
Abstract
The increase in body size of humans and other vertebrates requires a physiological infrastructure to provide adequate delivery of oxygen to tissues and cells to maintain oxygen homeostasis. The heart, lungs and the vasculature are all part of a highly regulated system that ensures the distribution of the precise amount of oxygen needed throughout the mammalian organism. Given its fundamental impact on physiology and pathology, it is no surprise that the response of cells to a lack of oxygen, termed hypoxia, has been the focus of many research groups worldwide for many decades now. The transcriptional complex hypoxia-inducible factor has emerged as a key regulator of the molecular hypoxic response, mediating a wide range of physiological and cellular mechanisms necessary to adapt to reduced oxygen.
Similar content being viewed by others
Molecular Biology of Hypoxia-Inducible Factor 1 (HIF-1)
Investigation of the molecular mechanisms of one of the most striking responses to hypoxia – the induction of the hematopoietic growth hormone erythropoietin (EPO) – paved the way for the first identification of a hypoxia-inducible transcription factor. When the blood oxygen content is reduced in anemia or at high altitude, the EPO production in renal interstitial fibroblasts is rapidly turned on. The up to several 100-fold induction of EPO mRNA and protein induces erythropoietic responses that directly increase blood oxygen transport. Studies of DNA–protein interactions at the 3′ enhancer of the EPO gene identified a protein complex that only bound in hypoxia. It was designated hypoxia-inducible factor 1 (HIF-1) by Semenza and Wang.1
It soon became obvious that the HIF system is a key regulator of a broad range of cellular and systemic responses to hypoxia and acts in all mammalian cells. Changes in gene expression directly or indirectly regulated by HIF extend to well over a 100 genes. HIF-mediated pathways influence metabolic adaptation, erythropoiesis, angiogenesis and vascular tone, cell growth and differentiation, survival and apoptosis, and thus are critical factors in development, physiology and disease (for review, see Maxwell et al.2).
HIF is a heterodimeric DNA-binding complex composed of two basic helix-loop-helix proteins of the PAS family (PER, AHR, ARNT and SIM family):3 the constitutive HIF-1β and one of either hypoxia-inducible α-subunits, HIF-1α or HIF-2α. In hypoxia, the α/β heterodimer binds to a core pentanucleotide sequence (RCGTG) in the hypoxia response elements (HREs) of target genes. HIF-β subunits are non-oxygen-responsive nuclear proteins that also have other roles in transcription, for example, in the xenobiotic response (for review, see Gu et al.4). In contrast, the HIF-α subunits are highly inducible by hypoxia.
Regulation of HIF-α Subunits by Protein Hydroxylation
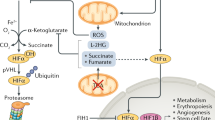
Under normoxic conditions, HIF-α subunits have a very short half-life.5 Cells continuously synthesize and degrade HIF-α protein. However, under decreasing concentrations of oxygen, the degradation of HIF-α is retarded.6 The interface between oxygen and the HIF-α subunit is provided by distinct enzymatic reactions: the hydroxylation of two prolyl residues (Pro402 and Pro564 in human HIF-1α) in the oxygen-dependent degradation domain (ODDD) of the α-subunits.7, 8 This oxygen-dependent hydroxylation regulates the interaction with the von Hippel–Lindau tumor suppressor protein (pVHL). pVHL is the recognition component of an E3 ubiquitin ligase complex that targets HIF-α for proteolysis by the ubiquitin–proteasome pathway.9, 10 Other components of the complex are elongin B, elongin C, Rbx1 and Cul2, which also participate in other E3 ubiquitin ligase complexes. Under hypoxic conditions, prolyl hydroxylation is suppressed, HIF-α protein escapes proteasomal destruction and can accumulate. It translocates to the nucleus and dimerizes with HIF-1β. The heterodimeric transactivating complex HIF then binds to the HRE in promoter or enhancer sequences of target genes (Figure 1).

Regulation of HIF-1α protein by prolyl hydroxylation and proteasomal degradation. There are three hydroxylation sites in the HIF-1α subunit: two prolyl residues in the oxygen-dependent degradation domain (ODDD) and one asparaginyl residue in the C-terminal transactivation domain (C-TAD). In the presence of oxygen, prolyl hydroxylation is catalyzed by the Fe(II)-, oxygen- and 2-oxoglutarate-dependent PHDs. The hydroxylated prolyl residues allow capture of HIF-1α by the von Hippel–Lindau protein (pVHL), leading to ubiquitination and subsequent proteasomal degradation. Asparaginyl hydroxylation is catalyzed by an enzyme termed as factor-inhibiting HIF (FIH) at a single site in the C-TAD. This hydroxylation prevents cofactor recruitment. In the absence of hydroxylation due to hypoxia or PHD inhibition, HIF-1α translocates to the nucleus, heterodimerizes with HIF-1β and binds to hypoxia-response elements (HREs) in the regulatory regions of target genes
The HIF-modifying enzymes were identified as related to egl-9 in Caenorhabditis elegans and termed prolyl hydroxylase domain (PHD) enzymes (PHD1–PHD3).11, 12 The PHDs are non-heme Fe(II)- and 2-oxoglutarate-dependent dioxygenases that split molecular oxygen. One oxygen atom is inserted into the HIF-α peptide at the prolyl residue, the other reacts with 2-oxoglutarate yielding succinate and CO2 as products. In hypoxia, the HIF prolines remain unmodified. The effects of hypoxia can be mimicked by iron chelation, use of 2-oxoglutarate analogs such as dimethyloxalylglycine or substitution of Fe(II) by metal ions such as cobalt.
The ODDD of the HIF-α subunit contains an N-terminal (N-ODDD) and a C-terminal portion (C-ODDD),13, 14 both of which can interact with pVHL independently.15 Furthermore, two transactivation domains (TADs) have been defined in HIF-1α and HIF-2α: an N-terminal TAD (which overlaps with the C-ODDD) and a C-terminal TAD.16 In contrast to regulation of HIF-α stability by proline modification in the ODDD, transcriptional activity is regulated by the hydroxylation of a C-terminal asparagine residue (Asn 803 in human HIF-1α).17 The hydroxylation reaction is carried out by an asparaginyl hydroxylase termed factor-inhibiting HIF (FIH) and this modification prevents interaction of the HIF-α C-TAD with the CH-1 domain of the transcriptional coactivator p300.18, 19 Thus, in contrast to the prolyl hydroxylation that enables protein–protein interaction, the asparaginyl hydroxylation prevents protein recruitment. FIH can also interact with pVHL,20 which fits well with the observation that an almost complete upregulation of HIF target genes can be found in cells devoid of functional pVHL.9, 21 This suggests that in the absence of pVHL both the oxygen-dependent controls of stability and activity of HIF-α are diminished and thus the functions of VHL might not be limited to those of an E3 ligase.
Additional HIF Isoforms
In keeping with the complexity of the hypoxic response, three principal isoforms of HIF-α exist (HIF-1α, HIF-2α and HIF-3α). All are encoded by distinct gene loci and further diversity is generated by alternative promoter usage and splicing patterns. HIF-1α and HIF-2α share a similar domain architecture and undergo similar proteolytic regulation; however, the tissue expression of HIF-2α seems to be more limited.22 There is mounting evidence, both in vivo from animals with targeted disruption of HIF2-α and also in vitro, that both isoforms have non-redundant functions in the regulation of gene expression. The complexity, however, is illustrated by the fact that a functional overlap between the two exists, which varies from one cell type to another. The molecular mechanisms for the target gene specificity are a matter of intense investigation and are now beginning to be understood in more detail.23, 24
HIF-3α is less closely related and its role is not yet fully understood. Interestingly, alternative splicing of HIF-3α generates an inhibitory PAS domain protein that is composed of the N-terminal basic helix-loop-helix and PAS domains but lacks TAD.25 It inhibits HIF response by forming transcriptionally inactive heterodimers with HIF-1α.
HIF-1α and Metabolic Adaptation
After the finding that the HIF system was widely operating in most cells and was not restricted to EPO regulation, critical HIF-1α-binding sites were identified in other genes encoding the glycolytic enzymes phosphoglycerate kinase-1 and lactate dehydrogenase A.26 Successive studies identified more enzymes involved in this metabolic pathway that are upregulated by hypoxia, as are glucose transporters and also enzymes of the gluconeogenesis. The metabolism of glucose to CO2 and water is oxygen dependent and very energy efficient. Glucose is transformed into pyruvate in the cytoplasm and, secondarily, pyruvate is catabolized through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) in the mitochondria. In contrast to OXPHOS, O2-deprived cells utilize the less energy-efficient metabolism of pyruvate to lactic acid, an effect described by Louis Pasteur in 1861. Interestingly, most cancer cells rely on this source of energy also in normoxia, as described by Otto Warburg et al.27 more than 80 years ago. Multiple enzymes responsible for shifting the metabolism toward anaerobic glycolysis are directly controlled by HIF-1α. Since HIF-1α overexpression is a frequent feature of many tumors, the contribution of HIF-1α to this characteristic metabolic phenotype of tumors seems very likely.
In addition, it was discovered recently that HIF-1α also has a significant influence on mitochondrial respiration. HIF-1α induces the expression of pyruvate dehydrogenase kinase (PDK), which inhibits the enzyme pyruvate dehydrogenase by phosphorylation. Thus, pyruvate is not converted into acetyl-CoA, preventing pyruvate entry into the TCA cycle.28, 29 As a consequence, the mitochondrial oxygen-consumption is downregulated and hypoxic ROS generation is attenuated. HIF-1 also fine-tunes mitochondrial respiration by changing the composition of the cytochrome oxidase complex in hypoxia: a more efficient isoform is upregulated by HIF-1, and the isoform predominant in normoxia is degraded by the HIF-1α-regulated LON protease.30 Finally, in VHL-defective renal carcinoma cell lines, mitochondrial biogenesis and metabolism are actively repressed in a HIF-1-dependent manner.30 Thus, HIF-1α modulates key metabolic pathways to optimize glucose and O2 utilization in hypoxia to generate sufficient amounts of ATP without producing excessive amounts of ROS by inhibition of the TCA cycle and mitochondrial respiration (Figure 2).

HIF-1α controls metabolic and pH-regulating pathways. Cells respond to hypoxia by HIF-1α-mediated upregulation of glucose transporters (Glut-1 and Glut-3) and enzymes of glycolysis. Conversion of pyruvate to lactic acid is facilitated by the induction of lactate dehydrogenase (LDH). HIF-1α also induces pyruvate dehydrogenase kinase-1 (PDK-1), which inhibits the conversion of pyruvate into acetyl-CoA by pyruvate dehydrogenase (PDH), thus preventing entry of pyruvate into the TCA cycle. Subunit composition of cytochrome c oxidase (COX4) is influenced by HIF-1α in hypoxia: COX4-2 is induced and COX4-1 is reciprocally reduced by induction of the protease LON that degrades COX4-1. Switching the COX subunits ensures optimal efficiency of mitochondrial respiration in hypoxia. Furthermore, pH homeostasis is maintained by induction of carbonic anhydrase IX (CAIX) and the monocarboxylate transporter MCT 4 and the Na+/H+ exchanger NHE1
Apart from controlling key glycolytic enzymes, HIF-1α is also implicated in the regulation of intracellular pH. One consequence of cytosolic glucose metabolism is an increase in intracellular lactic acid concentration. For tumor cells to survive and proliferate, it is important to extrude these acids. HIF-1α has been demonstrated to regulate at least one member of the H+/lactate co-transporter family that excretes lactic acid from the cytoplasm, the monocarboxylate transporter (MCT 4 31). In addition, H+ ions are transported out through the HIF-1α-regulated Na+/H+ exchanger NHE1.32 Furthermore, the carbonic anhydrases IX and XII are among the most highly induced HIF-1α targets: bound to the cell membrane they convert the metabolically generated CO2 into carbonic acid. The base HCO3− re-enters the cell and contributes further to intracellular alkalinization. As a consequence, the extracellular tumor microenvironment is acidotic, which has been described for many tumors and is correlated with poor prognosis in cancer patients, even though the intracellular pH in these cells is maintained at a level that allows survival and proliferation.
Taken together, the molecular mechanisms underlying the adaptation of cellular metabolism to hypoxia impact the metabolic phenotype of cancer cells, acting on the triad of increased glucose uptake, increased lactate production and decreased mitochondrial respiration.
HIF-1α and Angiogenesis
Beyond a certain size, simple diffusion of oxygen becomes inadequate to meet metabolic demands, especially for rapidly proliferating cells in embryos and in growing tumors. The three major processes involved in the formation of new blood vessels are referred to as vasculogenesis, angiogenesis and arteriogenesis. Early studies postulated that as the cell mass expands, angiogenic factors would be released,33 but the triggers for the so-called angiogenic switch, a phenomenon in which a tumor progresses from a non-angiogenic to an angiogenic phenotype, remained obscure. The hypoxic microenvironment caused by the increased oxygen consumption of hyperplasia and/or hypertrophy and the decreased oxygen delivery due to the increase in diffusion distance was assumed to contribute to the angiogenic switch. An important link between hypoxia and angiogenesis was the discovery that the expression of the potent vascular endothelial growth factor (VEGF) was induced by hypoxia.34
Angiogenesis is essential for development, wound healing, tissue or organ regeneration, but it is also part of pathological processes, such as cancer and certain retinopathies. It is an intricate multistep and temporally ordered process that involves a great number of genes, modifiers and pathways. Many of these genes are directly induced by HIF-1α, such as nitric oxide synthases, angiogenic and vascular growth factors (VEGF) and genes regulating matrix metabolism (urokinase-type plasminogen activator receptor; uPAR). Others are independently regulated by hypoxia and might be influenced by secondary mechanisms, but a central role of HIF-1α is well established: it is required for proper vascularization of the mouse embryo35 and for coordinating the complex cooperation of angiogenic growth factors. This was demonstrated in transgenic animals where the overexpression of VEGF alone led to hypervascularity with hyperpermeability in the skin,36 whereas in contrast the vessels induced by a stable HIF-1α transgene driven by the same skin-specific promoter were not leaky.37
The individual steps of angiogenesis require distinct changes to a variety of cells (e.g. endothelial cells or pericytes). Endothelial cells have to be transformed from a stable growth-arrested state to a plastic proliferative phenotype. The basement membrane has to be digested and the extracellular matrix remodeled so that the endothelial cells are able to migrate. HIF-1α signaling pathways have been demonstrated to influence factors such as uPAR, collagen prolyl-4-hydroxylases, matrix metalloproteinases (e.g. MMP-2) and tissue inhibitors of matrix metalloproteinases (TIMP-1) (Figure 3; for review, see Fukuda et al.30).

HIF-1α regulates factors involved in developmental and pathological angiogenesis. The steps of angiogenesis involve multiple gene products expressed by different cell types (e.g. endothelial cells and pericytes). A coordinated sequence of events is necessary with a tight balance of activators and inhibitors of angiogenesis. HIF-1α directly regulates genes involved in steps such as vasodilation, increased vascular permeability, extracellular matrix remodeling and proliferation (bold). Other genes (underlined) are also hypoxia-responsive; however, direct binding of HIF to regulatory regions in those genes has still to be defined
The hypoxic signaling is not restricted to mere target gene upregulation. A well-studied example of how multifaceted the hypoxic response can be is VEGF. VEGF is directly induced by HIF-1α38 and biological activity is further increased by the hypoxic upregulation of VEGF receptor-1 (VEGFR-1/Flt-1).39 VEGF mRNA stability is also increased under hypoxic conditions.40 Deletion of HIF-1α in endothelial cells disrupts an autocrine signaling loop for hypoxic induction of VEGFR-2 by VEGF signaling through VEGFR-1, which ultimately leads to impaired vascularization of xenografts.41
Despite the multitude of insights into individual molecular pathways involved in angiogenesis, such as increased migration and tube formation, which may be predicted to induce angiogenesis in vitro, these analyses in isolated systems clearly have their limitations, especially when the large scale of interconnections and complexity involved in the process of angiogenesis in vivo are considered. Genetic studies have so far not been successful to establish a simple and comprehensive model of how HIF-1α promotes tumor angiogenesis and ultimately tumor growth.42, 43 And in xenograft models, which rely more on angiogenesis than naturally occurring tumors, the extent of angiogenesis is dependent on the site of implantation of the xenografts.44
HIF-1α and Cancer
Many genes that are induced by HIF-1α are expressed at higher levels in cancer than in normal tissues, particularly angiogenic growth factors (such as VEGF) and enzymes of the glucose metabolism. As mentioned above, the hallmark of cancer metabolism is significantly influenced by HIF-1α: increased glucose uptake, lactate production and decreased respiration. HIF activation is a common feature of tumors,45, 46 is generally more pronounced in aggressive tumors47 and can be an independent predictor of poor prognosis in certain types of cancer.48 It has to be taken into account, however, that knowledge from developmental studies indicates that proliferation, hypoxia and angiogenesis are linked by physiological pathways originally designed to maintain oxygen supply.
In genetic models, HIF-1α has been identified as a positive factor for tumor growth43 and increased HIF-1α activation was also correlated with the development of a more aggressive phenotype in a model of epidermal carcinogenesis.49 Nevertheless, the mechanisms contributing to HIF activation in tumors are complex and difficult to dissect: HIF-α activity can be influenced by the hypoxic tumor microenvironment and also inactivation of tumor suppressor genes. The clearest example of the latter is loss of pVHL function with a subsequent activation of HIF-α (for review, see Ivan and Kaelin50). Recently, research on another hereditary renal cell carcinoma caused by inactivation of the TCA cycle enzyme fumarate hydratase (FH) opened an interesting link between hypoxic and metabolic signaling. The Krebs cycle intermediate fumarate inhibits 2-oxoglutarate-dependent dioxygenases such as the HIF hydroxylases. Using a genetic approach, it was demonstrated that FH deficiency can indeed upregulate HIF-α and that the animals develop renal cysts but not overt cancer.51 However, the role of HIF in causing this tumor predisposition is less clearly established. Other dioxygenases that are inhibited by rising fumarate concentrations also have long-established functions in matrix formations. Thus, precise understanding of the underlying mechanisms is far from being complete and the distinction of cause and effect remains problematic.
Other tumor suppressor genes that influence HIF-1α include p5352 and PTEN,53 which suppresses hypoxic HIF-α induction and target gene activation, possibly via modulation of AKT. In addition to the effects of tumor suppressor genes, growth factors have a positive influence on the HIF system. HIF-1α protein synthesis can be induced by many different growth factors and cytokines, such as insulin,54 insulin-like growth factors55 or PDGF.56 P42/44 mitogen-activated kinase (MAPK) has been implicated in phosphorylation of HIF-1α and activation of MAPK amplifies transcriptional response of HIF.57
The tumor microenvironment has been demonstrated to impact HIF-1α stability. Radiation can induce HIF-1α through enhanced translation of HIF-1α mRNA released from stress granules upon reoxygenation.58 This effect is dependent on free radical generation in vivo. Recently, it was reported that ionizing radiation requires NO produced from macrophages in the tumor microenvironment to stabilize HIF-1α through S-nitrosylation.59 Apart from the molecular insights about how HIF-α protein stability can be regulated, another important aspect of these studies is that certain tumors respond to therapy by activating HIF-1α.
HIF-1α activation does not only modulate significant metabolic or angiogenic pathways but has also been implicated in tumor invasiveness and metastasis. These are complex multistep processes where tumor cells have to break away from the tumor, cross the basement membrane, migrate through extracellular matrix, invade into the vessels, extravasate and proliferate at a suitable site. These steps require a coordinated expression and interaction of numerous genes. HIF-1α plays a central role, as was recently shown in a transgenic mouse model of tumor progression and metastasis: HIF-1α null tumors exhibit retarded growth and reduced pulmonary metastases.60 Multiple downstream genes might be involved in the development of this phenotype, which are under current investigation. In renal cancers, loss of the cell–cell adhesion molecule E-cadherin has been found to increase aggressiveness and invasiveness. HIF-1 appears to play a main role in mediating the downregulation of E-cadherin in the VHL-deficient background,61 however, HIF-2α also seems to be involved.62 The metastatic process is affected by several downstream hypoxia-induced genes: the chemokine-receptor CXCR4 is a direct HIF target.63 Its ligand SDF-1 is also induced by hypoxia and regulates adhesion, migration and homing of CXCR4-expressing cells; this indicates that both the receptor and its ligand play important roles in different steps of metastasis. More recently, the extracellular matrix protein lysyl oxidase was identified to be induced by hypoxia and associated with lower metastasis-free and overall survival rates.64 It regulates key steps such as invasion, migration and metastatic growth in distant organs.
Taken together, HIF-1α appears to be a highly involved factor in the development of a characteristic tumor phenotype influencing growth rate, invasiveness and metastasis. Investigation of xenograft tumors or genetically modified animals is indispensable, especially the former is influenced by the origin of the cells (e.g. renal or other), the genetic background (e.g. VHL status) or the site of implantation44 to name only a few. And it has to be taken into account that HIF-1α does not only regulate actively downstream processes but is itself influenced by the tumor microenvironment in many different ways (Figure 4).

HIF-1α in cancer. In malignant tissue, different stimuli activate HIF-1α: local hypoxia due to increased proliferation or insufficient oxygen supply, inactivation of tumor suppressors such as VHL, oncogenes, growth factors, accumulation of TCA intermediates such as fumarate or succinate and therapeutic irradiation. Together with other cell types such as macrophages these factors contribute to a tumor microenvironment that is capable of modulating the HIF response itself. These complex interactions together influence the phenotype and the behavior of the tumor in terms of progression, invasiveness or metastatic potential
HIF-1α Knockout Animals
To better understand the role of HIF-1α in vivo, knockout animals have been generated. Consistent with the central role of HIF in the hypoxic response, targeted inactivation of HIF-1α35, 65 or HIF-1β66 in the mouse leads to embryonic lethality due to abnormal vascular development. The defects in vasculature have been observed in the yolk sac as well as in the developing embryonic tissue and are associated with severe hypoxia in the HIF-1α−/− embryos. Embryos with inactivation of VHL also die in utero at about E12 due to defects in placental development.67 In contrast to the drastic effects, more subtle approaches utilizing mice with heterozygous defects or tissue-specific knockouts of HIF-1α provided fascinating insights into the physiology of HIF-1. Mice with heterozygous defects of HIF-1α have a reduced protective effect of hypoxic preconditioning in a model of cardiac ischemia,68 and a dramatic effect on carotid body neural activity and ventilatory adaptation to chronic hypoxia.69, 70 Another elegant approach is the investigation of tissue-specific loss of HIF-1α function by the use of the Cre recombinase-loxP technology. After the generation of mice carrying a loxP-flanked exon 2 of HIF-1α,43 various studies were performed to examine the consequences of the deletion of HIF-1α in different tissues. Taken together, the data demonstrate that HIF-1 is critical not only for hypoxic adaptation but also for physiological function in many cell types. Of particular interest is that HIF-1 is a key factor for the functional integrity and antimicrobial defense capacity of the immune system.71, 72 Other significant roles of HIF-1 were described in the myocardium,73 in the colonic epithelium,74 for chondrogenesis75 and for osteoblast development.76 Thus, using this genetic approach the physiology of the hypoxic response can be characterized elegantly in vivo with important implications on a wide range of different diseases.
Pharmacological Manipulation of HIF-1α
The central role of HIF-1α in physiology and pathology makes it an attractive yet intricate target for pharmacological manipulations. Inhibitors of HIF could have some potential as anticancer therapeutics, whereas activators of HIF might be useful for the treatment of ischemic disease. The potential caveats of such manipulations are obvious: the great level of complexity of the HIF system is further complicated by the multifaceted changes imposed by disease states. Thus, a highly specific targeting of the organ or tissue of interest is mandatory since a general inhibition or activation of HIF will almost certainly generate pronounced side effects. Many of the compounds inhibiting or activating HIF-α still lack isoform specificity; therefore, all manipulations will affect both isoforms depending on the targeted tissue.
HIF-1 is often detected in cancer and HIF-1-regulated genes have impact on the cancer phenotype. HIF inhibitors for cancer therapy target the HIF pathway on different levels: they decrease HIF mRNA or protein levels, inhibit DNA binding or decrease HIF-mediated transactivation (for review, see Fukuda et al.30). It is critical, however, to determine that HIF-1 is the cause for malignant predisposition in the particular tumor intended to be treated and not a marker of a physiological activation.
On the other hand, the physiological (and protective) HIF response is well documented in ischemic, hypoxic and inflammatory conditions. Different pharmacological compounds have been utilized to activate HIF, and the best studied so far are inhibitors of the prolyl-hydroxylases.77, 78
Thus, pharmacological approaches to exploit both aspects of the HIF response therapeutically are emerging from basic science laboratories. Some of them are currently under investigation in early-phase clinical trials. It will be critically important to select the right cohort of patients and the right regimen of the available substances to utilize the full potential of these compounds with the lowest possible side effects.
Conclusion
Although the understanding of the biology of the HIF system, and more precisely the biology of HIF-1α, at a molecular level has increased remarkably over the past years, the understanding of the physiological implications of the various pathways involved especially in angiogenesis and cancer has lagged behind. In contrast, the complexity of the system is reflected in sometimes unexpected results, and in some instances the mechanisms and interactions are even more intricate than originally thought: regulation of metabolic processes in hypoxia by HIF-1 go vastly beyond optimizing glucose utilization alone but influence biogenesis and function of mitochondria as well. The physiological role of HIF-1α in normoxia is more distinct than anticipated. However, the overlap of the extensive physiological responses with tumor development and progression complicates the distinction of cause and effect: tumor cells might share the physiologically linked pathways of HIF-1α activation and clearly more research is needed to determine the individual role of HIF in certain types of cancer. This is of crucial importance when the pharmacological manipulation of the HIF-pathway is intended as a therapeutical intervention.
Abbreviations
- EPO:
-
erythropoietin
- HIF:
-
hypoxia-inducible factor
- ODDD:
-
oxygen-dependent degradation domain
- pVHL:
-
von Hippel–Lindau tumor suppressor protein
References
Semenza GL, Wang GL . A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 1992; 12: 5447–5454.
Maxwell PH, Pugh CW, Ratcliffe PJ . Activation of the HIF pathway in cancer. Curr Opin Genet Dev 2001; 11: 293–299.
Wang GL, Jiang BH, Rue EA, Semenza GL . Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92: 5510–5514.
Gu YZ, Hogenesch JB, Bradfield CA . The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol 2000; 40: 519–561.
Jewell UR, Kvietikova I, Scheid A, Bauer C, Wenger RH, Gassmann M . Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J 2001; 15: 1312–1314.
Jiang BH, Semenza GL, Bauer C, Marti HH . Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol 1996; 271 (4 Pt 1): C1172–C1180.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292: 464–468.
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ et al. Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001; 292: 468–472.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399: 271–275.
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel–Lindau protein. Nat Cell Biol 2000; 2: 423–427.
Bruick RK, McKnight SL . A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001; 294: 1337–1340.
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR et al. C.elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001; 107: 43–54.
Huang LE, Gu J, Schau M, Bunn HF . Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin–proteasome pathway. Proc Natl Acad Sci USA 1998; 95: 7987–7992.
Pugh CW, O'Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ . Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J Biol Chem 1997; 272: 11205–11214.
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ . Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J 2001; 20: 5197–5206.
Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL . Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem 1997; 272: 19253–19260.
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML . Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002; 295: 858–861.
Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem 2002; 277: 26351–26355.
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK . FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 2002; 16: 1466–1471.
Mahon PC, Hirota K, Semenza GL . FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 2001; 15: 2675–2686.
Iliopoulos O, Levy AP, Jiang C, Kaelin Jr WG, Goldberg MA . Negative regulation of hypoxia-inducible genes by the von Hippel–Lindau protein. Proc Natl Acad Sci USA 1996; 93: 10595–10599.
Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J 2003; 17: 271–273.
Hu CJ, Sataur A, Wang L, Chen H, Simon MC . The N-terminal transactivation domain confers target gene specificity of hypoxia inducible factors HIF-1 alpha and HIF-2 alpha. Mol Biol Cell 2007; 18: 4528–4542.
Lau KW, Tian YM, Raval RR, Ratcliffe PJ, Pugh CW . Target gene selectivity of hypoxia-inducible factor-alpha in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer 2007; 96: 1284–1292.
Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001; 414: 550–554.
Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ . Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3′ enhancer. Proc Natl Acad Sci USA 1994; 91: 6496–6500.
Warburg O, Posener K, Negelein E . Ueber den Stoffwechsel der Tumoren. Biochem Z 1924; 152: 319–344.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV . HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3: 177–185.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC . HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3: 187–197.
Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL . HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007; 129: 111–122.
Ullah MS, Davies AJ, Halestrap AP . The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J Biol Chem 2006; 281: 9030–9037.
Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL . HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 2006; 291: L941–L949.
Folkman J, Merler E, Abernathy C, Williams G . Isolation of a tumor factor responsible for angiogenesis. J Exp Med 1971; 133: 275–288.
Shweiki D, Itin A, Soffer D, Keshet E . Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992; 359: 843–845.
Ryan HE, Lo J, Johnson RS . HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 1998; 17: 3005–3015.
Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999; 286: 2511–2514.
Elson DA, Thurston G, Huang LE, Ginzinger DG, McDonald DM, Johnson RS et al. Induction of hypervascularity without leakage or inflammation in transgenic mice overexpressing hypoxia-inducible factor-1alpha. Genes Dev 2001; 15: 2520–2532.
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 1996; 16: 4604–4613.
Gerber HP, Condorelli F, Park J, Ferrara N . Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem 1997; 272: 23659–23667.
Levy NS, Chung S, Furneaux H, Levy AP . Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem 1998; 273: 6417–6423.
Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP et al. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004; 6: 485–495.
Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998; 394: 485–490.
Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res 2000; 60: 4010–4015.
Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 2003; 4: 133–146.
Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 2000; 157: 411–421.
Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 1999; 59: 5830–5835.
Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL . Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer 2000; 88: 2606–2618.
Birner P, Schindl M, Obermair A, Plank C, Breitenecker G, Oberhuber G . Overexpression of hypoxia-inducible factor 1alpha is a marker for an unfavorable prognosis in early-stage invasive cervical cancer. Cancer Res 2000; 60: 4693–4696.
Elson DA, Ryan HE, Snow JW, Johnson R, Arbeit JM . Coordinate up-regulation of hypoxia inducible factor (HIF)-1alpha and HIF-1 target genes during multi-stage epidermal carcinogenesis and wound healing. Cancer Res 2000; 60: 6189–6195.
Ivan M, Kaelin Jr WG . The von Hippel–Lindau tumor suppressor protein. Curr Opin Genet Dev 2001; 11: 27–34.
Pollard PJ, Spencer-Dene B, Shukla D, Howarth K, Nye E, El-Bahrawy M et al. Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell 2007; 11: 311–319.
An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM . Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature 1998; 392: 405–408.
Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev 2000; 14: 391–396.
Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B . Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J 1998; 17: 5085–5094.
Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL . Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res 1999; 59: 3915–3918.
Richard DE, Berra E, Pouyssegur J . Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J Biol Chem 2000; 275: 26765–26771.
Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J . p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 1999; 274: 32631–32637.
Moeller BJ, Cao Y, Li CY, Dewhirst MW . Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004; 5: 429–441.
Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q et al. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell 2007; 26: 63–74.
Liao D, Corle C, Seagroves TN, Johnson RS . Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res 2007; 67: 563–572.
Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel–Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res 2006; 66: 2725–2731.
Esteban MA, Tran MG, Harten SK, Hill P, Castellanos MC, Chandra A et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res 2006; 66: 3567–3575.
Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W . Chemokine receptor CXCR4 downregulated by von Hippel–Lindau tumour suppressor pVHL. Nature 2003; 425: 307–311.
Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006; 440: 1222–1226.
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 1998; 12: 149–162.
Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC . Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature 1997; 386: 403–407.
Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci USA 1997; 94: 9102–9107.
Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H et al. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia–reperfusion injury. Circulation 2003; 108: 79–85.
Kline DD, Peng YJ, Manalo DJ, Semenza GL, Prabhakar NR . Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc Natl Acad Sci USA 2002; 99: 821–826.
Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 1999; 103: 691–696.
Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003; 112: 645–657.
Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V . Cutting edge: essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol 2007; 178: 7516–7519.
Huang Y, Hickey RP, Yeh JL, Liu D, Dadak A, Young LH et al. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J 2004; 18: 1138–1140.
Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH . Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 2004; 114: 1098–1106.
Provot S, Zinyk D, Gunes Y, Kathri R, Le Q, Kronenberg HM et al. Hif-1alpha regulates differentiation of limb bud mesenchyme and joint development. J Cell Biol 2007; 177: 451–464.
Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest 2007; 117: 1616–1626.
Warnecke C, Griethe W, Weidemann A, Jurgensen JS, Willam C, Bachmann S et al. Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB J 2003; 17: 1186–1188.
Kim WY, Safran M, Buckley MR, Ebert BL, Glickman J, Bosenberg M et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J 2006; 25: 4650–4662.
Acknowledgements
AW was supported by a scholarship of the German Research Foundation (DFG WE4275/1-1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by NS Chandel
Rights and permissions
About this article
Cite this article
Weidemann, A., Johnson, R. Biology of HIF-1α. Cell Death Differ 15, 621–627 (2008). https://doi.org/10.1038/cdd.2008.12
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.12
Keywords
This article is cited by
-
Regulation of genes involved in the metabolic adaptation of murine microglial cells in response to elevated HIF-1α mediated activation
Immunogenetics (2024)
-
Analogies between the periphery of cancer and the leading edge of pulmonary fibrosis
Journal of Translational Medicine (2023)
-
Methylglyoxal, a highly reactive dicarbonyl compound, as a threat for blood brain barrier integrity
Fluids and Barriers of the CNS (2023)
-
Pharmacological inhibition of neuropeptide Y receptors Y1 and Y5 reduces hypoxic breast cancer migration, proliferation, and signaling
BMC Cancer (2023)
-
A microfluidic-based PDAC organoid system reveals the impact of hypoxia in response to treatment
Cell Death Discovery (2023)
