
Abstract
Macroautophagy is an evolutionarily conserved vacuolar, self-digesting mechanism for cellular components, which end up in the lysosomal compartment. In mammalian cells, macroautophagy is cytoprotective, and protects the cells against the accumulation of damaged organelles or protein aggregates, the loss of interaction with the extracellular matrix, and the toxicity of cancer therapies. During periods of nutrient starvation, stimulating macroautophagy provides the fuel required to maintain an active metabolism and the production of ATP. Macroautophagy can inhibit the induction of several forms of cell death, such as apoptosis and necrosis. However, it can also be part of the cascades of events that lead to cell death, either by collaborating with other cell death mechanisms or by causing cell death on its own. Loss of the regulation of bulk macroautophagy can prime self-destruction by cells, and some forms of selective autophagy and non-canonical forms of macroautophagy have been shown to be associated with cell demise. There is now mounting evidence that autophagy and apoptosis share several common regulatory elements that are crucial in any attempt to understand the dual role of autophagy in cell survival and cell death.
Similar content being viewed by others


Autophagic Cell Death, Autophagy and Cell Death: Not just a Question of Terminology
The term ‘autophagy’ embraces several different mechanisms: microautophagy, macroautophagy and chaperone-mediated autophagy,1 all of which are involved in the lysosomal degradation of cellular components. In this review, we will focus essentially on macroautophagy, because of the large body of data available about its cross talk with cell death. Macroautophagy (hereafter called ‘autophagy’) is a mechanism conserved among eukaryotic cells that starts with the formation of a multi-membrane-bound vacuole, known as an autophagosome, which ultimately fuses with the lysosomal compartment and the degradation of the sequestered material.2 The seminal discovery of autophagy-related (ATG) genes initially in yeast and then in multicellular organisms3, 4 has led to an important breakthrough in the understanding of how autophagosomes are formed and of the part autophagy plays in cell physiology and human diseases.1, 5 To clarify how macroautophagy is modulated in response to stress situations, it is also absolutely essential to elucidate how the macroautophagy is regulated upstream of the Atg machinery.6
Macroautophagy occurs at a basal rate in most cells, where it acts as a cytoplasmic quality-control mechanism to eliminate protein aggregates and damaged organelles.1, 5 The physiological importance of basal autophagy in maintaining tissue homeostasis has recently been demonstrated in conditional brain and liver Atg knockout mouse models.7, 8, 9 These studies have demonstrated the role of autophagy in preventing the cytotoxic deposition of aggregate-prone proteins in the cytoplasm, and the contribution of autophagy to the elimination of ubiquitinated proteins that are efficient substrates for the proteasome.
On the other hand, when the supply of nutrients is limited, starvation-induced autophagy contributes to the lysosomal recycling of nutrients to maintain protein synthesis, to produce substrates for oxidation and for ATP synthesis in the mitochondria,10 and to contribute to the inhibition of apoptosis.11 In the newborn mouse, in vivo, autophagy in various tissues is required to maintain energy homeostasis and survival after the sudden interruption of the supply of nutrients by the maternal placenta.12 In tumors, autophagy has a cytoprotective role against cell death in environments in which the availability of oxygen and nutrients is poor.13 Moreover, autophagy also protects cancer cells against drug-induced apoptosis in experimental models of tumors.14 Interestingly, autophagy has been shown to protect mammary epithelial cells against anoikis (a form of cell-death triggered by the loss of interaction with the extracellular matrix) during the formation of acini in normal breast development.15 It is essential to find out whether autophagy is necessary for cancer cells to survive independently of their interaction with the extracellular matrix.
It is also interesting to note that when macroautophagy is inhibited, another form of autophagy, known as chaperone-mediated autophagy,16 a selective form of autophagy for proteins located in the cytoplasm1 is able to protect cells against some death-inducing stimuli such as reactive oxygen and UV.17 However, stimulating chaperone-mediated autophagy (CMA) is not able to compensate for the cytoprotective role of macroautophagy versus Fas/TNFα exposure. The differential effect of CMA in response to various cell death stimuli is probably due to the fact that this process cannot replace macroautophagy in eliminating damaged mitochondria in response to Fas/TNFα. In contrast, CMA actively promotes the removal of oxidized proteins in response to reactive oxygen species (ROS) and UV. This study has extended our awareness of the cross talk that exists between CMA and macroautophagy, which had previously been demonstrated for cell adaptation to starvation and to cell death stimuli.16
The protective role of autophagy against cell death is not limited to apoptosis, but also extends to necrosis. Inhibition of autophagy in apoptosis-deficient tumors sensitizes cancer cells to necrosis during shortages of nutrients and/or oxygen.13 In this context, inhibiting autophagy leads to a massive, necrosis-induced, inflammatory response that can influence tumor cell proliferation. It has recently been shown that autophagy of damaged mitochondria blocks the induction of cell death in the presence of caspase inhibitors through a glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-dependent mechanism.18 The effect of GAPDH depends on an increase in glycolysis, and on a nuclear effect that induces the expression of Atg12.
Three types of cell death have been identified: type-I cell death or apoptosis, type-II cell death or autophagic cell death, and type-III cell death, or necrosis.19 Here we will only attempt a brief description of these three forms of cell death.
Apoptosis (or type-I cell death) is characterized by the activation of a family of proteases, caspases, chromatin condensation and fragmentation, cell shrinkage, plasma membrane blebbing, and the formation of apoptotic bodies. Necrosis, or type-III cell death, is characterized by the swelling of cells, and abrupt plasma membrane rupture. Type-II cell death or autophagic cell death is defined as a cell death mechanism in which macroautophagy is the sole mechanism involved in the execution of cell death without any signature of type-I or type-III cell death. It should be noted that some other forms of cell death that do not match all the hallmarks of any of the three types originally defined are also observed in mammalian cells (entosis, oncosis, paraptosis). The complexity of the role of autophagy in cell death is revealed when cells with mixed phenotypes die, for instance when a given dying cell exhibits signs of both type-I and type-II cell death. In this situation, it is important to find out whether the cells are dying with macroautophagy or through macroautophagy. Multiple levels of dialog between macroautophagy and apoptosis have been identified.20, 21 The relationship between macroautophagy and apoptosis can be divided roughly into three categories. In the first, macroautophagy is not part of the cell death mechanism (in this situation, cells are dying with autophagy); in response to many events macroautophagy is triggered to counteract or delay cell death. In the second category, macroautophagy is part of the apoptotic program, acting either upstream of the apoptotic cascade or during the final stage of apoptosis to assist in eliminating the apoptotic corpse. In the third category, both macroautophagy and apoptosis are required in parallel pathways to contribute to cell death. In the second and third categories, cells can be described as dying through autophagy.
It is also important to consider two other criteria in the context of autophagy and cell death. First, one widely accepted concept is that excessive bulk self-digestion through macroautophagy is a cell-destructive mechanism. However, as discussed below, in mammalian cells selective forms of autophagy can lead to cell death without massive self-digestion. Second, two different forms of macroautophagy, canonical macroautophagy, and non-canonical macroautophagy can contribute to cell death. The term ‘non-canonical macroautophagy’ is used to designate a process that does not use the entire machinery of Atg proteins to form autophagosomes, which ultimately fuse with the lysosomal compartment.
An Overview of Autophagic Cell Death and Cell Death Through Autophagy
In the previous section, we discussed the cytoprotective role of macroautophagy in preventing the induction of apoptosis and necrosis. However, autophagy can also be a cell death mechanism, and autophagic cell death has been shown to occur during the development of Drosophila and Dictyostelium discoideum.22, 23 Moreover, excessive starvation-induced autophagy has been shown to have detrimental effects in Caenorhabditis elegans.24 So far there is no evidence that autophagy plays any role in cell death in mammals in vivo. However, in cultured mammalian cells a large body of evidence suggests that autophagy is involved in cell death (Table 1). This topic is under the spotlight in recent literature.20, 21
The aim of this section is not to provide an exhaustive review of the literature, but to give an overview of current knowledge about the role of autophagy during cell death. The term ‘autophagy-dependent cell death’ is used here to cover all the situations where autophagy triggers cell death either independently of apoptosis,22 or in combination with apoptosis (upstream of, alongside to, or during its final stage) to kill cells. For the sake of clarity, the term ‘apoptosis’ is used here synonymously with caspase-dependent cell death.
As discussed elsewhere,21 one must be cautious when considering the role of autophagy in cell death. Simply observing autophagic structures is not sufficient to demonstrate that autophagy is involved in cell death. In many situations, the autophagy observed in dying mammalian cells is a last desperate effort by the cells to survive a stressful situation. Moreover, the exclusive use of drugs to manipulate autophagy or apoptosis can lead to biased conclusions. 3-Methyladenine (3-MA), which inhibits the formation of autophagosomes by interfering with the activity of hVps34, also has other targets in cells, for example, the inhibition of class I phosphatidylinositol 3-phosphate kinase (PI3K), p38MAPK, C-Jun N-terminal kinase, and the mitochondrial permeability transition pore that can influence the cell death decision independently of autophagy.30 Similarly, the frequently used pan-caspase inhibitor zVAD is a potential inhibitor of the lysosomal cathepsins.31 The impairment of lysosomal function blocks the autophagic pathway, resulting in the accumulation of autophagic vacuoles and an incomplete degradative phenotype.
Autophagic Cell Death
As discussed in the preceding section, autophagic cell death is a type of cell death that occurs independently of the apoptotic machinery and of the activation of caspases. The first evidence that autophagy and Atg proteins can be responsible for cell death was reported for Bax−/−/Bak−/− MEF cells exposed to various different apoptotic inducers.26 Autophagy-dependent cell death is characterized by an increase in Beclin 1 and Atg5, whereas no rise in these Atg proteins is observed during starvation-induced autophagy. Another study32 recently reported that Bax−/−/Bak−/− cells have a greater sensitivity to cell death through autophagy than apoptosis-competent cells. Interestingly, the authors hypothesized that Bak/Bax could have an inhibitory effect on the activity of PTEN (a phosphatase and tensin homolog deleted from chromosome 10 that deactivates the PI3K/Akt/PKB pathway and so triggers autophagy),33 and that inactivating Bak/Bax consequently stimulates autophagy (Figure 1). In this context, Bak and Bax should be added to the growing list of members of the Bcl-2 family of proteins able to regulate autophagy.25, 35 Bak/Bax−/− cells are valuable tools for use in uncovering the mechanism of autophagic cell death in cultured cells. However, caution is called for in attempting to extrapolate these findings to the in vivo situation, because the double deficiency of Bax and Bak has not been identified in human tumors.

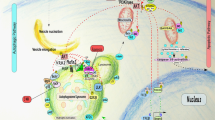
The diagram shows the common regulatory signaling elements of autophagy and apoptosis. Common cellular stress can trigger both autophagy and apoptosis by several signal transduction pathways. BNIP3, death-associated protein kinase (DAPK), and ceramide are all positive effectors of cell death by both caspase-dependent apoptosis and autophagy. PTEN is a tumor suppressor protein that activates autophagy by inhibiting PKB and subsequently mammalian target of rapamycin (mTOR) C1. In contrast, mTORC2 activates PKB and inhibits autophagy. mTOR is a constituent of two complexes: the rapamycin-sensitive mTORC1, which signals to S6K and 4EBP1 to control protein synthesis, and the rapamycin-insensitive mTORC2, which signals to FOXO3 by PKB. The inhibition of PKB upregulates the expression of autophagy genes in a Foxo3-dependent transcription mechanism. The proteins of the Bcl-2 family regulate both autophagy and apoptosis. The positive control of autophagy by Beclin 1 is impaired after association with the anti-apoptotic Bcl-2 protein. This inhibitory effect can be abolished by the BH3-only protein BAD. Bax/Bak, which are pro-apoptotic members of the Bcl-2 family, can inhibit autophagy by inhibiting PTEN. The transcription factor NF-κB inhibits autophagy and apoptosis in cancer cells. The cytoplasmic form of p53 (p53c) inhibits autophagy whereas the nuclear activity of p53 (p53n) stimulates autophagy and apoptosis by the induction of the expression of the lysosomal protein DRAM. The functional interaction of p53 with the proteins of the Bcl-2 family and its consequences on autophagy and apoptosis not indicated in the figure has been recently discussed in Tasdemir et al.34
Another seminal study in the discovery of autophagic cell death in mammalian cells was the unanticipated effect of caspase-8 blockade by pharmacological and RNAi methods.27 It was observed in several cell lines that caspase-8 inhibition triggered cell death through autophagy. U937 human cells were rescued from cell death by blocking autophagy through the knockdown of BECN1 and ATG7 expression genes. This study revealed that caspases can regulate both apoptotic and non-apoptotic cell death.
Autophagy and Apoptosis
Autophagy collaborates with the apoptotic machinery to produce cell demise in three situations: upstream of mitochondrial events and the activation of caspases 9 and 3, in parallel to the apoptotic cascade, or downstream of caspases during caspase-dependent cell death.
Autophagy upstream of apoptosis
Two examples of autophagy occurring upstream of apoptosis have been found: in CD4+T lymphocytes during human immunodeficiency virus infection36 and in human cancer cells in response to TNFα.37 When HIV envelop glycoproteins bind to their receptors, this triggers autophagy in uninfected bystander CD4T lymphocytes.36 Inhibiting the autophagy blocks apoptosis in bystander cells, whereas inhibiting caspases does not block cell death with autophagic features in bystander cells. This study shows that autophagy is required to prime apoptosis, and is also able to trigger caspase-independent cell death in the absence of caspase activation. The importance of autophagy in eliminating bystander CD4-T lymphocytes in vivo remains to be elucidated.
Dysregulation of the NF-κB pathway is associated with many human diseases, including the development and progression of cancer, viral infections and a number of diseases related to inflammation.38 When NF-κB is inhibited in cancer cells, TNFα triggers autophagy that contributes to TNFα-induced apoptotic signaling, including caspase-3 activation.37 Autophagy stimulation is dependent on the production of ROS and on the inhibition of mammalian target of rapamycin (mTOR). Whether autophagy is able to induce cell death when caspase 3 is inhibited remains to be investigated in this context. Interestingly, autophagy has been shown to regulate the activation of NF-κB by degrading activators of the canonical and non-canonical NF-κB pathways.39 Overall, the findings of these studies suggest that NF-κB plays an important part in regulating autophagy.
When autophagy and apoptosis signals converge to kill cells
DRAM (damaged-regulated autophagy modulator) is a target of p53.40 In response to stress, DRAM is expressed in a p53-dependent manner and triggers programmed cell death. DRAM is a lysosomal trans-membrane protein that controls the initiation of autophagy by an unknown mechanism. In the absence of p53, DRAM does not affect cell viability. Cell death induced by p53 in response to cell stress requires the induction of both autophagy and other pro-death signals (Bax, BH3-only proteins and mitochondrial depolarization). However DRAM does not regulate autophagy induced by p73, another member of the p53 family.41 It remains to be shown whether DRAM is involved in regulating autophagy in other circumstances, such as starvation; although this seems unlikely because starvation-induced autophagy is independent of the nuclear function of p53.34
Autophagy downstream of apoptosis
The last stage of apoptosis is the elimination of apoptotic cells by professional and nonprofessional phagocytic cells. Recently the role of autophagy in the translocation of phosphatidylserine to the outer leaflet of dying cells (the ‘eat me signal’) and in the secretion of lysophosphatidic acid (the ‘come and get me signal’) has been shown to lead to the elimination of apoptotic cells during embryo development and morphogenesis.42 Adding methylpyruvate stimulates ATP production and restores the externalization of phosphatidylserine in autophagy-defective cells, suggesting that autophagy maintains the level of ATP under these conditions.42 Interestingly sphingosine 1-phosphate, which is known to stimulate autophagy,43 is secreted by dying cells44 and has been identified as a ‘come and get me’ signal. These findings highlight the importance of autophagy in ensuring a clean death process by favoring the elimination of apoptotic bodies.
Cell Death Through Non-Canonical Autophagy
It has been shown that several drugs and chemotherapeutic agents stimulate autophagic cell death through Beclin 1.45 This is the classical or canonical autophagy pathway,1 in which Beclin 1 initiates the formation of the autophagosome by forming a multiprotein complex (Figure 2). Class III phosphatidylinositol-3-kinase or hVps34, which produces PtdIns3P, is a component of this complex.46 The roles of the PtdIns3P and Beclin 1 complex in the nucleation of the pre-autophagosomal membrane are not yet understood. Recently some evidence has emerged that suggests that autophagy and cell death can be induced independently of Beclin 1 and hVps34. In dopaminergic neuronal cells, the Parkinsonian neurotoxin 1-methyl-4-phenylpyridinium47 induced a mitochondrial injury involving extracellular signal-regulated protein kinase activation and an increase in autophagic vacuoles and cell death.47 Neither pharmacological inhibitors of PI3K nor knocking-down BECN1 gene expression reversed MPP+-induced autophagic cell death. Conversely, the inactivation of the ATG6, ATG7, and ATG8 genes did inhibit MPP+-induced autophagy. In breast cancer cells, resveratrol induces autophagic cell death in a Beclin 1-independent manner.48 The knockdown of ATG7 gene expression impairs the cell death elicited by resveratrol. Conversely, disabling BECN1 or hVPS34 gene expression, or adding the pharmacological inhibitor 3-MA does not inhibit resveratrol-induced death.

Schematic representation of canonical and non-canonical autophagy in mammalian cells. In canonical autophagy, the Beclin 1/hVps34 complex is required for the nucleation of the isolation membrane (phagophore), which initiates the formation of autophagosomes. Atg9, Atg1 complex, and WIPI-1(Atg18) proteins are also involved in the biogenesis of autophagosomes. In non-canonical autophagy, autophagosomes are formed independently of the Beclin 1/hVps34 complex. The role of other Atgs (??) and the origin of the phagophore or isolation membrane remain to be elucidated. However, the ubiquitin-like conjugation systems Atg12-Atg5 and light chain 3 (LC3)-PE are both required for autophagosome formation in both canonical and non-canonical autophagy. Whatever mechanism is responsible for autophagosome formation, fusion with the lysosomal compartment is observed to degrade the autophagic cargo. Canonical autophagy is involved in cell survival, and in both caspase-dependent and caspase-independent cell death. Non-canonical autophagy is involved in caspase-independent cell death, but whether it has any other functions remains to be investigated
However, both the canonical and non-canonical forms of autophagy are characterized by inhibition of mTOR-Raptor or mTORC1 signaling, suggesting that the Atg1 complex is involved in the formation of autophagosomes in non-canonical autophagy (see below). The role of other Atg proteins in non-canonical autophagy (Figure 2), and that of PtdIns3P, which can be formed independently of the activity of hVps34, remains to be investigated in non-canonical autophagy. Nevertheless, these findings show that at least two independent mechanisms can induce autophagic cell death and that the regulation of autophagy is more complex than hitherto believed.
Cell Death Through Selective Autophagy
There is now mounting evidence in the literature that autophagy can selectively target specific cell structures.49 Molecular events leading to the selective degradation of mitochondria, peroxisomes, and ribosomes have been identified in yeast.49 This selectivity is conserved in mammalian cells.50 Selective autophagy targeting enzymes, protein aggregates, and the endoplasmic reticulum has been also reported. In the following section, we briefly discuss the role of these different forms of selective autophagy in cell death.
Selective autophagy against proteins and protein aggregates
The elimination of cytoplasmic catalase is an example of the selective degradation of soluble proteins by macroautophagy. Catalase is a key enzyme involved in neutralizing intracellular ROS. The selective autophagic degradation of the antioxidant enzyme in zVad-treated mouse L929 cells caused a dramatic accumulation of ROS, peroxidation of lipids, loss of membrane integrity, and non-apoptotic cell death.28 However, caspase inhibition can also increase ROS production. It is possible that ROS regulating proteins may be directly modulated by caspases. However, little is known about the caspase substrates14 that control ROS regulation. In addition, when catalase levels decrease, cell death occurs even when cells have been treated with wortmannin, an inhibitor of autophagy. The molecular mechanism by which autophagy selectively degrades catalase is not yet understood, but this mechanism is an example of how autophagy, which was originally envisaged as promoting survival and cytoprotection, can in fact be a competent executor of cell death.
Inhibition of autophagy is associated with the accumulation of protein aggregates in mouse brain and liver.7, 8, 9 This accumulation is associated with the induction of cell death in various brain areas and leads to rapid neurodegeneration.7, 8 Accordingly, in models of Huntington's disease, stimulation of autophagy has been shown to protect cells against the apoptosis induced by aggregate-prone proteins.51 The protein p62/sequestrosome1 interacts with light chain 3 (LC3, the mammalian ortholog of Atg8) to recruit protein aggregates into autophagosomes.52 Genetic deletion of p62 suppresses the accumulation of ubiquitin-positive protein aggregates,53 whereas a loss of p62 markedly reduces the liver injury caused by the inhibition of autophagy, although it has little effect on neuronal cell death or neurodegeneration. This study highlights the tissue specificity of autophagy in pathological processes.53
Mitophagy and reticulophagy in cell death
The selectivity of autophagy toward mitochondria (mitophagy) and endoplasmic reticulum (reticulophagy), and its role in cell survival and cell death have been discussed in several recent reviews.20, 21, 54, 55 In hepatocytes, mitophagy can serve as a mechanism that limits the cell damage induced by the opening of the mitochondrial permeability transition pore.20 However, in other systems, mitophagy can have lethal consequences. In mammalian cells, cytotoxic stress (such as actinic damage, nutrient deprivation, actinomycin D, staurosporin, or metabolic conditions) can produce considerable mitochondrial damage leading to mitophagy.21 The excessive and uncontrolled removal of mitochondria by autophagy in response to increased stress may promote caspase activation and cell death by lysosomal/autolysosomal enzymes, due to lysosome disruption, and the release of lysosomal hydrolases, such as cathepsins.21
Reticulophagy is a process involving the selective formation of autophagosomes using the ER as a source for membranes, including their outer double membrane.55 ER stress can be induced by drugs, such as tunicamycin, thapsigargin, or brefeldin A. Reticulophagy could help to achieve homeostatic control of the cell, by preventing the accumulation of incorrectly folded proteins in the ER lumen, and reducing the distension of the ER that results from folding stress.
Recent data indicate that inhibition of ER stress-induced autophagy increases apoptosis in various cellular contexts.21, 55 It is not yet clear whether reticulophagy is essentially a mechanism intended to protect cells against the accumulation of unfolded proteins or whether it is involved in autophagy-induced cell death.
Macroautophagy and Cell Death Signaling: A Simplified View
Many recent studies have shown that autophagy and apoptosis share common regulatory elements (Figure 1; see, for recent review21). Physiological autophagy is a highly regulated mechanism that needs to be kept in a range compatible with cell survival. The mTORC1 and the Beclin 1 complexes are major regulators of autophagy.56 When the activity of these complexes is uncontrolled, this can lead to unchecked autophagy and self-destruction by the cell.25 Another aspect that is emerging is the regulation of the expression and activity of Atg proteins by post-translational modifications that may indicate that these proteins are implicated in cell death.
mTOR and the Atg1-mTOR signaling network both play a major role in the regulation of autophagy.56 In yeast, the TOR complex 1 (TORC1) occurs upstream of the Atg1 complex that plays an essential role in the elongation of pre-autophagosomal membrane during the formation of autophagosomes, and this paradigm is conserved in all eukaryotic cells. Inhibiting the TORC1 complex during nitrogen57 starvation or by the immunosuppressor rapamycin stimulates autophagy.57, 58
A recent study in Drosophila has shown that dAtg1 exercises negative feedback control on dTORC1.59 Interestingly, cells with a high level of Atg1-dependent autophagy are eliminated by apoptosis. In mammalian cells, the ULK1/2 proteins (ULKs are the mammalian homologs of the yeast Atg1) occur downstream from mTORC1 in the autophagic pathway,60, 61 and also have a negative feedback effect on mTORC1.62 We do not know whether an elevation of the expression of ULK1/2 would trigger a higher level of autophagy and cell death.
mTORC1 is characterized by its sensitivity to rapamycin, which in many settings stimulates autophagy. However, recently this view has been challenged by the observation that rapamycin inhibits autophagy in cancer cells.63 This study suggests that S6K, a direct substrate of mTORC1, may under some conditions be involved in the induction of autophagy by a feedback mechanism affecting signaling elements upstream of mTORC1. The role of S6K in the positive regulation of autophagy was originally reported in Drosophila64 and remains to be demonstrated in mammalian cells.65
The role of mTOR in regulating autophagy can also involve the mTORC2 complex, which is less sensitive to rapamycin.25 mTORC2 phosphorylates Akt/PKB, which negatively regulates the activity of the transcriptional factor FOXO3. Interestingly, FOXO3 has been shown to stimulate autophagy in muscle cells by increasing the expression of several of the proteins involved in autophagy.66
Downregulating the activity of mTORC2 stimulates autophagy in muscle cells independently of the activity of mTORC1. Overall, it can be assumed that both mTORC1 and mTORC2 are involved in the regulation of autophagy in response to various stimuli, and do so by controlling the signaling of autophagy (mTORC1), and the expression of the proteins required for the formation of autophagosomes.
The Beclin 1/Bcl-2 rheostat
Originally, Beclin 1 (the mammalian ortholog of the yeast Atg6) was discovered through its interaction with Bcl-2 and Bcl-xL using a yeast two-hybrid system.67 The functional significance of this interaction has been recently elucidated. The inhibition of autophagy by Bcl-2 has been demonstrated in an ex vivo yeast model, and in vivo in mice,25 and shown to be dependent on the Beclin 1/Bcl-2 interaction. Interestingly, this interaction is modulated by conditions known to regulate autophagy. Under nutrient-rich conditions, when autophagy is inhibited, Beclin 1 and Bcl-2 interact strongly. In contrast, during starvation, when autophagic rates are high, the interaction between Beclin 1 and Bcl-2 is weak. This suggests that Bcl-2 acts as a rheostat that turns autophagy on or off as required. The expression of Beclin 1 mutants that are unable to associate with Bcl-2 leads to a high level of autophagy and cell death. In this setting, knocking down the expression of Atg reduces autophagy and protects against cell death.
Recently, a BH3 domain was identified in the Beclin 1 sequence from amino acids 112–159.35 The BH3-only proteins BAD and BNIP3, and the pharmacological BH3 mimetic ABT737, both disrupt the Beclin 1/Bcl-2 (or Bcl-xL) complex and stimulate autophagy.35, 68
However, the role of autophagy in cell death has not been determined under these conditions. This point should be investigated in detail, because the expression of the protein BAD has been shown to be required for the induction of starvation-induced autophagy. Ceramide and short chain analogs of ceramide, which are well-known inducers of apoptosis, are also strong inducers of autophagy.69 It has been reported that the C2-ceramide analog triggers autophagic cell death through the induction of BNIP3 and BNIP3L.70 However, the role of ceramide on the Beclin 1/Bcl-2 complex was not considered in this study. Recent findings in our laboratory show that ceramide is able to induce the dissociation of the Beclin 1/Bcl-2 complex by directly affecting the association of the two proteins (Pattingre et al., in preparation). However, we cannot exclude the possibility that ceramide may induce the dissociation of the complex by BNIP3-dependent and -independent mechanisms. The regulation of the dissociation of the Beclin 1/Bcl-2 complex is probably an important aspect of keeping autophagy under control.
Reactive oxygen species and Atg4
Reactive oxygen species have been shown to regulate the induction of autophagy and its impact on cell survival and cell death. ROS control starvation-induced autophagy by regulating the activity of Atg4.71 This protein is involved in the deconjugation of LC3-II. A cysteine residue in the catalytic site of Atg4 is highly sensitive to ROS. The oxidized form of the protein is inactive toward LC3-II thus favoring the formation of autophagosomes. The induction of autophagy by ROS is also associated with the induction of autophagic cell death or with the priming of apoptosis. ROS may also trigger autophagic cell death through the selective elimination of the catalase.28 In response to TNFα, the inhibition of NF-κB is required to produce ROS to trigger autophagy upstream of apoptosis. Blockade of ROS production or of the inhibition of autophagy protects NF-κB-deficient cells from TNFα-induced apoptosis.37 It would be interesting to evaluate Atg4 activity under these circumstances.
Expression of Atg
As discussed above, the forced expression of Atg1 leads to excessive autophagy and triggers apoptosis.59 Interestingly, activation of transcription factor 1,72 which is frequently required in apoptosis, is also involved in the expression of some of the genes linked to the autophagic pathway.72 Activation of E2F1 by 4-hydroxytamoxifen upregulates the expression of LC3, Atg1, Atg5, and DRAM in U2OS cells containing an inducible E2F1. Moreover, E2F1 has been shown to be linked to the promoter of Beclin 1,73 even if the effect of E2F1 on Beclin 1 expression is still not clear. However, this finding supports the suggestion that autophagy may be regulated at the transcriptional level. An increase of Beclin 1 is frequently observed when autophagy is associated with a cell death program, but the significance of this accumulation is not clear. Beclin 1 is a BH3-only protein, but its forced expression does not trigger cell death and only one study has reported a pro-apoptotic function for Beclin 1 after its overexpression in MKN28 human gastric cancer cells treated with cis-diaminedichloroplatinum.74 The expression of Atg5 has been shown to be increased during autophagic cell death in Bax/Bak−/− MEFs.26 Recent studies have shown that the expression of some Atg proteins and proteins related to autophagy was increased during muscle wasting, that is, LC3, Atg4, hVps34, Beclin 1, Atg12, Bnip3, and Bnip3i,66 but this was not associated with the induction of cell death. Clearly, under these conditions, increased autophagy is a survival mechanism and generates amino acids for gluconeogenesis in the liver. Similarly, as discussed in the previous section, the expression of Atg12 is increased during protective autophagy against caspase-independent cell death.18 It can be assumed that an increase in some Atg proteins is required to maintain the ongoing autophagic pathway, whereas an increase in some others, such as Atg1, would lead to a loss of control of autophagy. This emerging aspect of the regulation of autophagy will probably yield important information about the impact of autophagy on cell homeostasis. One has to keep in mind that the effect on the autophagic flux of increasing a particular Atg protein is entirely dependent on the flux control coefficient of that particular Atg protein under the conditions being investigated in a particular cell type; this situation is entirely analogous to the behavior of enzymes in a metabolic pathway where control is shared among the various enzymes, and no single enzyme exercises complete control.75
Translational and post-translational modifications Atg5
The protein Atg5 is one of the Atg proteins involved in the two ubiquitin-like modification systems (the Atg12 and LC3 conjugation systems) required during the formation of autophagosomes.4 During the Atg5-Atg12 attachment, the C-terminal Gly of Atg12 forms an isopeptide bond with the ɛ-amino group of a lysine residue in Atg5. This linkage is required for the polar head of phosphatidylethanolamine, a component of the phospholipid bilayer, to attach to Atg8/LC3. The formation of the Atg5-Atg12 conjugate is constitutive, and occurs soon after the individual proteins have been synthesized. Besides its role in cytoprotective autophagy, an increase in Atg5 renders cells more sensitive to cell death inducers, and the interaction of Atg5 with FADD is probably required for the induction of caspase-dependent cell death independent of vacuolization.76 Recently, hitherto unknown cross talk was reported to occur between the autophagic machinery and apoptosis mediated by a calpain-dependent cleavage of Atg5 that is independent of its function in autophagy.77 The 24-kDa N-terminal fragment generated by calpain cleavage is translocated to the mitochondria, where its triggers the permeability of the outer mitochondrial membrane upstream of caspase activation. These new findings are conceptually important, as they demonstrate that Atg-dependent cell death is not necessarily associated with autophagosome formation.
p14ARF
The tumor suppressor protein ARF (p19ARF in mouse and p14ARF in humans) encoded by an alternative reading frame of the INK4a/ARF locus is an activator of p53.78 The tumor suppressor function of ARF is largely dependent on its capacity to antagonize the function of Mdm2, a p53 inhibitor, either by sequestrating Mdm2 in the nucleolus or by inhibiting its E3 protein ligase activity. A short form ARF, designated smARF, can be generated by internal initiation of translation.29 This short form of ARF is translocated to the mitochondria and triggers caspase-independent cell death. Cell death is attenuated when the expression of ATG5 and BECN1 is knocked down by siRNA. The stimulation of autophagy and dissipation of the mitochondrial membrane potential seem to depend on the stabilization of smARF in the mitochondrial membrane by its interaction with the p32 protein.79
Conclusions
Autophagy can be a cell death mechanism in mammalian cells either by selectively targeting key cell survival elements or as a result of excessive bulk self-digestion.25, 28 Identifying what determines selective autophagy in mammalian cells would provide important information about the impact of selective autophagy on cell survival.50 Moreover, recent studies of the regulation of Atg gene expression provide an important line of research to understanding the consequences of elevated Atg for cell survival.66, 72
More generally, one may wonder whether cell death ‘through’ signaling pathways controlling autophagy does really exist. Such a signaling pathway cannot be stimulated during starvation-induced autophagy or in other settings where autophagy is cytoprotective. To the best of our knowledge, most of the signaling pathways and second messengers (Ca2+, ceramide, cAMP, ROS) so far described as regulating autophagy are effective in both cytoprotective and death-dedicated autophagy.6, 21 This would seem to make it difficult to identify which form of signaling is dedicated to the cell death aspect of autophagy. It seems more likely that the robustness and the loss of control of autophagy signaling are probably involved in balancing the roles of autophagy in cell survival and cell death.
Regarding autophagic cell death, it is generally assumed that excessive autophagy leads to self-destruction of mammalian cells. At this point, it is interesting to consider another concept that has emerged from some ‘unusual but informative and beautiful’ organisms.80 In the slime mold, D. discoideum, Golstein and co-workers23 demonstrated that autophagic cell death is dependent on two pathways: starvation-induced autophagy and a pathway involving a differentiation factor, DIF. We do not know whether this paradigm has been conserved during evolution, but this provocative hypothesis deserves investigation in mammalian cells.
One intriguing aspect of autophagy is the increasing number of regulators now known to be shared with apoptosis.6, 21 Many of these common regulators act by anti-apoptotic and anti-autophagic pathways (e.g. Bcl-2 and NF-κB). Under certain circumstances when cells execute a program such as entering the cell cycle, the negative pressure of anti-apoptotic molecules is released to check whether cells are able to execute the cellular program correctly (Figure 3). If the program cannot be successfully executed, the incompetent cells will undergo apoptosis. What would be the implications of abolishing the inhibitory effect on autophagy under these conditions? According to this scenario, the abolition of autophagic inhibition may act as a bodyguard to protect more apoptosis-sensitized cells against adverse, external, nonprogrammed stimuli (changes in nutritional state, cytotoxic agents). If cells cannot overcome the unanticipated stress situation, then autophagy may contribute to their demise (Figure 3).

Hypothetical functional significance of the co-regulation of autophagy and apoptosis. Some molecules, such as Bcl-2 and NF-κB, are both anti-apoptotic and anti-autophagic. During the execution of a genetic program, the cell can either survive or die by apoptosis depending on its ability to execute the program correctly. During this ‘quality control’ phase, the cell's anti-apoptotic and anti-autophagic capacities are reduced. If the check shows that the cell is capable of implementing the program, the cell will survive, and in this case, autophagy contributes to inhibiting the induction of apoptosis by cleaning up the cytoplasm (Cqc: quality control of the cytoplasm). In this situation, it is assumed that the cell is not dependent on autophagy for metabolism and ATP production (Met/ATP). If the check indicates that the cell is not capable of implementing the program, then the cell will die by apoptosis. In this case, autophagy may be a simple bystander or may delay the induction of apoptosis. However, while executing the cell program without any internal errors (program check OK), the cell may receive ‘nonprogrammed’ stimuli from the extracellular milieu (UV, shortage of nutrients, drugs). In this context, autophagy may contribute to cell survival by overcoming the stress situation. However, if the stress is too severe, autophagy can turn into a cell death mechanism either by contributing to apoptosis or through autophagic cell death
However, the evidence that autophagy acts through autophagic cell death remains to be demonstrated in mammals. The major challenge for future studies is to fully elucidate the physiological role of autophagy, and its complex relationships with many human diseases.
Abbreviations
- 3-MA:
-
3-methyladenine
- ATG:
-
autophagy-related
- CMA:
-
chaperone-mediated autophagy
- DAPK:
-
death-associated protein kinaseGAPDH
- glyceraldehyde-3-phosphate dehydrogenase∣LC3:
-
light chain 3
- mTOR:
-
mammalian target of rapamycin
- PI3K:
-
phosphatidylinositol 3-phosphate kinase
- ROS:
-
reactive oxygen species
References
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451: 1069–1075.
Levine B, Klionsky DJ . Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6: 463–477.
Klionsky DJ . Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8: 931–937.
Ohsumi Y . Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2: 211–216.
Levine B, Kroemer G . Autophagy in the pathogenesis of disease. Cell 2008; 132: 27–42.
Codogno P, Meijer AJ . Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005; 12 (Suppl 2): 1509–1518.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441: 885–889.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441: 880–884.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169: 425–434.
Lum JJ, DeBerardinis RJ, Thompson CB . Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 2005; 6: 439–448.
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–1040.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432: 1032–1036.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51–64.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117: 326–336.
Fung C, Lock R, Gao S, Salas E, Debnath J . Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19: 797–806.
Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM . Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci USA 2006; 103: 5805–5810.
Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem 2008; 283: 4766–4777.
Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell 2007; 129: 983–997.
Clarke PG . Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990; 181: 195–213.
Gozuacik D, Kimchi A . Autophagy and cell death. Curr Top Dev Biol 2007; 78: 217–245.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Berry DL, Baehrecke EH . Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131: 1137–1148.
Lam D, Kosta A, Luciani MF, Golstein P . The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol Biol Cell 2008; 19: 691–700.
Kang C, You YJ, Avery L . Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 2007; 21: 2161–2171.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122: 927–939.
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6: 1221–1228.
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304: 1500–1502.
Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 2006; 103: 4952–4957.
Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell 2006; 22: 463–475.
Tolkovsky AM, Xue L, Fletcher GC, Borutaite V . Mitochondrial disappearance from cells: a clue to the role of autophagy in programmed cell death and disease? Biochimie 2002; 84: 233–240.
Wu YT, Tan HL, Huang Q, Kim YS, Pan N, Ong WY et al. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy 2008; 4: 457–466.
Kim KW, Mutter RW, Cao C, Albert JM, Freeman M, Hallahan DE et al. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J Biol Chem 2006; 281: 36883–36890.
Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem 2001; 276: 35243–35246.
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10: 676–687.
Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26: 2527–2539.
Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 2006; 116: 2161–2172.
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 2006; 281: 30373–30382.
Karin M . Nuclear factor-kappaB in cancer development and progression. Nature 2006; 441: 431–436.
Xiao G . Autophagy and NF-kappaB: fight for fate. Cytokine Growth Factor Rev 2007; 18: 233–243.
Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126: 121–134.
Crighton D, O'Prey J, Bell HS, Ryan KM . p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ 2007; 14: 1071–1079.
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128: 931–946.
Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R et al. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem 2006; 281: 8518–8527.
Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a ‘come-and-get-me’ signal. FASEB J 2008; advance online 24 March 2008.
Kondo Y, Kanzawa T, Sawaya R, Kondo S . The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5: 726–734.
Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T . Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001; 2: 330–335.
Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT . Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 2007; 170: 75–86.
Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R . Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 2008; advance online 18 April 2008.
van der Vaart A, Mari M, Reggiori F . A picky eater: exploring the mechanisms of selective autophagy in human pathologies. Traffic 2008; 9: 281–289.
Beau I, Esclatine A, Codogno P . Lost to translation: when autophagy targets mature ribosomes. Trends Cell Biol 2008; advance online 27 May 2008.
Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC . Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet 2006; 15: 1209–1216.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131–24145.
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131: 1149–1163.
Scherz-Shouval R, Elazar Z . ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 2007; 17: 422–427.
Hoyer-Hansen M, Jaattela M . Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 2007; 14: 1576–1582.
Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P . Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008; 90: 313–323.
Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ . Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 1995; 270: 2320–2326.
Noda T, Ohsumi Y . Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273: 3963–3966.
Scott RC, Juhasz G, Neufeld TP . Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol 2007; 17: 1–11.
Chan EY, Kir S, Tooze SA . siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 2007; 282: 25464–25474.
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181: 497–510.
Lee SB, Kim S, Lee J, Park J, Lee G, Kim Y et al. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep 2007; 8: 360–365.
Zeng X, Kinsella TJ . Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res 2008; 68: 2384–2390.
Scott RC, Schuldiner O, Neufeld TP . Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 2004; 7: 167–178.
Klionsky DJ, Meijer AJ, Codogno P . Autophagy and p70S6 kinase. Autophagy 2005; 1: 59–60; discussion 60–51.
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6: 458–471.
Pattingre S, Levine B . Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res 2006; 66: 2885–2888.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283: 10892–10903.
Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem 2004; 279: 18384–18391.
Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S . Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res 2004; 64: 4286–4293.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z . Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26: 1749–1760.
Polager S, Ofir M, Ginsberg D . E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; advance online 14 April 2008.
Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ . Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol Cell Biol 2001; 21: 6820–6832.
Furuya D, Tsuji N, Yagihashi A, Watanabe N . Beclin 1 augmented cis-diamminedichloroplatinum induced apoptosis via enhancing caspase-9 activity. Exp Cell Res 2005; 307: 26–40.
Fell DA, Thomas S . Physiological control of metabolic flux: the requirement for multisite modulation. Biochem J 1995; 311 (Part 1): 35–39.
Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280: 20722–20729.
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8: 1124–1132.
Sherr CJ . The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2001; 2: 731–737.
Reef S, Shifman O, Oren M, Kimchi A . The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 2007; 26: 6677–6683.
Golstein P . Cell death in unusual but informative and beautiful model organisms. Semin Cancer Biol 2007; 17: 91–93.
Acknowledgements
We apologize to authors whose work could not be cited in the paper because of space limitation. We thank Dr. Ezio Ghigo, University of Turin, for scientific support for this review. The study in P. Codogno's laboratory is supported by institutional funding from The Institut National de la Santé et de la Recherche Médicale (INSERM), grants from the Association pour la Recherche sur le Cancer and Agence Nationale de la Recherche (ANR). This study was supported by FIN Ex-60% 2007 research grants (to RG).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Kroemer
Rights and permissions
About this article
Cite this article
Scarlatti, F., Granata, R., Meijer, A. et al. Does autophagy have a license to kill mammalian cells?. Cell Death Differ 16, 12–20 (2009). https://doi.org/10.1038/cdd.2008.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.101
Keywords
This article is cited by
-
Cyclic mechanical strain with high-tensile triggers autophagy in growth plate chondrocytes
Journal of Orthopaedic Surgery and Research (2022)
-
AMP-activated protein kinase: a potential therapeutic target for triple-negative breast cancer
Breast Cancer Research (2019)
-
Adaptive responses to low doses of radiation or chemicals: their cellular and molecular mechanisms
Cellular and Molecular Life Sciences (2019)
-
TAK1 mediates convergence of cellular signals for death and survival
Apoptosis (2019)
-
Dysregulated autophagy contributes to caspase-dependent neuronal apoptosis
Cell Death & Disease (2018)
