infusions contribute to improve T-cell recovery. Blood 113(19):4771–4779
Ricci F et al (2009) Fludarabine in the treatment of chronic lymphocytic leukemia: a review. Ther Clin Risk Manag 5(1):187–207
Abruzzo LV et al (2002) Epstein-Barr virus-positive B-cell lymphoproliferative disorders arising in immunodeficient patients previously treated with fludarabine for low-grade B-cell neoplasms. Am J Surg Pathol 26(5):630–636
de Leval L et al (2004) Distinct clonal origin in two cases of Hodgkin’s lymphoma variant of Richter’s syndrome associated With EBV infection. Am J Surg Pathol 28(5):679–686
Fong D et al (2005) Hodgkin’s disease variant of Richter’s syndrome in chronic lymphocytic leukaemia patients previously treated with fludarabine. Br J Haematol 129(2):199–205
Thornton PD et al (2005) Richter’s transformation of chronic lymphocytic leukemia. The possible role of fludarabine and the Epstein-Barr virus in its pathogenesis. Leuk Res 29(4):389–395
Mao Z et al (2007) IgVH mutational status and clonality analysis of Richter’s transformation: diffuse large B-cell lymphoma and Hodgkin lymphoma in association with B-cell chronic lymphocytic leukemia (B-CLL) represent 2 different pathways of disease evolution. Am J Surg Pathol 31(10):1605–1614
Ascani S et al (1997) Pyothorax-associated lymphoma: description of the first two cases detected in Italy. Ann Oncol 8(11):1133–1138
Petitjean B et al (2002) Pyothorax-associated lymphoma: a peculiar clinicopathologic entity derived from B cells at late stage of differentiation and with occasional aberrant dual B- and T-cell phenotype. Am J Surg Pathol 26(6):724–732
Childs CC, Parham DM, Berard CW (1987) Infectious mononucleosis. The spectrum of morphologic changes simulating lymphoma in lymph nodes and tonsils. Am J Surg Pathol 11(2):122–132
Lustberg MB, Aras O, Meisenberg BR (2008) FDG PET/CT findings in acute adult mononucleosis mimicking malignant lymphoma. Eur J Haematol 81(2):154–156
Macsween KF, Crawford DH (2003) Epstein-Barr virus—recent advances. Lancet Infect Dis 3(3):131–140
Otteman LA et al (1985) Infectious mononucleosis mimicking a B cell immunoblastic lymphoma associated with an abnormality in regulatory T cells. Am J Med 78(5):885–890
Vrsalovic MM et al (2004) T- and B-cell clonality and frequency of human herpes viruses-6, -8 and Epstein Barr virus in angioimmunoblastic T-cell lymphoma. Hematol Oncol 22(4):169–177
Zhou Y et al (2007) Angioimmunoblastic T-cell lymphoma: histological progression associates with EBV and HHV6B viral load. Br J Haematol 138(1):44–53
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Quintanilla-Martinez, L., de Jong, D., de Mascarel, A. et al. Gray zones around diffuse large B cell lymphoma. Conclusions based on the workshop of the XIV meeting of the European Association for Hematopathology and the Society of Hematopathology in Bordeaux, France. J Hematopathol 2, 211–236 (2009). https://doi.org/10.1007/s12308-009-0053-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-009-0053-9