
Abstract
Recent studies show that in Alzheimer's disease (AD), alterations in neurogenesis contribute to the neurodegenerative process. Neurodegeneration in AD has been associated with aberrant signaling through the cyclin-dependent kinase-5 (CDK5) pathway via its activators p35/p25; however, the role of CDK5 in the mechanisms of defective adult neurogenesis in AD is unknown. First, to study AD-like abnormal activation of CDK5 signaling in an in vitro model of neurogenesis, neuronal progenitor cells (NPCs) were infected with a viral vector expressing p35, and exposed to amyloid-β protein (Aβ1−42). These conditions resulted in impaired maturation and neurite outgrowth in vitro, and these effects were reversed by pharmacological or genetic inhibition of CDK5. Similarly, neurogenesis was impaired in a transgenic mouse model of AD that expresses high levels of amyloid precursor protein (APP), and this effect was reversed in transgenic mice crossed with a CDK5 heterozygous-deficient mouse line. A similar rescue effect was observed in APP transgenic mice treated with Roscovitine, a pharmacological inhibitor of CDK5. Taken together, these data suggest that the CDK5 signaling pathway has a critical role in maintaining the integrity of NPCs and neuronal maturation in the adult hippocampus. Moreover, potential therapeutic approaches could focus on modulating the aberrant activity of CDK5 to target the neurogenic and neurodegenerative alterations in AD.
Similar content being viewed by others


Main
During aging and in the progression of Alzheimer's disease (AD), synaptic plasticity and neuronal integrity are disturbed.1 In AD, this has been linked to aberrant activation of cyclin-dependent kinase-5 (CDK5) and its activators p35 and p25,2, 3 and with increased levels of CDK5 in the brains of AD patients.4
CDK5 is the predominant CDK found in the brain, is highly expressed in neurons, and has an important role in synaptic plasticity, dendritic growth,5, 6 and neuronal development7 in mature neurons. Many of the diverse functions of CDK5 activity occur via phosphorylation of numerous substrates, including cytoskeletal proteins (e.g., nestin, doublecortin (DCX)), synaptic proteins (e.g., synapsin I, PSD95), and transcription factors (e.g., MEF2). Unlike canonical CDK signaling, CDK5 is activated by the non-cyclin proteins p35 and p39.8 Although CDK5 activation via p35 is associated with physiological activity of CDK5, the truncated p25 form – which in AD can be generated by amyloid-β (Aβ)-mediated calpain cleavage of p359– abnormally stimulates CDK5 signaling and leads to aberrant phosphorylation of substrates such as tau.10 This process contributes to neurotoxicity and cell death in AD, possibly via improper cell cycle re-entry in mature neurons.7, 11
In addition to alterations in synaptic plasticity and neuronal cell survival in AD, recent studies have revealed that the neurodegenerative process in this disorder might include dysregulation of adult neurogenesis.12, 13, 14 In the brains of AD patients and in animal models, there may be a decrease in the generation of new neurons from neural progenitor cells (NPCs) in the subgranular zone (SGZ) of the adult hippocampal dentate gyrus (DG).14, 15, 16, 17, 18 Consistent with these observations, we have recently found that markers of neurogenesis are reduced in a stage-dependent manner in the hippocampus of patients with AD.19
Although the mechanisms of neurogenesis in the fetal brain have been extensively studied, less is known about the signaling pathways regulating adult neurogenesis and their role in neurodegenerative disorders. During embryogenesis, CDK5 is a critical factor in neuronal development20 and migration.21 Homozygous ablation of the Cdk5 gene is lethal in mice;21, 22 however, heterozygous knockdown of one gene copy results in relatively normal brain development.22 More recently, CDK5 has been shown to be essential for adult neurogenesis.6, 23 In one in vivo model, conditional ablation of the Cdk5 gene resulted in reduced numbers of immature SGZ neurons through both cell-intrinsic and cell-extrinsic mechanisms.23 In a separate study, retrovirus-mediated inhibition of CDK5 activity using a dominant-negative kinase mutant (DNcdk5) altered dendritic morphology and orientation of hippocampal NPCs.6 These studies suggest that physiological CDK5 activity is critical for maturation of progenitor cells in the adult hippocampus; however, the effects of abnormal CDK5 activity associated with the pathogenesis of AD have not been explored in the adult neurogenic niches.
It is possible that the alterations in adult hippocampal neurogenesis associated with AD might be related to abnormal CDK5 activity. In support of this possibility, we have previously shown in a mouse model of AD that a neurotrophic therapy that promotes neurogenesis also modulates CDK5 activity.18, 24 Moreover, in other in vitro models of AD, Aβ has been shown to impair neurogenesis via calpain activation and p35 dysregulation;25 however, the effects of CDK5 activation on adult neurogenesis remain unclear. In this study we investigated the role of aberrant CDK5 signaling in defective neurogenesis in AD, and we report that in in vitro and in vivo models, deficits in neuronal maturation can be reversed by genetic or pharmacological modulation of CDK5.
Results
Characterization of the CDK5 signaling pathway in an in vitro model of adult hippocampal neurogenesis
To begin to investigate the role of CDK5 in adult neurogenesis in AD, we first examined the expression patterns of the CDK5 pathway, and markers of differentiation, in an in vitro model of adult hippocampal neurogenesis. For this purpose, NPCs derived from adult rat hippocampus were differentiated toward a neuronal phenotype over a period of 4 days essentially as previously described.26 (Supplementary Figure 1A).
NPCs were grown in NPC differentiation media for 4 days and analyzed by western blot, quantitative real-time PCR (qRT-PCR), and immunocytochemistry. By western blot, the levels of early glial (S100) and immature neuronal markers (β-III tubulin (Tuj1) and DCX) increased during differentiation, and the levels of progenitor cell markers (nestin, proliferating cell nuclear antigen (PCNA), and brain lipid-binding protein (BLBP)) decreased. Expression of markers of other mature lineages (glial fibrillary acidic protein (GFAP), astroglial marker) remained low (Supplementary Figure 1B). Immunoblot and qRT-PCR analyses showed that protein and mRNA expression levels of CDK5 increased approximately twofold during differentiation, and levels of p35/p25 remained relatively low at all stages studied (Supplementary Figure 1B). During neuronal induction, cells acquired a neuron-like phenotype (Supplementary Figure 1C–E). Immunocytochemical analysis confirmed that levels of CDK5 and β-III tubulin increased concurrently over the 4-day differentiation period (Supplementary Figure 1F–K).
Modeling AD-associated aberrant activation of CDK5 in adult neurogenesis in vitro
Given the potential role of abnormal CDK5 activity in neurodegeneration in AD,2, 3, 4 we sought to develop an in vitro model to examine whether this kinase might also contribute to defective neurogenesis in AD models. For this purpose, we stimulated the CDK5 pathway in the adult rat hippocampal NPC model by expressing the CDK5 activator, p35, utilizing a viral vector. Infection of NPCs with an adenoviral (adv) construct expressing p35 resulted in high levels of p35 expression after 4 days of differentiation, and generation of the p25 fragment was detectable with high levels of p35 expression (Figure 1a).

Characterization of an in vitro model of adult neurogenesis in AD, and impaired neurite outgrowth with aberrant CDK5 activation. Differentiating NPCs were infected at day 2 with an adenoviral vector expressing p35 or GFP as a control, and 24 h later, cells were exposed to 1 μM Aβ1−42. Cells were analyzed by live cell microscopy, or lysed for immunoblot analysis, or briefly extracted, and then fixed with glutaraldehyde and processed for β-tubulin immunofluorescence. (a) Immunoblot analysis of the expression levels of CDK5, p35/p25, β-III-tubulin (Tuj1), GFP, and actin (loading control) in NPC-derived neural progeny expressing adenovirus-delivered p35 or GFP (vector control) and treated with Aβ1−42 for 24 h. Graph showing quantitative analysis of p35/p25 immunoreactivity demonstrates increased p25 generation in p35-expressing cells after Aβ treatment. *Nonspecific immunoreactive band detected with the p35/p25 antibody. (b–g) Live cell imaging showing the morphology of NPC-derived neural progeny expressing p35 or GFP control and treated with Aβ. (h) No significant cytotoxicity (measured by LDH assay) was detected in cells infected with p35 adenovirus or GFP control, or with Aβ treatment alone or in combination with p35 expression. (i) Increased kinase activity of CDK5 in p35 and Aβ alone and p35/Aβ-treated NPC-derived neural progeny. (j–m) Compared with vehicle-treated controls (Ctrl), shorter β-tubulin-immunoreactive neurites were detected in NPC-derived neural progeny with p35 or Aβ treatment alone or in combination. Cell nuclei were co-stained with DAPI reagent. (n) Quantitative image analysis showing reduced neurite lengths in NPC-derived neural progeny with p35 or Aβ treatment alone or in combination. Scale bar=10 μm. *P<0.05 compared with vehicle-treated controls by one-way ANOVA with post hoc Dunnett's test (N=3). **P<0.01 compared with vehicle-treated controls by one-way ANOVA with post hoc Dunnett's test (N=3)
Phase contrast microscopy of live cell cultures showed no significant morphological differences in adv-p35-infected cultures compared with uninfected or adv-GFP-infected controls (Figures 1b–d). As Aβ has been shown to abnormally regulate the activity of this pathway, following adv-p35 infection, cultures were treated with 1 μM Aβ1−42 for 24 h (Figures 1e–g). Compared with infection with adv-p35 alone, combined p35/Aβ treatment resulted in an ∼20% increase in p25 immunoreactivity, and a 30–40% reduction in p35 immunoreactivity (Figure 1a). This supports the contention that combined p35/Aβ treatment promotes cleavage of p35 and generation of the p25 fragment. Although the production of p25 is associated with neurotoxicity in mature neurons, no significant cell toxicity was detected in the cultured NPCs under these conditions (Figure 1h). This allowed subsequent analyses to focus on the effects of aberrant CDK5 activity on cell maturation. Analysis of kinase activity confirmed that in cells expressing p35 or treated with Aβ alone, or in combination, CDK5 activity was significantly increased compared with controls (Figure 1i).
Abnormal activation of CDK5 signaling impairs NPC maturation in vitro
Under baseline conditions, by the end of the 4-day differentiation period, the majority (>90%) of NPC-derived progeny were immunopositive for β-III-tubulin (Tuj1) (Figures 2a–c, Supplementary Figure 1H), suggesting that these cells represent predominantly immature neurons. In order to determine whether activation of CDK5 was associated with any measurable differences in neuronal maturation, neurite outgrowth was assessed in p35/Aβ-treated NPC-derived neural progeny (Figures 1j–n). β-Tubulin immunofluorescence and neurite outgrowth studies revealed that p35 and Aβ treatment alone or in combination resulted in shorter processes in NPC-derived neural progeny compared with vehicle-treated controls (Figures 1j–n); however, other controls for Aβ (reverse Aβ42−1 peptide) and aggregating proteins (transthyretin (TTR)) had no significant effects on neurite outgrowth (not shown).

Morphological and maturation alterations in NPC-derived neural progeny expressing p35 and treated with Aβ. Differentiating NPCs were infected with an adenoviral vector expressing p35 or vector control on day 2 of differentiation, and then treated with 1 μM Aβ or vehicle control on day 3 for 24 h, followed by fixation with paraformaldehyde. Samples were processed for double-immunolabeling with antibodies against p35 and markers of proliferative and neuronal differentiation status (neuronal, β-III tubulin (Tuj1), and doublecortin (DCX); progenitor cells, nestin). (a–f) Double-labeling analysis with Tuj1 and p35 antibodies in vector-infected NPC-derived neural progeny treated with vehicle control (a–c) or Aβ (d–f) for 24 h. (g–l) Double-labeling analysis with Tuj1 and p35 antibodies in p35-expressing NPC-derived neural progeny treated with vehicle control (g–i) or Aβ (j–l) for 24 h. (j–r) In combined p35/Aβ-treated cultures, progeny acquired an abnormal stellate morphology and cells were co-immunoreactive for markers of proliferation stages (nestin) and neuronal (Tuj1 and DCX) lineage (arrows). (s) Quantitative analysis of proportion of NPC-derived neural progeny co-labeled with Tuj1 and nestin antibodies. Scale bar=15 μm. *P<0.05 compared with vehicle-treated controls by one-way ANOVA with post hoc Dunnett's test (N=3)
To further characterize the effects of CDK5 activation with p35/Aβ, double-labeling immunocytochemical analysis was performed (Figure 2). Immunolabeling with an antibody against p35/p25 confirmed that p35 was expressed at high levels in >95% of the adv-p35-infected cells after 4 days under differentiating conditions (Figure 2h). In cultures exposed to p35/Aβ, NPC-derived neural progeny displayed considerable co-immunoreactivity for markers of neuronal lineage (Tuj1 and DCX) and progenitor cell markers (nestin) (Figures 2j–r). Although in vector-infected, vehicle-treated control cultures, ∼10% of cells coexpressed β-III tubulin and nestin, in p35/Aβ-treated progeny, this proportion was increased to 30% (Figure 2s). This suggests that the neuronal maturation process under these conditions may be impaired, resulting in hybrid-like cells arrested in an early phase of neuronal development. These results support the possibility that constitutive activation of CDK5 signaling might promote the generation of cells that are unable to fully complete neuronal maturation.
Pharmacological inhibition of abnormal CDK5 activity, or siRNA knockdown of CDK5, rescues neuronal maturation in NPC-derived neural progeny
In order to determine whether CDK5 contributes directly to alterations in neuronal maturation (Figure 1), we used a pharmacological inhibitor of CDK5 (Roscovitine), or an siRNA-based approach, to inhibit CDK5 activity or expression, respectively. First, to confirm that these conditions were not overtly toxic, and to characterize the efficacy of CDK5 siRNA (siCDK5) treatment in this system, we performed a series of control experiments with NPCs treated on day 2 of differentiation with Roscovitine or siCDK5 (Supplementary Figure 2). Following Roscovitine treatment, CDK5 and p35 protein levels were comparable with vehicle-treated controls (Supplementary Figure 2A), and these conditions showed no significant toxicity (Supplementary Figure 2B). Western blot, qRT-PCR, and LDH assays demonstrated that transfection with siCDK5 resulted in a marked reduction in CDK5 protein and mRNA levels (∼50%) without significant toxicity (Supplementary Figures 2C–E). Immunocytochemical analysis confirmed that CDK5 levels were significantly reduced in differentiating NPCs transfected with siCDK5 (Supplementary Figures 2F–H).
Next, we sought to determine whether pharmacological or genetic inhibition of CDK5 might protect against the maturation defects observed in NPC-derived neural progeny exposed to p35/Aβ. For this purpose, on day 2 of neuronal induction, differentiating NPCs were pretreated with Roscovitine, or transfected with siCDK5 for 6 h, followed by infection with adv-p35, and Aβ treatment (Figure 3). Neurite outgrowth studies of β-tubulin-positive processes revealed that modulating CDK5 with Roscovitine or siCDK5 reversed the neurite outgrowth deficits observed after p35/Aβ treatment (Figures 3a–g). Taken together, these results support the possibility that AD-related aberrant activation of CDK5 signaling impairs the maturation of adult hippocampal NPCs, and this effect can be reversed by modulation of CDK5 by pharmacological or genetic means.

Downmodulation of abnormal CDK5 activity in vitro rescues neuronal maturation. Differentiating NPCs were pretreated on day 2 with the pharmacological inhibitor Roscovitine (Rosco) or transfected with siRNA against CDK5 (siCDK5, 5 nM), and then infected 6 h later with adenovirus expressing p35, followed by treatment with Aβ on day 3 for 24 h. On day 4, NPC-derived neural progeny were briefly extracted and fixed with glutaraldehyde for β-tubulin immunofluorescence and neurite outgrowth analysis. Cell nuclei were co-stained with DAPI reagent. (a–c) Compared with controls (a), uninfected NPC-derived neural progeny treated with Rosco (b) or siCDK5 (c) showed only a mild reduction in the lengths of β-tubulin-positive neurites. (d–f) NPC-derived neural progeny with p35/Aβ treatment showed a notable decrease in the lengths of β-tubulin-positive neurites (d); this was rescued by treatment with Rosco (e) or siCDK5 (f). (g) Neurite outgrowth measurements using NeuronJ showed a 50% reduction in neurite lengths in p35/Aβ-treated cells, and this effect was reversed in cultures pretreated with Rosco or transfected with siCDK5. Scale bar=10 μm. *P<0.05 compared with vehicle-treated controls by one-way ANOVA with post hoc Dunnett's test (N=3). #P<0.05 compared with p35/Aβ-treated NPCs by one-way ANOVA with post hoc Tukey–Kramer test (N=3)
Alterations in neurogenesis in APP transgenic mice are rescued by knockdown of CDK5 expression
To further elucidate the role of AD-related aberrant CDK5 activation in adult neurogenesis in vivo, we first characterized the expression patterns of members of the CDK5 signaling pathway in the DG of a transgenic (tg) mouse line expressing high levels of neuronal amyloid precursor protein (APP). We have previously shown that hippocampal neurogenesis is impaired in the brains of these animals.19, 24 In support of a role for CDK5 in the adult hippocampus, double-labeling immunocytochemical analyses demonstrated that APP, CDK5, and p35 were expressed in the DG of APP tg mice (Figures 4a–i). Moreover, CDK5 was detected in cells surrounding and within a subset of SGZ cells that are immunopositive for the developing neuronal marker DCX (Figures 4j–l).

Characterization of expression patterns of the CDK5 signaling pathway in the neurogenic niche of the adult hippocampus in APP tg mice. Brain sections from APP tg mice (9 months old) were double-labeled with antibodies against APP and CDK5 or p35, and CDK5 and the mature neuron marker microtubule-associated protein-2 (MAP2) or the immature neuron marker doublecortin (DCX). All images are from the dentate gyrus (DG) of the hippocampus. (a–c) Double-labeling analysis of APP and CDK5, showing coexpression in cells of the DG (arrows), particularly in the granular cell layer (GCL). (d–f) Double-labeling analysis of APP and p35, showing coexpression of these markers throughout the neuropil of the DG. (g–i) Double-labeling analysis of MAP2 and CDK5, showing CDK5 expression localized primarily to the GCL and subgranular zone (SGZ). MAP2 was detected more predominantly in the molecular layer (ML). (j–l) Double-labeling analysis of CDK5 and DCX in the SGZ and GCL regions, showing expression in and around cells in the SGZ. A subset of cells were co-immunolabeled with both CDK5 and DCX (arrows). Scale bar=20 μm
To investigate whether there were any alterations in CDK5 signaling in the hippocampus of APP tg mice, additional immunohistochemical studies were performed. Compared with non-tg littermate controls, CDK5 (Figures 5a, c, and e) and p35/p25 (Figures 5f, h, and j) immunoreactivity were increased in the DG of APP tg mice. Furthermore, immunoblot analysis showed that levels of phosphorylated CDK5 (Tyr15) – a modification that upregulates CDK5 activity,27 – were increased in the brains of APP tg mice compared with non-tg controls (Supplementary Figures 3A and B). Consistent with previous studies,17, 18, 19 there was a reduction in the numbers of DCX-positive (Figures 5k, m, and o) and 5-bromo-2-deoxyuridine (BrdU)-positive (Figures 5p, r, and t) cells in the DG of APP tg mice compared with non-tg controls.

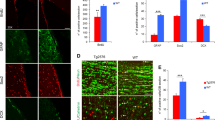
In vivo genetic modulation of CDK5 rescues neurogenic deficits in the hippocampus of APP tg mice. Nine-month-old wild-type control mice (non-tg), CDK5 heterozygous deficient (CDK5+/−), APP tg mice, or crosses (CDK5+/−/APP) were treated with BrdU. Brain sections were processed for immunohistochemical analysis with antibodies against CDK5, p35/p25, doublecortin (DCX), or BrdU. All images are from the dentate gyrus of the hippocampus. (a–e) Immunohistochemical analysis of the levels of CDK5 immunoreactivity in wild-type, CDK5+/−, APP tg mice, or CDK5+/−/APP crosses. (f–j) Immunohistochemical analysis of the levels of p35/p25 immunoreactivity in wild-type, CDK5+/−, APP tg mice, or CDK5+/−/APP crosses. (k–o) Immunohistochemical analysis of the numbers of DCX-immunoreactive cells (arrows) in wild-type, CDK5+/−, APP tg mice, or CDK5+/−/APP crosses. (p–t) Immunohistochemical analysis of the numbers of BrdU-immunoreactive cells (arrows) in wild-type, CDK5+/−, APP tg mice, or CDK5+/−/APP crosses. Scale bar=50 μm. *P<0.05 compared with wild-type controls by one-way ANOVA with post hoc Dunnett's test. #P<0.05 compared with APP tg mice by one-way ANOVA with post hoc Tukey–Kramer test. N=4 animals per group
In addition to the reduction in the total numbers of DCX-positive cells in the DG of APP tg mice compared with non-tg mice, the DCX-positive cells detected in the APP tg mice also displayed significantly shorter DCX-positive processes extending from the SGZ (Figures 6a, c, and e). These results are consistent with our in vitro results showing that aberrant CDK5 activation resulted in impaired neurite outgrowth in cultured NPCs (Figure 1), and support the contention that abnormally high levels of CDK5 activity might be detrimental to neuronal maturation. Thus, we hypothesized that in vivo modulation of CDK5 expression or activity might reverse the neurogenic deficits in the brains of APP tg mice with abnormal CDK5 activation.

In vivo genetic modulation of CDK5 rescues neurite outgrowth defects in the hippocampus of APP tg mice. Brain sections from 9-month-old wild-type control mice (non-tg), CDK5 heterozygous deficient (CDK5+/−), APP tg mice, or crosses (CDK5+/−/APP) were processed for immunohistochemical analysis with an antibody against doublecortin (DCX) and developed with DAB. All images are from the dentate gyrus of the hippocampus. (a–d) Immunohistochemical analysis of DCX-immunoreactive cells in the dentate gyrus of non-tg, CDK5+/−, APP tg mice, or CDK5+/−/APP crosses. Brackets denote representative DCX-immunoreactive processes extending from cells in the SGZ that were measured. (e) Quantitative image analysis of the lengths of DCX-immunoreactive processes extending from DCX-immunoreactive cells located in the SGZ. *P<0.05 compared with wild-type controls by one-way ANOVA with post hoc Dunnett's test. #P<0.05 compared with APP tg mice by one-way ANOVA with post hoc Tukey–Kramer test. N=4 animals per group
To investigate this possibility using a genetic approach, APP tg mice were crossed with a line of mice that is heterozygous deficient for endogenous murine Cdk5 gene (CDK5+/−). As reported previously,22 immunohistochemical and immunoblot analyses showed that CDK5+/− mice express ∼50% lower brain levels of CDK5 than wild-type controls (Figures 5a, b, and e and Supplementary Figures 3A and C). In support of previous studies using different models of CDK5 ablation in the adult neurogenic niche,23 the CDK5 heterozygous deficient animals also exhibited a reduction in DCX- and BrdU-positive cells in the DG of the hippocampus compared with controls (Figures 5k, l, o, p, q, and t). These results are consistent with the possibility that both abnormally low or high levels of CDK5 activity are detrimental to neuronal development.
In the offspring generated by crossing APP tg mice with CDK5+/− mice (CDK5+/−/APP tg), the levels of CDK5 were reduced in the brains of CDK5+/−/APP tg mice compared with APP tg mice, and were similar to levels in CDK5+/− mice (Figures 5a–e and Supplementary Figures 3A and C). Although immunohistochemical analysis revealed some localized changes in p35/p25 immunoreactivity in the hippocampus (Figures 5f–j), no differences were observed in total brain levels of p35 immunoreactivity by immunoblot (Supplementary Figures 3A and D). Supporting the possibility that reduced CDK5 activity was related to the reversal of neurogenic deficits in the CDK5+/−/APP animals compared with APP tg mice, immunoblot analysis demonstrated that CDK5 activity in CDK5+/−/APP tg mice was downregulated to levels similar to wild-type controls (Supplementary Figure 3B).
Remarkably, in the CDK5+/−/APP tg mice, there was a significant recovery of numbers of DCX- and BrdU-positive cells compared with APP tg mice (Figures 5m–o and r–t). Consistent with the in vitro studies (Figure 3), genetic knockdown of CDK5 in APP tg mice resulted in a recovery of DCX-positive processes extending from the SGZ of the hippocampal DG (Figures 6a–e). Thus, normalization of CDK5 activity levels may be beneficial to the maintenance of neuronal maturation in the adult hippocampus.
As CDK5-mediated neurotoxicity in AD has been linked to abnormal cell cycle re-entry in mature neurons, we sought to determine whether the neurogenesis effects in vivo might be related to other cell cycle defects. Therefore, we assessed the levels of other components of the cell cycle (cyclin D1 and CDK2) in the brains of these animals by immunoblot. Although there was a slight trend toward increased cyclin D1 levels in APP tg animals, the differences were not significant, and no differences were detected in CDK2 levels between the groups (Supplementary Figures 3A, G, and H).
Next, to determine whether rescued neurogenesis in CDK5+/−/APP tg mice might be because of neuropathological differences, additional immunoblot and immunohistochemical studies were performed. We found that APP tg mice and CDK5+/−/APP tg mice showed similar levels of APP expression and Aβ production (Supplementary Figures 3A, E, and F and Supplementary Figure 4). To examine general neuronal integrity at lower power, immunolabeling studies were performed with an antibody against NeuN, followed by stereological image analysis (Supplementary Figure 5). Neuronal density in the neocortex and hippocampus was comparable among non-tg, CDK5+/−, and CDK5+/−/APP tg mice, whereas in APP tg mice there was a reduction in the numbers of NeuN-positive cells in layers II and III of the cortex (Supplementary Figures 5A–E), and in the DG of the hippocampus (Supplementary Figures 5F–J). Behavioral studies showed that normalization of CDK5 activity in CDK5+/−/APP tg mice was accompanied by improved performance in the water maze test compared with APP tg mice (Figures 7a and b). These results show that knockdown of CDK5 in APP tg mice rescued both the neurogenic and neurodegenerative alterations in APP tg mice.

Improved cognitive performance in the water maze behavioral test in mice with genetically reduced CDK5 levels. (a and b) Nine-month old wild-type control mice (non-tg), CDK5 heterozygous deficient (CDK5+/−), APP tg mice, or crosses (CDK5+/−/APP tg) were trained in the water maze, beginning with a 3-day exposure to the visible platform. Mice were then tested for learning and memory with the hidden platform for 4 days, and measurements were taken to determine the time (in seconds) that mice took to reach the platform (a), and distance swum before reaching the platform (b). *P<0.05 compared with wild-type controls by one-way ANOVA with post hoc Dunnett's test. #P<0.05 compared with APP tg mice by one-way ANOVA with post hoc Tukey–Kramer test. N=4 animals per group
CDK5 inhibition with Roscovitine in vivo ameliorates the defects in neurogenesis in APP tg mice
To investigate the possibility that modulation of CDK5 using a pharmacological approach might have similar protective effects on adult neurogenesis in APP tg mice, we performed additional in vivo experiments where APP tg mice were treated with the CDK5 inhibitor, Roscovitine. APP tg mice that received intracerebral infusion with Roscovitine or vehicle control displayed levels of CDK5 or p35/p25 expression similar to vehicle-treated non-tg animals (Figures 8a–h). Compared with non-tg controls, vehicle-treated APP tg mice displayed a significant reduction in DCX-positive and BrdU-positive cells in the SGZ of the DG (Figures 8i, j, l, m, n, and p). Remarkably, inhibition of CDK5 activity with Roscovitine recovered the numbers of DCX-positive and BrdU-positive cells in APP tg mice (Figures 8i–p). These results support the possibility that aberrant activation of CDK5 in AD contributes to defective neurogenesis, and inhibition of CDK5 might be useful in therapeutic approaches aimed at rescuing the neurogenic alterations associated with AD.

Roscovitine treatment rescues neurogenic deficits in the hippocampus of APP tg mice. Brain sections from 9-month-old APP tg mice and non-tg controls treated with Roscovitine or vehicle control and BrdU were processed for immunohistochemical analysis with antibodies against CDK5, p35/p25, doublecortin (DCX), or BrdU. All images are from the dentate gyrus of the hippocampus. (a–d) Analysis of CDK5 immunoreactivity in non-tg and APP tg mice treated with vehicle control or Roscovitine. (e–h) Analysis of p35/p25 immunoreactivity in non-tg and APP tg mice treated with vehicle control or Roscovitine. (i–l) Analysis of DCX immunoreactivity and the numbers of positive cells in non-tg and APP tg mice treated with vehicle control or Roscovitine. (m–p) Analysis of BrdU immunoreactivity and numbers of positive cells in non-tg and APP tg mice treated with vehicle control or Roscovitine. Scale bar=50 μm. *P<0.05 compared with vehicle-treated non-tg controls by one-way ANOVA with post hoc Dunnett's test. N=4 animals per group. #P<0.05 compared with APP tg mice by one-way ANOVA with post hoc Tukey–Kramer test. N=4 animals per group
Discussion
In this study we found that in an in vitro model system of adult hippocampal neurogenesis in AD, abnormal stimulation of the CDK5/p35/p25 signaling pathway resulted in impaired maturation of NPCs, as reflected by reduced neurite outgrowth in NPC-derived neural progeny. These effects were rescued by downmodulation of CDK5 activity by Roscovitine or siRNA specific for CDK5. Similarly, in the APP tg mouse model of AD, we found that deficits in neurogenesis in the adult hippocampus were reversed by genetic or pharmacological inhibition of CDK5.
Previous studies have implicated CDK5 in embryonic neuronal development20 and neuroblast migration in the cerebral cortex.21, 22 In healthy mature neurons, CDK5 regulates synaptic plasticity28 and neurite outgrowth;20 however, less is known about its role in adult neurogenesis. Several recent studies have implicated CDK5 in the maintenance of a non-cycling state in postmitotic neurons.29 This function appears to be dependent on the subcellular localization of CDK5, which is regulated by cell cycle stage29 and interactions with the cell cycle inhibitor p27,30 among other factors. Consistent with previous observations,29 in this study we have detected some CDK5 expression in both the nuclear and cytoplasmic compartments in both in vitro and in vivo models, which is likely a reflection of variability in cell cycle stage of cycling NPCs and immature neural progeny.
Recent studies have implicated CDK5 in neuronal maturation and dendritic development during the process of adult neurogenesis.6, 23 Although loss of CDK5 activity does not appear to alter neuronal fate, it severely disrupts dendritic development of NPCs in vivo.6, 23 Targeted ablation of CDK5 expression in adult hippocampal NPCs reduces the numbers of DCX-positive immature neurons in the SGZ.23 Similarly, the results of this study show in an alternative model of CDK5 deficiency that the numbers of hippocampal DCX- and BrdU-positive cells were reduced.
It is important to note that although too little CDK5 activity is detrimental to embryonic and adult neurogenesis,6, 22, 23 pathological activation of CDK5 associated with AD has a significant negative impact as well. Notably, we observed similar defects in neurogenesis in the brains of APP tg mice that were rescued by CDK5 inhibition, suggesting that abnormal CDK5 activation could reflect a loss of normal function and/or a gain of atypical function; however, future studies will be necessary to elucidate the mechanisms involved. A previous review paper aptly labeled CDK5 as a ‘Jekyll and Hyde kinase’,2 reflecting both the important physiological role of CDK5 in development22, 28 and the abnormal CDK5 activity associated with neurodegeneration in the pathogenesis of AD.9
Although physiological CDK5 activity is required for the critical functions noted above, abnormal stimulation of CDK5 in the pathogenesis of AD has been shown to contribute to the neurodegenerative process and mature neuronal cell death.9 In this neurotoxic pathway, Aβ triggers calcium influx, which activates calpain activity and subsequent cleavage of p35 to p25, a stable activator of CDK5 that promotes the hyperphosphorylation of downstream substrates of CDK5.9 In this context, for this study we focused on the activation of this pathway to further probe the role of CDK5 in adult hippocampal neurogenesis and neuronal maturation in AD. Our studies in an in vitro model of aberrant CDK5 activation in adult neurogenesis in AD show that p35 overexpression combined with Aβ treatment promoted the generation of abnormal neural progeny and impaired neurite outgrowth.
It was an unexpected finding that under conditions of abnormal CDK5 activation in our in vitro NPC model, the proportions of progeny coexpressing markers traditionally associated with proliferation or neuronal differentiation stages were more prominent than under control conditions. We hypothesize that these disease-like conditions might arrest cells in an early transitional state where expression of progenitor cell markers might be prolonged or atypical, reflecting an impairment in the neuronal maturation process. In support of this possibility, a previous study has shown that in the DG of AD brains, progenitor cells fail to differentiate into mature microtubule-associated protein-2 (MAP2)-positive neurons.14
Further studies will be necessary to delineate the precise mechanisms and downstream targets through which abnormal CDK5 activation interferes with the maturation of NPCs in AD. Several substrates of CDK5 are worth further investigation because a number of these are involved in cytoskeletal development, and could be functionally related to neurite outgrowth. Taken together, this study supports a role for abnormal activation of CDK5 in impaired maturation of NPCs in the adult hippocampus in AD. Therapeutic approaches targeting at normalizing, but not eradicating, CDK5 activity may prove useful in promoting the appropriate maturation of hippocampal NPCs in AD.
Materials and Methods
NPC culture, neuronal differentiation assay, and live cell imaging
Adult rat hippocampal NPCs (provided by F Gage, Salk Institute, La Jolla, CA, USA) provide a useful model for studying adult hippocampal neurogenesis. This in vitro model has been extensively characterized in several previous studies where the phenotype and expression patterns of a variety of lineage markers of neuronal progeny derived from these NPCs have been examined in detail.31, 32 Moreover, their survival and differentiation has been confirmed after in vivo transplantation in the brain,32 suggesting that these cells are capable of undergoing neuronal maturation similar to endogenous NPCs.
Adult rat hippocampal NPCs were cultured routinely for expansion as previously described,26 with some modifications. Briefly, cells were grown as monolayers on poly-ornithine/laminin-coated plates in NPC basal media (DMEM/F-12 (Mediatech, Manassas, VA, USA), 2 mM L-glutamine, and 1% penicillin–streptomycin (all from Invitrogen, Carlsbad, CA, USA)) supplemented with 1 × B27 supplement (without vitamin A) and 20 ng/ml fibroblast growth factor-2 (FGF2; Millipore, Temecula, CA, USA). This media (NPC Growth media) was used to enhance growth conditions for maximal proliferation of cells. For differentiation, cells were plated onto poly-ornithine/laminin (Sigma-Aldrich, St. Louis, MO, USA)-coated plates or coverslips in NPC growth media, and transferred the following day (day 0) to NPC differentiation media (NPC basal media supplemented with 1 × N2 supplement (Invitrogen), 1 μM all-trans retinoic acid (Sigma-Aldrich), 5 μM forskolin (Sigma-Aldrich), and 1% FBS). Cells were differentiated for 4 days, with fresh NPC differentiation media added at day 2. Live cell imaging of NPCs and NPC-derived neural progeny in culture was performed on an Olympus (Center Valley, PA, USA) IX70 inverted microscope. It should be noted that this differentiation procedure generates heterogeneous cultures comprising predominantly immature neuronal progeny with a minor fraction of cells expressing progenitor or glial cell markers, and therefore we refer to the cells derived from the differentiation process as ‘NPC-derived neural progeny’.
Adaptation of the adult hippocampal NPC neural induction protocol to model CDK5 activation in AD
To investigate the role of the CDK5 signaling pathway in the differentiation of NPCs, we studied the effects of activating this pathway in the adult rat hippocampal NPC model by utilizing a viral vector expressing the CDK5 activator, p35. As Aβ has been shown to abnormally stimulate the activity of this pathway, p35-expressing differentiating cells were treated in combination with exogenous Aβ peptide. For this purpose, cells were infected on day 2 of differentiation with adenovirus expressing human p35 or GFP control (Vector Biolabs, Philadelphia, PA, USA) at a multiplicity of infection (MOI) of 30. Although p35 expression reflected a several-fold increase over endogenous levels (Figure 1a), this is consistent with a previous study investigating the effects of plasmid-driven p35 overexpression in a mature neuronal cell line, where similar high levels of p35 were demonstrated with no off-target effects.33 Following p35 expression, cells were treated on day 3 with freshly solubilized human Aβ1−42 (1–5 μM; American Peptide, Sunnyvale, CA, USA), or reverse human Aβ42−1 peptide (1 μM; American Peptide) as a control. Additional controls included vehicle alone or a non-Aβ amyloidogenic protein, human TTR (1 μM; EMD Chemicals, San Diego, CA, USA). At the conclusion of the in vitro experiments, cells were lysed for immunoblot analysis, or cells on coverslips were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for immunocytochemical analysis.
For neurite outgrowth studies, a subset of cultured cells on coverslips were fixed in a solution containing glutaraldehyde, which better preserves the cytoskeleton for optimal visualization of neurites using immunofluorescence with an antibody against β-tubulin. For this purpose, a fixation procedure was used essentially as previously described by Desai and Mitchison (http://mitchison.med.harvard.edu/protocols/gen1.html). Briefly, media was gently aspirated from cells growing on glass coverslips, and then cells were extracted for 30 s in cytoskeletal buffer (CB, 80 mM PIPES pH 6.8, 1 mM MgCl2, 4 mM EGTA) containing 0.5% freshly added Triton-X 100. Glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) was immediately added to the CB on the coverslips at a final concentration of 0.5%. Coverslips were incubated for 10 min at 37°C. Fixative was then removed and a freshly prepared solution of 0.1% NaBH4 in PBS was added and samples were incubated for 7 min at room temperature to quench free glutaraldehyde. Coverslips were washed at least three times in PBS to remove the NaBH4, and samples were then processed for β-tubulin immunofluorescence analysis.
Pharmacological treatments, siRNA knockdown, and analysis of cell toxicity
For inhibition of the CDK5 signaling pathway, cells were treated at day 2 of differentiation with either the pharmacological CDK5 inhibitor Roscovitine (1–10 μM, Calbiochem, San Diego, CA, USA) or transfected with siRNA against CDK5 or control nontargeting fluorescent-tagged siRNA (37.5–150 ng; Qiagen, Valencia, CA, USA) using the HiPerfect transfection reagent (Qiagen) according to the manufacturer's protocol. Additional experiments were performed in NPC-derived neural progeny after 4 days of differentiation to assess potential toxicity of the viral expression of p35 and Aβ treatment using the LDH-based CytoTox Assay according to the manufacturer's instructions (Promega, Madison, WI, USA).
Animal models, treatments, and tissue processing
For studies of CDK5 activation and neurogenesis in an animal model of AD, tg mice expressing high levels of mutated human (h)APP751 under the control of the mThy-1 promoter (mThy1-hAPP751)34 were used. These tg mice produce high levels of Aβ1−42, and amyloid plaques are found in the brain at an early age (beginning at 3 months).34 To study the effects of genetic CDK5 inhibition in vivo, Cdk5 heterozygous deficient mice were used (CDK5+/−).22 Full ablation of both gene copies of Cdk5 (CDK5−/−) in mice is embryonic lethal, and hence in order to study Cdk5 gene knockdown in the adult mouse brain, the CDK5+/− animals were used as a model of reduced CDK5 activity. CDK5+/− mice were kindly provided by the laboratory of Dr. Joseph Gleeson (University of California at San Diego (UCSD)).
For in vivo studies of neurogenesis, 8-month-old non-tg, CDK5+/−, APP tg, or CDK5+/−/APP tg crossed mice (N=8 per group) received 5 injections (one per day) with BrdU (Sigma-Aldrich) at 50 mg/kg or vehicle control (saline) (N=4 per group, per treatment). After 1 month, mice were killed and their brains were prepared for immunoblot and immunohistochemical analyses, and stereological assessment of neurogenesis.
For pharmacological treatments, 8-month-old non-tg and APP tg animals (N=8 per group) received intracerebral infusions of Roscovitine (Calbiochem) at a concentration of 20 mg/kg into the lateral ventricle (N=4 per group, per treatment). For this purpose, mice were anesthetized, and under sterile conditions a 26-gauge stainless steel cannula was implanted stereotaxically into the lateral ventricle using the bregma as a reference (0.5 mm; 1.1 mm lateral; depth 3 mm) and secured to the cranium using superglue. The cannula was connected via a 5 mm coil of V3 vinyl tubing (Biolab Products, Lake Havasu City, AZ, USA) to a model 1007D osmotic minipump (Alzet, Cupertino, CA, USA) surgically placed subcutaneously beneath the shoulder. The solutions were delivered at a flow rate of 0.5 μl/h for 2 weeks. The pump was left for an additional 2 weeks and mice were killed at 1 month after the initiation of the infusions.
Behavioral studies
Briefly, as previously described,35 in order to evaluate learning and memory in the CDK5+/−/APP tg mice, the animals were trained in the water maze, beginning with a 3-day exposure to the visible platform. Mice were then tested for learning and memory with the hidden platform for 4 days, followed by one Visible Test trial and 1-day probe testing. In the memory portion of the water maze behavioral test, the platform was removed (Probe test) to evaluate the time spent (Time) and distance traveled (Distance) swimming in the target quadrant where the platform was previously located. All experiments described were approved by the animal subjects committee at the UCSD and were performed according to the NIH recommendations for animal use.
Tissue processing
In accordance with the NIH guidelines for the humane treatment of animals, mice were anesthetized with chloral hydrate and flush-perfused transcardially with 0.9% saline. Brains were removed and divided sagitally. One hemibrain was post-fixed in phosphate-buffered 4% PFA at 4°C for 48 h and sectioned at 40 μm with a Vibratome 2000 (Leica Microsystems, Bannockburn, IL, USA), whereas the other hemibrain was snap frozen and stored at −70°C for protein analysis. All experiments described were approved by the animal subjects committee at the UCSD and were performed according to the NIH recommendations for animal use.
Immunoblot analysis
For preparation of total cell lysates from cultured adult rat hippocampal NPCs and NPC-derived neural progeny, adherent cells in culture were lysed in buffer composed of 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA (TNE) containing 1% Triton-X 100 and fresh protease, and phosphatase and calpain inhibitors to obtain total cell lysates. For preparation of homogenates from mouse brains, briefly as previously described,36 0.1 g of frozen tissue was homogenized in 500 μl of a detergent-free HEPES-based lysis buffer (1.0 mM HEPES, 5.0 mM benzamidine, 2.0 mM 2-mercaptoethanol, 3.0 mM EDTA, 0.5 mM magnesium sulfate, 0.05% sodium azide; final pH 8.8) that facilitates separation of membrane and cytosolic fractions. Fresh protease, phosphatase, and calpain inhibitor cocktails (Calbiochem) were added to all lysis buffers. Following a brief centrifugation step to clear nuclei and cell debris, total homogenates were then centrifuged for 1 h at 100 000 r.p.m. at 4°C. Supernatants were saved (cytosolic fraction) and the pellets (membrane fraction) were resuspended in 500 μl HEPES lysis buffer.
For immunoblot analysis of total cell lysates from NPC-derived neural progeny, 20 μg of total protein from each sample was separated by gel electrophoresis on 4–12% Bis-Tris gels (Invitrogen) and blotted onto 0.45 μm PVDF membranes (Millipore). Immunoblots were blocked in a solution containing 5% BSA in PBS with 0.05% Tween-20 (BSA-PBST). Blots were then probed in primary antibody solutions made up in BSA-PBST containing polyclonal antibodies against CDK5 (1 : 500, C-8, rabbit; Santa Cruz Biotechnology, Santa Cruz, CA, USA), p35/p25 (1 : 500, C-19, rabbit; Santa Cruz Biotechnology), or the immature neuronal marker DCX (1 : 500, goat; Santa Cruz Biotechnology), or mouse monoclonal antibodies against the immature neuronal marker β-III tubulin (1 : 10 000, Tuj1 clone; Covance, Princeton, NJ, USA), the NPC/radial glia marker S100-β (1 : 1000; Sigma-Aldrich), the astroglial marker GFAP (1 : 1000; Millipore), the NPC marker nestin (1 : 1000; Millipore), the proliferating cell marker PCNA (1 : 1000, PC10 clone; Santa Cruz Biotechnology), the radial glial marker BLBP (1 : 1000; Millipore), or actin (1 : 2000, C4 clone; Millipore) as a loading control as previously described.34 In agreement with several previous studies,9, 10, 37 by immunoblot it was difficult to consistently detect the band corresponding to p25 in the in vitro NPC model system. Under basal conditions, only background immunoreactivity was detected in the region of 25 kDa; however, when cells were infected with adv-p35, a faint, diffuse band was visible at a molecular weight of 25 kDa (Figure 1a).
For immunoblot analysis of cytosolic and membrane fractions from brain homogenates, 20 μg of total protein from each sample was separated by gel electrophoresis and immunoblotted and blocked as described for lysates from NPC-derived neural progeny. Blots were then probed with rabbit polyclonal antibodies against CDK5 (1 : 500, C-8; Santa Cruz Biotechnology), phosphorylated CDK5 (Tyr15, 1 : 500; Santa Cruz Biotechnology), p35/p25 (1 : 500, C-19; Santa Cruz Biotechnology), cyclin D1 (1 : 500; Santa Cruz Biotechnology), CDK2 (1 : 500; Santa Cruz Biotechnology), or mouse monoclonal antibodies against full-length human APP (1 : 1000, 6E10 clone; Signet Laboratories, Dedham, MA, USA), Aβ (1 : 1000, 82E1 clone; Signet Laboratories), the immature neuronal marker β-III tubulin (1 : 10 000, Tuj1 clone, Covance), or actin (1 : 2000, C4 clone; Millipore) as a loading control. All blots were then washed in PBS, incubated with secondary species-specific antibodies (1 : 5000 in BSA-PBST), and visualized with enhanced chemiluminescence (ECL; PerkinElmer, Waltham, MA, USA). Images were obtained and semiquantitative analysis was performed with the VersaDoc gel imaging system and Quantity One software (Bio-Rad, Hercules, CA, USA). All signals were normalized to actin as a loading control, and expression levels were calculated as ratios compared with vehicle-treated or wild-type controls.
Immunoprecipitation and kinase activity assay
For analysis of CDK5 activity levels in cultured cells, a kinase activity assay was performed essentially as previously described.38 For this purpose, 300 μg of total cell lysate from NPC-derived neural progeny was incubated with 1 μg of rabbit polyclonal antibody against CDK5 (C-8; Santa Cruz Biotechnology) in a total volume of 500 μl CDK5 IP buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, and 1 mM EGTA) with freshly added protease, calpain, and phosphatase inhibitors for 1 h at 4°C with gentle rotation. Then, 20 μl of protein A (Roche, Basel, Switzerland) prewashed beads slurry was added to the lysate, and samples were incubated for 3 h at 4°C with gentle rotation. Samples were then centrifuged (12 000 r.p.m. for 30 s in a microcentrifuge) and washed three times with 1 ml CDK5 IP buffer, with gentle rotation for 10 min at 4°C between each wash. All supernatant was carefully removed after the final wash. Then, for the kinase activity assay, beads were resuspended in a final volume of 50 μl of CDK5 kinase assay buffer (25 mM Tris pH 7.5, 10 mM MgCl2) containing freshly added 90 μM ATP and 0.1 mM histone H1 peptide (Promega). Samples were incubated for 30 min at 30°C, and then transferred to wells in an all-white 96-well plate. To each sample, 50 μl of Kinase-Glo Plus reagent (Promega) was added, and luminescence values were measured on a DTX 880 Multimode plate reader (Beckman Coulter, Brea, CA, USA). This reagent reacts with the remaining ATP in each sample, and a standard curve of serial dilutions of ATP in kinase buffer alone with Kinase-Glo reagent allowed determination of the concentration of remaining ATP after the kinase reaction.
For assessment of CDK5 activity in mouse brain homogenates, levels of phosphorylated (Tyr15) CDK5 were analyzed as a marker of CDK5 activation. Previous studies have demonstrated that this modification results in dramatically increased CDK5 activity.27 Therefore, relative kinase activity in mouse brains was expressed as fold change in the levels of pCDK5 (Tyr15).
Immunocytochemical analysis
For analysis of levels of components of the CDK5 signaling pathway and markers of neurogenesis in NPC-derived neural progeny, adherent cells on coverslips were fixed in 4% PFA in PBS, and then processed for immunofluorescence or immunocytochemical analysis with diaminobenzidine (DAB). For immunofluorescence, coverslips were blocked in 10% serum (Vector Laboratories, Burlingame, CA, USA), and then immunolabeled with the rabbit polyclonal antibodies against CDK5 (1 : 1000, C-8; Santa Cruz Biotechnology), p35 (1 : 1000, C-19; Santa Cruz Biotechnology), or the mouse monoclonal antibodies against nestin (1 : 200; Millipore) or S100-β (1 : 250; Sigma-Aldrich) detected with the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences, Boston, MA, USA). For double-immunolabeling analysis, some samples were co-labeled with a mouse monoclonal antibody against β-III tubulin (1 : 250, Tuj1 clone; Covance) or a goat polyclonal antibody against the immature neuronal marker DCX (1 : 100; Santa Cruz Biotechnology) detected with FITC-conjugated secondary antibodies (1 : 75; Vector Laboratories). For immunocytochemical analysis with DAB, coverslips were blocked and incubated in primary mouse monoclonal antibody against β-III-tubulin (1 : 250, Tuj1 clone; Covance) or goat polyclonal antibody against DCX (1 : 100; Santa Cruz Biotechnology), and incubated with biotinylated secondary antibodies detected with DAB.
For analysis of the levels of components of the CDK5 signaling pathway and markers of neurogenesis in the brains of non-tg, CDK5+/− mice, or APP tg mice, brain sections were processed for double-labeling immunofluorescence or immunocytochemical analysis with DAB. For immunofluorescence, sections were blocked in 10% serum (Vector Laboratories), and then immunolabeled with the rabbit polyclonal antibodies against CDK5 (1 : 1000, C-8; Santa Cruz Biotechnology) or p35/p25 (1 : 1000, C-19; Santa Cruz Biotechnology), detected with the Tyramide Signal Amplification-Direct (Red) system (NEN Life Sciences). Sections were co-labeled with mouse monoclonal antibodies against human APP (8E5, 1 : 2000, courtesy of ELAN Pharmaceuticals, San Francisco, CA, USA), or the mature neuronal marker MAP2 (1 : 250; Millipore), or the goat polyclonal antibody against DCX (1 : 100; Santa Cruz Biotechnology), a marker of migrating neuroblasts, detected with FITC-conjugated secondary antibodies (1 : 75; Vector Laboratories). Single-labeling immunofluorescence analysis was performed on brain sections with antibodies against CDK5 and DCX as described for double-labeling without a second antibody step. The Aβ plaques in sections from the brains of mice studied were analyzed with an antibody against Aβ (4G8, 1 : 500; Signet Laboratories) detected with FITC-conjugated secondary antibodies (1 : 75; Vector Laboratories). Fluorescent-labeled sections were mounted under glass coverslips with ProLong Gold antifade reagent and imaged with a Zeiss × 63 (N.A. 1.4) objective on an Axiovert 35 microscope (Zeiss, Oberkochen, Germany) with an attached MRC1024 laser scanning confocal microscope system (Bio-Rad).36
For immunocytochemical analysis with DAB, sections were blocked and incubated in rabbit polyclonal antibodies against p35/p25 (1 : 500, C-19; Santa Cruz Biotechnology), rat antibody against BrdU (1 : 100, rat monoclonal; AbD Serotec, Raleigh, NC, USA), or primary mouse monoclonal antibody NeuN (1 : 500; Millipore), and incubated with biotinylated secondary antibodies detected with DAB. For sections immunostained for BrdU, pretreatments were performed essentially as previously described.39 DAB-immunostained sections were mounted with Entellan and imaged with a digital Olympus microscope.
All sections were processed simultaneously under the same conditions and the experiments were performed twice to assess reproducibility. To confirm the specificity of primary antibodies, control experiments were performed where sections were incubated overnight in the absence of primary antibody (deleted). For all immunolabeling studies, assessment of the levels of immunoreactivity was performed utilizing the Image-Pro Plus program (Media Cybernetics, Silver Spring, MD, USA). For each mouse, a total of three sections (10 images per section) were analyzed in order to estimate the average number of immunolabeled cells per unit area (mm2) or the average intensity of the immunoreactive signal (corrected pixel intensity).
Neurite outgrowth studies
For β-tubulin immunofluorescence and neurite outgrowth studies, NPC-derived neural progeny growing on coverslips were fixed with glutaraldehyde and incubated with a mouse monoclonal primary anti-β-tubulin antibody (1 : 250, clone B2.1; Sigma-Aldrich) for 1 h at room temperature and detected with FITC-conjugated secondary antibodies (1 : 75; Vector Laboratories). β-Tubulin-labeled coverslips were mounted under glass coverslips with ProLong Gold antifade reagent with DAPI (Invitrogen) and imaged with a Zeiss × 63 (N.A. 1.4) objective on an Axiovert 35 microscope (Zeiss) with an attached MRC1024 laser scanning confocal microscope system (Bio-Rad).36 For analysis of neurite outgrowth of NPC-derived neural progeny immunolabeled with β-tubulin or DCX-positive cells in the DG of mouse brain sections, processes were traced and lengths were measured using the ImageJ Program (NIH, Bethesda, MD, USA) with the NeuronJ Plugin.40
Quantitative analysis of neurogenesis in the hippocampus
For this purpose, a systematic, random counting procedure, similar to the optical disector, was used as described previously.18 For the purpose of this study, the morphometric analysis was focused on the SGZ of the DG. This area corresponds to the layer of NPCs located directly under the first layer of mature granular cells in the DG, which in addition to the SGZ, includes the granular cell layer (GCL) and the molecular layer (ML). The analysis was centered on the SGZ because a previous study has shown that this is the area most consistently affected in APP tg mice.17 To determine the number of BrdU-positive or DCX-positive cells in the SGZ of the hippocampus, every sixth section (200-μm interval) of the left hemisphere was selected from each animal and processed for immunohistochemistry. The reference volume was determined by tracing the areas using a semiautomatic stereology system (Stereoinvestigator; MicroBrightField, Colchester, VT, USA). Positive cells were counted within a 60 × 60 μm counting frame, which was spaced in a 300 × 300 μm counting grid. Positive profiles that intersected the uppermost focal plane (exclusion plane) or the lateral exclusion boundaries of the counting frame were not counted. The total counts of positive profiles were multiplied by the ratio of reference volume to sampling volume in order to obtain the estimated number of positive cells for each structure. Results were expressed as fold change compared with controls so that the scale is comparable in the studies of neurogenesis in both human and mouse brains.
qRT-PCR analysis
RNA was purified from cultured cells or the hippocampus from control and tg mice using the RNeasy mini kit (Qiagen). Total RNA was reverse transcribed using iScript cDNA Synthesis kit (Bio-Rad) with 1 μg of total RNA in accordance with the manufacturer's instructions. Quantitative PCR was performed with primers specific for CDK5 or GAPDH as a housekeeping gene control (primer assays; Qiagen) using the iCycler iQ Real-Time PCR Detection System (Bio-Rad). Reactions were performed in a volume of 25 μl using the iQ SYBR Green Supermix (Bio-Rad) according to the manufacturer's instructions.
Statistical analysis
All experiments were performed blind coded and in triplicate. Values in the figures are expressed as means±S.E.M. To determine the statistical significance, values were compared using Student's t-test, or one-way ANOVA with post hoc Dunnett's or Tukey–Kramer test. The differences were considered to be significant if P-values were <0.05.
Abbreviations
- AD:
-
Alzheimer's disease
- CDK5:
-
cyclin-dependent kinase-5
- Aβ:
-
amyloid-β protein
- APP:
-
amyloid precursor protein
- DCX:
-
doublecortin
- SGZ:
-
subgranular zone
- DG:
-
dentate gyrus
- PCNA:
-
proliferating cell nuclear antigen
- BLBP:
-
brain lipid-binding protein
- GFAP:
-
glial fibrillary acidic protein
- adv:
-
adenoviral
- tg:
-
transgenic
- BrdU:
-
5-bromo-2-deoxyuridine
- FGF2:
-
fibroblast growth factor-2
- PFA:
-
paraformaldehyde
- PBS:
-
phosphate-buffered saline
- TTR:
-
transthyretin
- DAB:
-
diaminobenzidine
- MAP2:
-
microtubule-associated protein-2
- GCL:
-
granular cell layer
- ML:
-
molecular layer
- NPC:
-
neural progenitor cell
- MOI:
-
multiplicity of infection
- CB:
-
cytoskeletal buffer
- ECL:
-
enhanced chemiluminescence
- qRT-PCR:
-
quantitative real-time PCR
References
Lee HG, Moreira PI, Zhu X, Smith MA, Perry G . Staying connected: synapses in Alzheimer disease. Am J Pathol 2004; 165: 1461–1464.
Cruz JC, Tsai LH . A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr Opin Neurobiol 2004; 14: 390–394.
Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH . Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999; 402: 615–622.
Lee KY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN . Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res 1999; 34: 21–29.
Jessberger S, Gage FH, Eisch AJ, Lagace DC . Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci 2009; 32: 575–582.
Jessberger S, Aigner S, Clemenson Jr GD, Toni N, Lie DC, Karalay O et al. Cdk5 regulates accurate maturation of newborn granule cells in the adult hippocampus. PLoS Biol 2008; 6: e272.
Cicero S, Herrup K . Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci 2005; 25: 9658–9668.
Tsai LH, Delalle I, Caviness Jr VS, Chae T, Harlow E . p 35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994; 371: 419–423.
Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH . Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000; 405: 360–364.
Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA 2000; 97: 2910–2915.
Lopes JP, Oliveira CR, Agostinho P . Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-beta and prion peptides. Cell Cycle 2009; 8: 97–104.
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC et al. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci USA 2004; 101: 343–347.
Boekhoorn K, Joels M, Lucassen PJ . Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis 2006; 24: 1–14.
Li B, Yamamori H, Tatebayashi Y, Shafit-Zagardo B, Tanimukai H, Chen S et al. Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol 2008; 67: 78–84.
Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG . Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience 2004; 127: 601–609.
Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP . Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromolecular Med 2002; 1: 125–135.
Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ . Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease. J Comp Neurol 2006; 495: 70–83.
Rockenstein E, Mante M, Adame A, Crews L, Moessler H, Masliah E . Effects of cerebrolysintrade mark on neurogenesis in an APP transgenic model of Alzheimer's disease. Acta Neuropathol (Berl) 2007; 113: 265–275.
Crews L, Adame A, Patrick C, Delaney A, Pham E, Rockenstein E et al. Increased BMP6 levels in the brains of Alzheimer's disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J Neurosci 2010; 30: 12252–12262.
Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH . The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 1996; 10: 816–825.
Hirota Y, Ohshima T, Kaneko N, Ikeda M, Iwasato T, Kulkarni AB et al. Cyclin-dependent kinase 5 is required for control of neuroblast migration in the postnatal subventricular zone. J Neurosci 2007; 27: 12829–12838.
Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 1996; 93: 11173–11178.
Lagace DC, Benavides DR, Kansy JW, Mapelli M, Greengard P, Bibb JA et al. Cdk5 is essential for adult hippocampal neurogenesis. Proc Natl Acad Sci USA 2008; 105: 18567–18571.
Rockenstein E, Torrance M, Mante M, Adame A, Paulino A, Rose JB et al. Cerebrolysin decreases amyloid-beta production by regulating amyloid protein precursor maturation in a transgenic model of Alzheimer's disease. J Neurosci Res 2006; 83: 1252–1261.
Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP . Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem 2002; 83: 1509–1524.
Ray J, Raymon HK, Gage FH . Generation and culturing of precursor cells and neuroblasts from embryonic and adult central nervous system. Methods Enzymol 1995; 254: 20–37.
Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM et al. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 2000; 26: 633–646.
Johansson JU, Lilja L, Chen XL, Higashida H, Meister B, Noda M et al. Cyclin-dependent kinase 5 activators p35 and p39 facilitate formation of functional synapses. Brain Res Mol Brain Res 2005; 138: 215–227.
Zhang J, Cicero SA, Wang L, Romito-Digiacomo RR, Yang Y, Herrup K . Nuclear localization of Cdk5 is a key determinant in the postmitotic state of neurons. Proc Natl Acad Sci USA 2008; 105: 8772–8777.
Zhang J, Li H, Herrup K . Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation. J Biol Chem 2010; 285: 14052–14061.
Palmer TD, Takahashi J, Gage FH . The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci 1997; 8: 389–404.
Gage FH, Coates PW, Palmer TD, Kuhn HG, Fisher LJ, Suhonen JO et al. Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proc Natl Acad Sci USA 1995; 92: 11879–11883.
Utreras E, Maccioni R, Gonzalez-Billault C . Cyclin-dependent kinase 5 activator p35 over-expression and amyloid beta synergism increase apoptosis in cultured neuronal cells. Neuroscience 2009; 161: 978–987.
Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E . Early formation of mature amyloid-b proteins deposits in a mutant APP transgenic model depends on levels of Ab1-42. J Neurosci Res 2001; 66: 573–582.
Rockenstein E, Adame A, Mante M, Moessler H, Windisch M, Masliah E . The neuroprotective effects of cerebrolysin trade mark in a transgenic model of Alzheimer's disease are associated with improved behavioral performance. J Neural Transm 2003; 110: 1313–1327.
Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 2000; 287: 1265–1269.
Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I et al. Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis 2001; 8: 32–44.
Hsiao YH, Chen PS, Yeh SH, Lin CH, Gean PW . N-acetylcysteine prevents beta-amyloid toxicity by a stimulatory effect on p35/cyclin-dependent kinase 5 activity in cultured cortical neurons. J Neurosci Res 2008; 86: 2685–2695.
Winner B, Lie DC, Rockenstein E, Aigner R, Aigner L, Masliah E et al. Human wild-type alpha-synuclein impairs neurogenesis. J Neuropathol Exp Neurol 2004; 63: 1155–1166.
Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M . Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A 2004; 58: 167–176.
Acknowledgements
This work was supported by the NIH Grants AG10435, AG18440, AG022074, AG11385, NS057096, AG03197, and MH62962, and the NIA Training Grant AG00216.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Crews, L., Patrick, C., Adame, A. et al. Modulation of aberrant CDK5 signaling rescues impaired neurogenesis in models of Alzheimer's disease. Cell Death Dis 2, e120 (2011). https://doi.org/10.1038/cddis.2011.2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.2
Keywords
This article is cited by
-
The role of the DFF40/CAD endonuclease in genomic stability
Apoptosis (2021)
-
Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid beta toxicity
Journal of Neuroinflammation (2018)
-
The Role of Cdk5 in Alzheimer’s Disease
Molecular Neurobiology (2016)
-
Activation of HIF-1α (hypoxia inducible factor-1α) prevents dry eye-induced acinar cell death in the lacrimal gland
Cell Death & Disease (2014)
-
The cyclin-dependent kinase family in the social amoebozoan Dictyostelium discoideum
Cellular and Molecular Life Sciences (2014)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}