
Abstract
Autophagy is a homoeostatic process necessary for the clearance of damaged or superfluous proteins and organelles. The recycling of intracellular constituents also provides energy during periods of metabolic stress, thereby contributing to cell viability. In addition, disruption of autophagic machinery interferes with embryonic development in several species, although the underlying cellular processes affected remain unclear. Here, we investigate the role of autophagy during the early stages of chick retina development, when the retinal neuroepithelium proliferates and starts to generate the first neurons, the retinal ganglion cells. These two developmental processes are accompanied by programmed cell death. Upon treatment with the autophagic inhibitor 3-methyladenine, retinas accumulated numerous TdT-mediated dUTP nick-end labelling-positive cells that correlated with a lack of the ‘eat-me’ signal phosphatidylserine (PS). In consequence, neighbouring cells did not engulf apoptotic bodies and they persisted as individual cell corpses, a phenotype that was also observed after blockade of phagocytosis with phospho-L-Serine. Supplying the retinas with methylpyruvate, a cell-permeable substrate for ATP production, restored ATP levels and the presentation of PS at the cell surface. Hence, engulfment and lysosomal degradation of apoptotic bodies were also re-established. Together, these data point to a novel role for the autophagic machinery during the development of the central nervous system.
Similar content being viewed by others
Main
Autophagy is a degradative pathway by which cells break down and recycle cytoplasmic components, such as long-lived proteins and damaged organelles and proteins. Thus, this process helps prevent the accumulation of deleterious products, as well as providing a supply of energy and amino acids.1, 2 The recent identification of regulatory molecules implicated in autophagy, the Atg proteins, has led to a resurgence of interest in this process.3 The Atg proteins were first discovered in yeast and the subsequent identification of orthologues in all the eukaryotes studied to date indicates that autophagy has been conserved throughout evolution. Besides its role in maintaining cell homoeostasis, autophagy fulfils additional roles in multicellular organisms. Indeed, the mutation of Atg proteins in Drosophila, Arabidopsis, and Caenorhabditis produces developmental defects.1, 4 In mammals, the loss of Beclin (the Atg6 orthologue) produces lethality at embryonic day 7.5 (E7.5) due to a failure in visceral endoderm formation.5 Recently, Ambra-1 was identified as a novel Beclin-interacting protein that is expressed only in vertebrates and that regulates autophagy by forming a multiprotein complex with Beclin and Vps34.6 The embryos of mice with Ambra-1 disruption die at E14.5 and display defects in neural tube closure, as well as increased proliferation and cell death.6 Thus, Ambra-1 seems to play a role in controlling cell proliferation and promoting cell survival during the development of the nervous system. In addition, knockout mice for Atg5 also display increased cell death in the retina at E18.57 and hence, it is clear that deficiencies in autophagy notably affect nervous system development.
In addition, autophagy has long been considered a type of programmed cell death, initially described associated with embryonic development in several species.8 In the nervous system, programmed cell death occurs from earliest embryonic stages up to adulthood, both under physiological and pathological conditions.9, 10 However, the links between autophagy and other types of programmed cell death, such as apoptosis, remain unclear.2, 11, 12 Hereby, we have used the embryonic chick retina, a well-characterised model of neural development and cell death, to study the role of autophagy in a developing multicellular system.13 In the chick retinal neuroepithelium at E4, the retinal ganglion cells (RGCs) are the first neuronal cell types to differentiate, following a centroperipheral gradient.14 This differentiation coexists with the intense proliferation of neuroepithelial cells and with cell death in the central part of the retina.15, 16 Thus, the early embryonic chick retina provides a good model system to characterise the possible relationship between autophagy and developmental processes such as proliferation, neural differentiation, and cell death. Furthermore, the neuroretina can be manipulated in organotypic cultures, conditions under which all these developmental processes are faithfully reproduced.15, 17 This permits short-term pharmacological manipulations to be used in an attempt to define the hierarchy and interconnections between the ongoing processes. We found that in association with the cell death that occurs during the development of the retina, inhibition of autophagy with 3-methyladenine (3-MA) induces the accumulation of apoptotic bodies in the neuroepithelium, as visualised by TdT-mediated dUTP nick-end labelling (TUNEL). This accumulation of apoptotic cell bodies correlates with a reduction in ATP levels necessary for exposure of phosphatidylserine (PS) on the membrane of apoptotic cells, so as to induce their engulfment and lysosomal degradation by neighbouring cells. The inhibition of autophagy can be bypassed by supplying the retina with methylpyruvate (MP), a cell-permeable substrate that increases ATP production. In the presence of MP, the presentation of PS is restored, as is cell engulfment and the degradation of cell corpses. Thus, during neurogenesis, the authophagic machinery provides the retina with the energy required for proper cell corpse removal and further degradation of apoptotic cells.
Results
The autophagic machinery is present in the chick retina
To characterise the possible role of autophagy in retinal development, we first studied whether the autophagic machinery was indeed present and active in the E4 chick retina. RT-PCR analysis identified transcripts of several genes involved in autophagy, namely, atg5, beclin, lc3a, and lc3b (Figure 1A). In addition, cytosolic punctate immunostaining for Beclin and Ambra-1 could be seen in the retina at this age (Figure 1B) and both partially colocalise (Figure 1B, panel d), as it has been recently shown.6 Accordingly, the E4 chick retina was able to respond to an autophagic stimulus. Amino-acid deprivation for 6 h in organotypic culture induced the conversion of LC3-I to the autophagosomal form LC3-II (Figure 1C). Moreover, exposure to 10 mM 3-MA, a classical inhibitor of autophagy, blocked this conversion. In conjunction, these data indicate that autophagy is active in the E4 chick retina and that it can be inhibited by exposure to 3-MA.

The autophagic machinery is active in E4 chick retina and can be inhibited by 3-MA. (A) Autophagy genes are expressed in the retina. Representative RT-PCR of a pool of E4 chick retinas demonstrating the expression of atg5, beclin-1, lc3a, lc3b, and gapdh as a control. (B) Beclin-1 (a) and Ambra-1 (b) immunostaining in the E4 chick retinal neuroepithelium, merged in (c). Scale bar, 20 μm. (d) Detail of neuroepithelial cells stained with Beclin-1 (green) and Ambra-1 (red). Scale bar, 5 μm. (C) Exposure to 3-MA inhibits autophagy in E4 retinas. E4 retinas were cultured for 6 h in EBSS or EBSS plus 10 mM 3-MA, and the LC3-I and LC3-II forms were identified in extracts by immunoblotting
3-MA treatment increases the number of TUNEL-positive cells
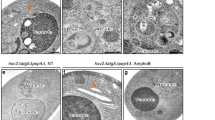
The potential role of autophagy in retinal development was studied in organotypic culture using a short-term pharmacological approach. E4 chick retinas were cultured for 6 h in the presence or absence of 3-MA and subsequently, the retinas were flat-mounted and apoptosis was visualised by TUNEL. As described previously,15, 16 TUNEL-positive cells were found throughout the E4 chick retina (Figure 2A, panel a). Exposure to 3-MA markedly increased the number of TUNEL-positive cells that were clustered in a well-defined area of the dorsotemporal retina (Figure 2A, panel b). We have previously demonstrated that this area corresponds to the region where prominent cell death is associated with neural differentiation.16 Higher magnification revealed the presence of unfragmented TUNEL-stained nuclei that were bigger than the apoptotic nuclei found in the untreated retina (Figure 2A, panels c and d). Hence, the cell morphology after 3-MA treatment was further characterised by ultrastructural electron microscopy of retinal sections (Figure 2B). In the 3-MA-treated retinas, there were numerous neuroepithelial cells that manifested apoptotic morphologies typical of different stages of the cell death process. Some cells displayed mild chromatin condensation at the periphery of the nucleus with an almost normal cytoplasm (Figure 2B, panel b, asterisks). Other dead cells had a reduced and condensed cytoplasm and highly condensed nuclei, whereas highly electron-dense cells with small nuclei and empty vesicles in the cytosol were also observed (Figure 2B, panel d). In the untreated retina, apoptotic cells were less frequent and presented highly condensed nuclei with slightly degraded cytosol (Figure 2B, panel c).

Exposure to 3-MA induces the accumulation of cell corpses in the E4 chick retina. (A) E4 retinas were cultured for 6 h in control medium (a and c) or in the presence of 10 mM 3-MA (b and d), whole-mounted, fixed, and stained for TUNEL followed by confocal microscopy. Perspective of the whole retina (a and b). Dotted lines represent the limits of the retina and the asterisks mark the optic nerve head. Scale bar, 300 μm. 3-MA exposure induces the accumulation of unfragmented TUNEL-stained nuclei (d) in comparison with untreated retinas (c). Scale bar, 20 μm. (B) Representative sections of the untreated (a and c) and 3-MA-treated (b and d) retinas visualised by electron microscopy. The asterisks (b) mark cells with perinuclear chromatin condensation and the arrows point to highly electrodense apoptotic cells. Photomicrograph of a representative dead cell in an untreated retina (c) and of several apoptotic cells in a 3-MA-treated retina (d). Scale bar, 20 μm (a and b) and 5 μm (c and d)
Treatment with 3-MA blocks phosphatidylserine exposure, engulfment, and lysosomal degradation of apoptotic cells
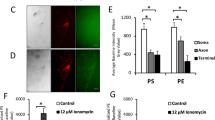
During apoptosis, PS is transferred to the outer leaflet of the plasma membrane of dying cells, where it is recognised by professional phagocytes or neighbouring cells that engulf the apoptotic bodies and degrade them in lysosomes.18, 19, 20 To assess whether the increase in TUNEL-labelled cells detected in the presence of 3-MA in our model system was due to the failure to present PS, we stained retinas with Annexin-V during the last half an hour of the culture, before performing the TUNEL reaction. Although the untreated retina presented a similar level of Annexin-V-labelled and TUNEL-positive cells (Figure 3A, panels a–c), exposure to 3-MA reduced the overall intensity of Annexin-V staining and prevented the appearance of Annexin-V-labelled cells in the dorsotemporal region of the retina, in striking contrast with the observed accumulation of TUNEL-positive cells (Figure 3A, panels d–f). The staining of Annexin-V at the periphery of the cells was compatible with the presentation of PS in the outer leaflet of the plasma membrane (Figure 3B). This extreme was confirmed by double in vivo labelling with Annexin-V and propidium iodide (PI), to check membrane integrity (Supplementary Figure 1). The large majority of Annexin-V-positive cells were not permeable to PI, indicating that they were exposing PS in the outer leaflet of the plasma membrane. Quantitative analysis confirmed that the increase in TUNEL-positive cells, when autophagy was inhibited, was completely dissociated from the presence of Annexin-V-labelled cells (Figure 3C and D). Thus, the increase in TUNEL-labelled cells appears to be a consequence of dying cells failing to present PS. The exposure of PS enables dead cells to be recognised and then engulfed by neighbouring cells.18, 19, 20 Engulfment was visualised in control retinas, where pyknotic TUNEL-positive nuclei were surrounded by F-actin from a neighbouring cell (Figure 4A, panel a).21 Conversely, relatively unfragmented TUNEL-stained nuclei in the area of prominent cell death induced by 3-MA were never seen to be surrounded by F-actin (Figure 4A, panel b). Similarly, although dead cells engulfed by neighbouring cells were often observed by electron microscopy in the control retinas (Figure 4B, panel a), we were unable to find a single dead cell engulfed by a neighbouring cell in the retinas exposed to 3-MA (Figure 4B, panel b).

Treatment with 3-MA blocks Annexin-V exposure. (A) E4 retinas were cultured for 6 h in the absence (a–c) or presence of 3-MA (d–f), and in presence of Annexin-V-biotin during the last half an hour in culture. The retinas were flat-mounted, fixed, and double stained for TUNEL (green, a and d) and avidin-Alexa 546 (red, b and e), followed by confocal microscopy. Merged images are shown in (c) and (f). The dorsotemporal part of the retina is shown. Scale bar, 150 μm. (B) Representative staining for Annexin-V (red), TUNEL (green), and DAPI (blue) in the E4 chick retina. Scale bar, 30 μm. (C and D) Quantification of TUNEL and Annexin-V-positive cell density in E4 retinas as those shown in (A) (C, untreated; 3-MA, 3-MA-treated). Quantification of maximal projection was performed on confocal series under a × 40 objective

Exposure to 3-MA prevents the engulfment of dead cells. (A) E4 retinas were cultured for 6 h in the absence (a) or presence of 3-MA (b), flat-mounted and fixed. The retinas were stained for TUNEL (green), Rhodamine-conjugated Phalloidin to visualise F-actin (red), and DAPI (blue). The retinas were analysed by confocal microscopy and a series of three sections from 0.5 μm confocal planes is shown. Note the apoptotic nuclei (a) surrounded by a ring of F-actin from an engulfing cell in the middle panel (e). Scale bar, 5 μm. (B) Electron microscopy photomicrograph of a representative apoptotic cell inside another cell in an untreated retina (a) and a representative individual dead cell in a 3-MA-treated retina (b). Scale bar, 5 μm
After engulfment, apoptotic bodies are degraded inside lysosomes, which can be visualised using acid lysosomotropic probes such as lysotracker (LTR).19, 22 Apoptotic bodies labelled by both LTR and TUNEL were present in control retinas confirming that lysosomes were associated with the engulfed apoptotic bodies (Figure 5A, panels a–c). In agreement with our previous observations, in the dorsotemporal region where cell death accumulates in the retinas exposed to 3-MA, we were unable to detect TUNEL-labelled apoptotic bodies in acidic organelles (Figure 5A, panels d–f). Quantitative analysis demonstrated an increase in the density of TUNEL-positive cells in the 3-MA-treated retinas (Figure 5B), again dissociated from the presence of LTR-positive cells (Figure 5C). Indeed, although 80% of the TUNEL-positive cells were also positive for LTR staining in untreated retinas, barely 5% of the TUNEL-positive cells were associated with LTR staining in retinas exposed to 3-MA in the dorsotemporal area of the retina (Figure 5D). To confirm that lysosomes were present in 3-MA-treated retinas and that the observed LTR labelling in untreated retinas was associated to lysosomes, we performed double staining for the chick lysosomal protein LEP-100 in combination with TUNEL or LTR. LEP-100 immunostaining was more sensitive than LTR to visualise lysosomes and revealed small lysosome spots in all cases (Supplementary Figures 2a and b; panels a and d), as well as lysosomes associated to TUNEL-positive pyknotic bodies in the untreated retinas (Supplementary Figure 2a, panels a–c). Confirming our previous observations, LEP-100 immunostaining was not associated to TUNEL-positive pyknotic bodies in 3-MA-treated retinas (Supplementary Figure 2a, panels d–f).

Exposure to 3-MA prevents degradation of dead cells inside lyosomes. (A) E4 retinas cultured for 6 h in the absence (a–c) or presence of 3-MA (d–f) were incubated with 1 μM LTR for the last 15 min in culture, flat-mounted, fixed, stained for TUNEL, and visualised by confocal microscopy. In control retinas, LTR (red, b) colocalised with TUNEL (green, a; merge in yellow, c). A representative field in the dorsotemporal part of the retina is shown. Scale bar, 20 μm. (B–D) Quantification of the density of TUNEL-positive (B), LTR-positive (C), and double-labelled cells (D) in E4 retinas as those shown in (A)
To check whether cultured E4 retinas were able to respond normally to a pro-apoptotic stimulus, we deprived the culture medium of insulin, a physiological survival signal at these stages of retinal development in the chick.15, 16 As previously reported, growth factor deprivation over 6 h augmented TUNEL labelling, which in this case was accompanied by increased Annexin-V and LTR staining (Figure 6). Thus, the accumulation of TUNEL-positive, Annexin-V-negative, and LTR-negative dead cells appears as a selective lack of phagocytosis induced by 3-MA treatment.

E4 retinas respond to apoptotic stimuli by increasing TUNEL, Annexin-V, and LTR staining. (A) E4 retinas were cultured for 6 h in the presence (a, c, e, g) and absence of insulin (b, d, f, h), and with Annexin-V-biotin and LTR (red) during the last half an hour of culture. The retinas were then flat-mounted, fixed, and double stained for TUNEL (green) and avidin-Alexa 546 (cyan) before confocal microscopy analysis. Merged images of Annexin-V, TUNEL, and LTR are also shown. Scale bar, 50 μm. Quantification of TUNEL, Annexin-V, LTR-positive cell densities (B–D) and percentage of double-stained TUNEL and LTR (E) as those shown in (A) (C, untreated retinas; −GF, retinas cultured without insulin)
Methylpyruvate restores ATP levels, PS presentation, engulfment, and degradation after inhibiting autophagy
It has recently been demonstrated that autophagy provides energy for PS presentation as an external source of ATP bypasses the inhibition of autophagy.7 We first checked whether inhibiting autophagy affected the ATP levels in the E4 chick retinal neuroepithelium. When compared to untreated retinas, there was a moderate but significant reduction in ATP levels in individual retinas exposed to 3-MA (Figure 7A). Certainly, autophagy is neither the single nor the major metabolic pathway to provide ATP. We further explored the possibility that the restricted availability of ATP may be the cause of our observations by supplying the cultured retinas with MP, a cell-permeable substrate for ATP production by mitochondria. As expected, MP restored the levels of ATP after exposure to 3-MA (Figure 7A) and, remarkably, it completely abolished the accumulation of TUNEL-positive cells upon exposure to 3-MA (Figure 7B and C, panel d). Moreover, MP supplementation restored the engulfment of apoptotic cells after the inhibition of autophagy (Figure 7D). Altogether, these results indicate that an external source of ATP overcomes the inhibition of autophagy, permitting correct PS exposure and engulfment of apoptotic bodies.

MP restores ATP level, TUNEL-staining pattern, and engulfment after exposure to 3-MA. (A) MP restores ATP level after 3-MA treatment. E4 retinas were cultured for 6 h in the presence or absence of 10 mM MP, 10 mM 3-MA, or both, and then the levels of ATP were determined. Bars represent the mean±S.E.M. of at least 10 retinas. *P<0.05 versus all other treatments. (B) An external source of ATP prevented the accumulation of TUNEL-positive cells. E4 retinas were cultured as above, dissociated, fixed, and stained for TUNEL. The percentage of TUNEL-positive cells was determined by counting at least 400 cells in six different fields. *P<0.05 versus all other treatments. (C) E4 retinas were cultured for 6 h without treatment (a), in the presence of 3-MA (b and d), or in the presence of 10 mM MP (c and d). Subsequently, the retinas were flat-mounted, fixed, and stained for TUNEL, and analysed by confocal microscopy. Perspective of the whole retina is shown. Dotted lines represent the limits of the retina and the asterisks mark the optic nerve head. Scale bar, 300 μm. (D) MP restores the engulfment of dead cells after 3-MA treatment. Electron microscopy photomicrograph of a representative individual dead cell in a 3-MA-treated retina (a), and a representative apoptotic cell inside a neighbouring cell in a retina treated with 10 mM 3-MA supplemented with 10 mM MP (b). Scale bar, 2 μm
TUNEL-positive cells accumulate after interference with phagocytosis
To further confirm that inhibition of phagocytosis causes accumulation of apoptotic cells, we tested whether a different type of inhibition would result in accumulation of TUNEL-positive cells as that observed with 3-MA. Phospho-L-Serine (P-L-Ser) is a PS mimetic molecule that binds to PS receptor thereby blocking the recognition sites in the engulfing cell and preventing phagocytosis.23 Incubating the retinas with 20 mM P-L-Ser induced an increase in TUNEL-positive cells (Figure 8A). Moreover, after phagocytosis inhibition with P-L-Ser, nuclear morphologies of condensed unfragmented nuclei were also found (Figure 8A, panel c) similar to the morphologies observed after exposure to 3-MA (Figure 2A, panel d and Figure 8A, panel b). The levels of Annexin-V were unchanged after phagocytosis blockage (Figure 8A, panel f), and as expected, LTR co-staining was completely absent from the treated retinas (Figure 8A, panel i). Quantitative determinations confirmed the significant increase in TUNEL staining (Figure 8B), a decrease in LTR staining (Figure 8C), and a reduction in the colocalisation of TUNEL and LTR staining (Figure 8D). In conclusion, blockage of PS receptor abolished phagocytosis and induced an accumulation of cell corpses in the retinal neuroepithelium, an observation that parallels 3-MA treatment. Thus, the accumulation of cell corpses after 3-MA exposure appears to be related to phagocytosis blockage and not to an increase in overall cell death.

PS receptor blockade by P-L-Ser in E4 retinas increases the number of TUNEL-positive apoptotic cells in the dorsotemporal region and inhibits LTR staining. (A) E4 retinas were cultured for 6 h without treatment (a, d, g, j), in the presence of 10 mM 3-MA (b, e, h, k) and in the presence of 20 mM P-L-Ser (c, f, i, l), incubated with Annexin-V-biotin and LTR (red) during the last half an hour of culture, flat-mounted, fixed, and double stained for TUNEL (green) and avidin-Alexa 546 (cyan) followed by confocal microscopy analysis. Merge staining is shown (j–l). Scale bar, 25 μm. (B–D) Quantification of the density of TUNEL-positive (B), LTR-positive (C) and double-labelled cells (D) in E4 retinas as those shown in (A)
Discussion
Our data demonstrate that the removal of dying cells at early stages of chick retinal development is deeply affected by inhibition of the autophagic machinery. Exposure to 3-MA inhibits PS presentation, engulfment and degradation inside lysosomes, and as a consequence, induces the accumulation of TUNEL-positive apoptotic bodies in these retinas. All these effects appear to be the result of diminished ATP levels, as they are reverted when an external source of ATP is available.
The E4 chick retina expresses several autophagic genes and is able to display an autophagic response to amino-acid starvation. Remarkably, the observed increase in the lipidation of LC3 to form LC3II associated to autophagy was prevented by 3-MA. These results provide the first molecular evidence that autophagy is functional at early stages of chick neural development, as has been found in other species.1
The dramatic increase in TUNEL-positive cells after 3-MA treatment occurs selectively in the dorsotemporal region of the retina and was accompanied by reduced fragmentation of the TUNEL-labelled nuclei. This observation that may reflect a blockage in the apoptotic process was confirmed by ultrastructural analysis of the 3-MA-treated retinas, which revealed many different, distinct phenotypes of dead cells, which are all compatible with an arrest in the culmination of the apoptotic process. Interestingly, a similar phenotype of cell corpse accumulation was observed in the retinas after blocking the PS receptor in the engulfing cell with P-L-Ser. Both 3-MA exposure and phagocytosis blockage with P-L-Ser induce a fully comparable accumulation of cell corpses during retinal neurogenesis, discarding the toxic effect of 3-MA treatment.
Our results concur with the recent observation that inhibition of the autophagic machinery blocks the expression of the eat-me signals and prevents the elimination of dead cells during embryoid body formation.7 The retina shows a highly structured three-dimensional cytoarchitecture and, similar to embryoid bodies, autophagy-dependent clearance of apoptotic cells seems to be a feature unique to three-dimensional tissues. Similarly, cell corpse accumulation is observed when autophagy is inhibited during the development of the chick otic vesicle, another three-dimensional structure (unpublished observations in collaboration with I. Varela-Nieto). Conversely, no accumulation of corpses was observed after inhibiting autophagy in cultures of dissociated retinal neuroblasts (our unpublished observations). Beyond these parallel findings, our short-term treatment approach also allows for establishing a hierarchy of biochemical and cellular processes because compensatory mechanisms, often found in mouse knockout and long-term studies, have little time to occur.
In the developing E4 chick retina, exposure to 3-MA significantly reduced the levels of ATP. MP is a cell-permeable analogue of pyruvate that serves as an alternative substrate for mitochondrial ATP generation24 and it has previously been used to restore ATP levels after autophagy inhibition.7, 25 In the retina, MP restored ATP levels in the presence of 3-MA, reduced the number of TUNEL-positive cells to basal numbers, and re-established Annexin-V and LTR staining (data not shown). Ultimately, engulfed apoptotic cells were often found in the 3-MA-treated retinas after supplementation with MP, indicating that ATP deficiency is indeed the primary effect of inhibiting autophagy, and that all the other effects appear as a consequence of ATP insufficiency. An interesting question that deserves further studies is whether a certain level of basal autophagy is taking place in all retinal cells or autophagy is active only in cells undergoing PCD.
Mammalian cells have an asymmetric distribution of phospholipids such that most of the PS is located in the inner leaflet of the plasma membrane bilayer. This asymmetry is maintained by the activity of specific lipid transporters such as the ATP-binding cassette (ABC) transporter family of proteins.26 These transporters use the energy from ATP to translocate specific substrates across the membrane.26 ABC1 and ABC7, structural orthologues of the Caenorhabditis elegans ced-7, are necessary for the proper engulfment of corpses during programmed cell death.27, 28, 29 ABC7 knockout mice are embryonically lethal and ABC1 null mice accumulate TUNEL-positive cells in areas of prominent cell death (e.g., the interdigital tissue of the developing limb), which frequently remain isolated and unengulfed. These phenotypes closely resemble our observations in the chick neuroretina when the autophagic machinery is inhibited. Hence, it is tempting to speculate that the decrease in ATP may affect normal function of the ABC transporters during cell death associated with retinal neurogenesis.
The 3-MA effects are exquisitely restricted to the dorsotemporal area of the E4 chick retina. This regional selectivity supports the specific effect of 3-MA treatment on cell death that occurs during retinal neurogenesis. In the developing chick retina, programmed cell death is closely associated with the generation of RGCs. Blocking cell-survival signals decreases the number of RGCs whereas interference with caspases increases their number.16, 30, 31 The depletion of growth factors induces an increase in TUNEL-positive cells, which is accompanied by an increase in Annexin-V staining and lysosomal degradation. Conversely, 3-MA treatment causes the accumulation of TUNEL-positive cells with no increase in Annexin-V staining or lysosomal degradation in the region where neurons are being generated.14, 15 In conclusion, autophagy inhibition reveals the area of prominent naturally occurring cell death associated with neurogenesis and extends previous observations on autophagy requirement during morphogenesis (i.e., the formation a hollow lumen7) to the process of neural differentiation.
In untreated retinas, most TUNEL-positive cells were found inside the acidic organelles of neighbouring cells, a data that reveal the magnitude and efficacy of the engulfing process during naturally occurring cell death. In addition, our data underlie the importance of optimal autophagy-dependent clearance of apoptotic cells during development. We have previously shown that a great number of cells die by apoptosis during neurogenesis in the retina.32 Thus, effective autophagy-driven clearance of apoptotic cells could avoid the initiation of a detrimental inflammatory response, as well as producing physical space for new proliferating neuroblasts and recently differentiated neurons. In addition, other interesting roles for engulfment have also recently been described. Competition among cells may be a way to adjust and select cell types during tissue development.33 As such, evidence from Drosophila shows that engulfment genes are necessary for cell competition and that even wild-type cells can themselves be killed by cells with elevated engulfing activity.34 Thus, engulfment may be a more relevant process in shaping tissue homoeostasis than previously recognised. The autophagy-dependent clearance of apoptotic cells may also underlie the observed increase in apoptosis in Ambra-1 knockout mice at stages when neurogenesis is taking place.6 The authors’ interpretations, however, favour other interesting alternatives, such as autophagic turnover of regulatory intracellular components and removal of specific key cells by autophagic cell death. This type of cell death has been suggested to play a role during development in several species8 and, indeed, has been observed during retinal development in the chick retina.35
Our short-term pharmacological approach (a mere 6 h exposure) suggests that autophagy has a primary and fundamental role during neuronal development in providing ATP for correct removal of apoptotic cells. However, this does not preclude additional, specific roles of autophagy in development. We believe that in the future, the developing neuroretina may provide further clues to understand this intriguing process.
Materials and Methods
Chick embryos
Fertilised White Leghorn eggs from Rodríguez-Serrano Farm (Alba de Tormes, Salamanca, Spain) were incubated at 38°C in a humidified incubator. Embryonic age, referred to in days of embryonic development (E), was staged according to Hamburger and Hamilton.36 All animal experimentation follows European Union guidelines and the CSIC bioethics committee approved the experiments.
Neuroretina organotypic culture
After removal of the eyes, neuroretinas were dissected free of other tissues and incubated in a chemically defined medium of DMEM/F12+N2 supplement (Sigma, St Louis, MI, USA) for 6 h at 37°C in a 5% CO2 atmosphere.17 Where indicated, 10 mM 3-MA (Sigma), 10 mM MP (Sigma) or 20 mM O-P-L-Ser (Sigma) were added to the medium. Physiological pH after MP and O-P-L-Ser addition was restored with sodium bicarbonate (Sigma). After culture, the retinas were washed twice with phosphate-buffered saline (PBS) and flat-mounted onto nitrocellulose membranes, fixed overnight in 4% paraformaldehyde (w/v) in 0.1 M phosphate buffer (pH 7.4), and processed.
Detection of apoptosis
TUNEL of fragmented DNA was performed on whole-mount retinas according to previous methods using FITC-dUTP15 and following the manufacturer's instructions (Apoptosis Detection System; Promega, Madison, WI, USA). Fixed neuroretinas were permeated for 1 h at RT with 1% Triton X-100 (w/v; Fluka, Buchs, Switzerland), treated with 20 μg/ml collagenase (Sigma) for 1 h at 37°C, and processed for TUNEL staining. At the end of the assay, the retinas were mounted with DABCO 4% (w/v, Sigma), glycerol 70% (v/v), and visualised on a confocal microscope (LEICA DMRE2, Heidelberg, Germany). For more accurate quantification of the apoptotic cells, the retinas were dissociated with 0.05% (w/v) trypsin in PBS-BSA (1 mg/ml) for 10 min, and fixed in 4% PFA while agitating for 1 h. Cells were stained by TUNEL and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA, USA) as previously described. The percentage of apoptotic cells was determined by counting TUNEL-positive cells with a × 40 objective on a Zeiss inverted microscope (Zeiss Axioplan, Oberkochen, Germany).
Cryosections
After culture, the retinas were washed twice with PBS, flat-mounted on nitrocellulose membranes, fixed overnight in 4% paraformaldehyde (w/v) in 0.1 M phosphate buffer (pH 7.4), washed with PBS, and cryoprotected in 15% sucrose-PBS for 6 h and 30% sucrose-PBS for 1 week. Retinas were embedded in OCT (Tissue Tek, Sakura Finetek, Tokyo, Japan) and stored at −20°C until use. Cryostat sections were performed (10 μm; LEICA, CM 1800), re-fixed in 4% paraformaldehyde (w/v) in 0.1 M phosphate buffer (pH 7.4) for 20 min, washed in PBS, permeated, and stained overnight for Beclin-1 (1/100; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and Ambra-1 (1/100; Covance, Paris, France). The retinal sections were then incubated for 1 h with an Alexa 546 secondary antibody (Invitrogen), washed in PBS, stained with DAPI, and mounted with DABCO. Sections were visualised by confocal microscopy.
Staining in whole-mount retinas
Annexin-V staining (Chemicon, Pittsburg, MA, USA) was performed following the manufacturer's instructions. Briefly, Annexin-V-biotin was added to the wells for the last half an hour of organotypic retinal culture. The retinas were then washed, fixed overnight with 4% PFA, permeated, and stained for TUNEL as described above. After the TUNEL reaction, the retinas were further incubated for 1 h with Alexa 546 conjugated to avidin (Invitrogen), mounted with DABCO, and visualised by confocal microscopy. LEP-100 immunostaining was performed with an LEP-100 antibody (1/100; Developmental Studies Hybridoma Bank, Iowa, EE UU) overnight. The retinas were then washed and incubated for 1 h with Alexa 546 or 488 (Invitrogen). The retinas were stained with DAPI, mounted in DABCO, and visualised by confocal microscopy. For live Annexin-V/PI staining, retinas were dissected and incubated for 15 min with Annexin-V-fluorescein. The retinas were then incubated for 1 min with PI solution (Trevigen, Gaithersburg, MD, USA), mounted in glycerol 70% in PBS, and immediately visualised by confocal microscopy. F-actin staining was performed after the TUNEL reaction by incubation with Phalloidin-conjugated Rhodamine (Invitrogen) for 3 h at 4°C. The retinas were stained with DAPI, mounted in DABCO, and visualised by confocal microscopy. LysoTracker Red (Invitrogen) staining was performed during the last 15 min of retina culture by adding 1 μM at 37°C. Subsequently, the retinas were washed, permeated, and stained with TUNEL as described above.
Electron microscopy
Retinas were fixed for 4 h at 4 °C in Karnowsky buffer (4% paraformaldehyde (w/v) and 2.5% glutaraldehyde (v/v) in 0.1 M phosphate buffer pH 7.14), washed and fixed again in aqueous 5% (w/v) osmium tetroxide, and embedded in Epon. Electron microscopy was performed with a Zeiss EM 902 transmission electron microscope, at 90 kV, on ultra-thin sections (50 nm) stained with uranyl acetate and lead citrate.
Western blotting
Retinas were lysed in a buffer containing 50 mM Tris-HCl pH 6.8, glycerol 10% (v/v), 2% SDS (w/v), 10 mM DTT, and 0.005% blue bromophenol. Forty micrograms of protein was resolved on a 15% SDS-PAGE gel. The proteins were then transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA) that were blocked for 1 h in PBS-Tween 20 (0.05% (v/v)) containing 5% non-fat milk and probed with antibodies against LC3 (MBL Internacional, Woburn, MA, USA) and tubulin (Sigma). The antibodies were detected with the appropriate horseradish peroxidase-labelled secondary antibodies (Pierce, Rockford, IL, USA) and were visualised with the SuperSignal West Pico chemioluminiscent substrate (Pierce).
RT-PCR of atg genes
RNA was isolated from a pool of eight retinas using Trizol reagent (Invitrogen). Reverse transcription was performed on 5 μg of total RNA using Oligo(dT)18–20 and the Superscript III enzyme (Invitrogen) following the manufacturer's instructions. Semiquantitative PCR was performed using Taq Polymerase (Invitrogen) under similar conditions: an initial step of denaturation at 94°C for 2 min; followed by 20–25 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 45 s, and extension at 72°C for 45 s; and a final step at 72°C for 5 min. The primer sequences used were atg5F 5′-GAGATAACTGAGAGGGAAGC-3′, atg5R 5′-ACTGTGATGCTCCAAGGAAG-3′; beclin-1F 5′-TGATGTCAACAGAAAGCGCC-3′, beclin-1R 5′-GCCACAATCAAGCGGTTCTT-3′; lc3aF 5′-AAGTACAGCAGATCCGAGAG-3′, lc3aR 5′-CTCGTAGATCTCTGAGATGG-3′; lc3bF 5′-GTACGACTGATCCGAGATCAG-3′,lc3bR5′-GAGGCATACACCATGTACAG-3′; and gapdhF 5′-GCAATGCATCGTGCACCACC-3′, gapdhR 5′-TGTGATGGCATGGACAGTGG-3′.
ATP determination
ATP was quantified by using the ATP Bioluminescent Assay Kit HS II (Roche, Basel, Switzerland) following the manufacturer's instructions. The retinas were dissected individually and ATP was extracted and measured in a luminometer via a luciferase reaction (Td-20/2 Turner Designs, Sunnyvale, CA, USA). The protein concentration was measured in the same extract with the BCA Protein Assay Kit (Thermo Scientific, Waltham MA, USA) to correlate the ATP concentration with the protein concentration in each neuroretina.
Statistical analysis
The data shown in the figures are the means±S.E. of each treatment group. The differences between treatments were analysed with Student's t-test using the JMPIN software.
Abbreviations
- 3-MA:
-
3-methyladenine
- DAPI:
-
4′,6-diamidino-2-phenylindole
- E:
-
embryonic day
- LTR:
-
lysotracker
- MP:
-
methylpyruvate
- PI:
-
propidium iodide
- PS:
-
phosphatidylserine
- p-L-Ser:
-
phospho-L-Serine
- RGC:
-
retinal ganglion cell
- TUNEL:
-
TdT-mediated dUTP nick-end labelling
References
Levine B, Klionsky DJ . Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6: 463–477.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Klionsky DJ . Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8: 931–937.
Patel S, Dinesh-Kumar SP . Arabidopsis ATG6 is required to limit the pathogen-associated cell death response. Autophagy 2008; 4: 20–27.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672–676.
Maria Fimia G, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447: 1121–1125.
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128: 931–946.
Clarke PG . Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990; 181: 195–213.
Mattson MP . Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 2000; 1: 120–129.
Boya P, de la Rosa EJ . Cell death in early neural life. Birth Defects Res C Embryo Today 2005; 75: 281–293.
Baehrecke EH . Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 2005; 6: 505–510.
Boya P, Gonzalez-Polo RA, Casares N, Perfettini J, Dessen P, Larochette N et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25: 1025–1040.
Marquardt T, Gruss P . Generating neuronal diversity in the retina: one for nearly all. Trends Neurosci 2002; 25: 32–38.
Prada C, Puga J, Perez-Mendez L, Lopez R, Ramire G . Spatial and temporal patterns of neurogenesis in the chick retina. Eur J Neurosci 1991; 3: 559–569.
Diaz B, Pimentel B, de Pablo F, de la Rosa EJ . Apoptotic cell death of proliferating neuroepithelial cells in the embryonic retina is prevented by insulin. Eur J Neurosci 1999; 11: 1624–1632.
Diaz B, Serna J, De Pablo F, de la Rosa EJ . In vivo regulation of cell death by embryonic (pro)insulin and the insulin receptor during early retinal neurogenesis. Development 2000; 127: 1641–1649.
de la Rosa EJ, Diaz B, de Pablo F . Organoculture of the chick embryonic neuroretina. Curr Top Dev Biol 1998; 36: 133–144.
Erwig LP, Henson PM . Clearance of apoptotic cells by phagocytes. Cell Death Differ 2008; 15: 243–250.
Schaefer KS, Doughman YQ, Fisher SA, Watanabe M . Dynamic patterns of apoptosis in the developing chicken heart. Dev Dyn 2004; 229: 489–499.
Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM . A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000; 405: 85–90.
Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, Schnabel H et al. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature 2005; 434: 93–99.
Erwig LP, McPhilips KA, Wynes MW, Ivetic A, Ridley AJ, Henson PM . Differential regulation of phagosome maturation in macrophages and dendritic cells mediated by Rho GTPases and ezrin-radixin-moesin (ERM) proteins. Proc Natl Acad Sci USA 2006; 103: 12825–12830.
Kurosaka K, Takahashi M, Watanabe N, Kobayashi Y . Silent cleanup of very early apoptotic cells by macrophages. J Immunol 2003; 171: 4672–4679.
Jijakli H, Nadi AB, Cook L, Best L, Sener A, Malaisse WJ . Insulinotropic action of methyl pyruvate: enzymatic and metabolic aspects. Arch Biochem Biophys 1996; 335: 245–257.
Lum J, Bauer D, Kong M, Harris M, Li C, Lindsten T et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120: 237–248.
Higgins CF, Linton KJ . The ATP switch model for ABC transporters. Nat Struct Mol Biol 2004; 11: 918–926.
Luciani MF, Chimini G . The ATP binding cassette transporter ABC1, is required for the engulfment of corpses generated by apoptotic cell death. EMBO J 1996; 15: 226–235.
Wu YC, Horvitz HR . The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 1998; 93: 951–960.
Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol 2006; 174: 547–556.
Pimentel B, Sanz C, Varela-Nieto I, Rapp UR, De Pablo F, de la Rosa EJ . c-Raf regulates cell survival and retinal ganglion cell morphogenesis during neurogenesis. J Neurosci 2000; 20: 3254–3262.
Mayordomo R, Valenciano AI, de la Rosa EJ, Hallbook F . Generation of retinal ganglion cells is modulated by caspase-dependent programmed cell death. Eur J Neurosci 2003; 18: 1744–1750.
Chavarría T, Valenciano AI, Mayordomo R, Egea J, Comella JX, Hallböök F et al. Differential, age-dependent MEK-ERK and PI3K-Akt activation by insulin acting as a survival factor during embryonic retinal development. Dev Neurobiol 2007; 67: 1777–1788.
Morata G, Martin FA . Cell competition: the embrace of death. Dev Cell 2007; 13: 1–2.
Li W, Baker NE . Engulfment is required for cell competition. Cell 2007; 129: 1215–1225.
Guimarães CA, Benchimol M, Amarante-Mendes GP, Linden R . Alternative programs of cell death in developing retinal tissue. J Biol Chem 2003; 278: 41938–41946.
Hamburguer V, Hamilton H . A series of normal stages in the development of the chock embryo. J Morphol 1951; 88: 49–92.
Acknowledgements
This research was supported by grants from the Spanish Ministerio de Educación y Ciencia (BFU2006-00508 to PB and SAF2007-66175 to EJdlR) and Comunidad de Madrid (CCG06-CSIC/SAL-0821 to PB). MAM is a FPU Fellow and PB is a Ramón y Cajal Fellow (both Ministerio de Educación y Ciencia programs). We thank the members of our lab and Aviva Tolkovsky for helpful discussions and comments on the manuscript. We also thank the Servicio de Microscopía Electrónica at the Universidad Complutense, and Mayte Seidsedos and Silvia Hernández for their excellent assistance with confocal microscopy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by E Baehrecke
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Mellén, M., de la Rosa, E. & Boya, P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ 15, 1279–1290 (2008). https://doi.org/10.1038/cdd.2008.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.40
Keywords
This article is cited by
-
Research progress on the mechanism of curcumin in cerebral ischemia/reperfusion injury: a narrative review
Apoptosis (2023)
-
The Emerging Role of Astrocytic Autophagy in Central Nervous System Disorders
Neurochemical Research (2022)
-
Autophagy in animal development
Cell Death & Differentiation (2020)
-
Age related retinal Ganglion cell susceptibility in context of autophagy deficiency
Cell Death Discovery (2020)
-
Autophagy protects against retinal cell death in mouse model of cytomegalovirus retinitis
BMC Ophthalmology (2019)
