
Abstract
Caspase-1, formerly known as interleukin (IL)-1-converting enzyme is best established as the protease responsible for the processing of the key pro-inflammatory cytokine IL-1β from an inactive precursor to an active, secreted molecule. Thus, caspase-1 is regarded as a key mediator of inflammatory processes, and has become synonymous with inflammation. In addition to the processing of IL-1β, caspase-1 also executes a rapid programme of cell death, termed pyroptosis, in macrophages in response to intracellular bacteria. Pyroptosis is also regarded as a host response to remove the niche of the bacteria and to hasten their demise. These processes are generally accepted as the main roles of caspase-1. However, there is also a wealth of literature supporting a direct role for caspase-1 in non-infectious cell death processes. This is true in mammals, but also in non-mammalian vertebrates where caspase-1-dependent processing of IL-1β is absent because of the lack of appropriate caspase-1 cleavage sites. This literature is most prevalent in the brain where caspase-1 may directly regulate neuronal cell death in response to diverse insults. We attempt here to summarise the evidence for caspase-1 as a cell death enzyme and propose that, in addition to the processing of IL-1β, caspase-1 has an important and a conserved role as a cell death protease.
Similar content being viewed by others


Facts
-
Caspase-1 cleaves the pro-inflammatory cytokine pro-interleukin (IL)-1β to an active secreted molecule in monocytes and macrophages.
-
Pyroptosis is a caspase-1-mediated macrophage cell death following infection by intracellular pathogens as part of the host response.
-
The inflammasome is a molecular scaffold that forms in response to pathogen, or damage-associated signals to activate caspase-1.
-
The caspase-1 cleavage site is present only in mammalian pro-IL-1β and is absent in the sequences of other vertebrates.
-
Caspase-1 inhibitors inhibit cell death in mammals and other vertebrates.
Open Questions
-
How is caspase-1 regulated during cell death processes in non-immune cells?
-
Is caspase-1-dependent cell death a conserved mechanism across vertebrates?
-
Have the non-caspase-1-dependent mechanisms of IL-1β secretion in sterile inflammation been underestimated?
Caspase-1 is an enzyme involved in the processing of pro-IL-1β to active secreted IL-1β, a key inflammatory mediator driving the host response to infection, injury, and disease. During disease, IL-1β-driven inflammation has often disastrous consequences, and thus represents a therapeutic target.1 Independent of IL-1β there is unheralded, yet convincing, evidence to suggest that caspase-1 can execute cell death processes. The most extensive evidence for both the inflammatory and direct cell death activities of caspase-1 exists in neuronal injury. Thus, caspase-1 represents a therapeutic target for the treatment of brain injury/disease, conditions for which there is considerable unmet clinical need because of limited clinical options available, and because of the limited regenerative capacity of the brain.2
IL-1β is the best characterised of the 11 IL-1 family members. It is produced by numerous cell types, although the majority of studies focus on its production by cells of the innate immune system, such as monocytes and macrophages.3 It is produced in response to ‘pathogen-associated molecular patterns’ (PAMPs), or ‘damage-AMPs’ (DAMPs) as an inactive 31-kDa precursor, called pro-IL-1β. PAMPs and DAMPs function through pattern recognition receptors (PRRs) on macrophage membranes to regulate pathways that control gene expression.4, 5 Stimuli that control pro-IL-1β expression are, however, generally inefficient as secretion stimuli, but render the cell ‘primed’ for subsequent exposure to more secretion competent stimuli. These further stimuli are additional PAMPs or DAMPs that function on cytosolic PRRs, commonly of the NOD-like receptor (NLR) family.3
An IL-1 processing activity was initially identified in the lysates of LPS-activated monocytes and ascribed as an IL-1 convertase, or IL-1-converting enzyme (ICE).6, 7 Significant homology of ICE with the cell death gene, ced-3, from Caenorhabditis elegans was identified that subsequently led, along with the discovery of multiple related proteases involved in mammalian apoptosis, to the reclassification of ICE as a member of the caspase family of proteases (caspase-1).8, 9, 10 Members of the caspases fall into one of two sub-families; apoptotic or inflammatory; caspase-1 is considered to belong to the inflammatory group.11 Thus, the vast majority of the literature on caspase-1 has focussed on its role in inflammation, with a wider role in cell death rarely considered, except for the pyroptotic cell death of macrophages associated with infection by intracellular pathogens.12 Here, we discuss evidence that caspase-1-dependent cell death is important beyond pyroptosis.
The Activation of Caspase-1
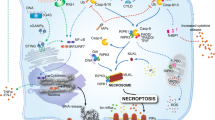
Caspase-1 is activated by recruitment to a molecular platform called an inflammasome.13 The known caspase-1-activating inflammasomes are composed of a PRR of the NLR family such as NLRP1, NLR family pyrin domain (PYD) containing 3 (NLRP3), NLRP6, NLRP7, NLR family CARD domain-containing protein 4 (NLRC4), or the DNA-sensing absent in melanoma 2 (AIM2) and RIG-1 receptors.14, 15, 16 The best-characterised inflammasomes to date are formed by PRRs of the NLR family, NLRP3 and NLRC4. These are composed of several domains including a leucine-rich repeat, important for PAMP/DAMP sensing, a nucleotide-binding domain required for oligomerisation, and a caspase activation and recruitment (CARD) and/or a PYD, for recruitment to caspase-1 directly, or via the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), respectively.14 NLRP3 is activated in response to a variety of structurally diverse PAMPs and DAMPs, and is thought to be the main sensor for sterile inflammatory stimuli (i.e., in response to injury/disease in the absence of infection). NLRC4 is thought to function mainly as a sensor of bacterial infection by sensing flagellin, but also the type III secretion system rod protein PrgJ.14 Although regulation of the inflammasome is an area of enormous interest currently, there are still many outstanding questions regarding the mechanisms of its activation.
The most reported consequence of caspase-1 activation is the rapid secretion of IL-1β. An additional consequence of caspase-1 activation in macrophages following infection by NLRC4-activating pathogens is a rapid, and caspase-1-dependent cell death called pyroptosis.12 Pyroptosis is a pro-inflammatory form of cell death that causes an infected macrophage to kill itself, and at the same time release IL-1β.12 The rapid, caspase-1-dependent pyroptotic cell death caused by Salmonella typhimurium,17 and the caspase-1-dependent clearance of NLRC4-activating pathogens in vivo18 do not depend upon IL-1β processing. Pyroptosis is suggested to serve principally to eliminate the intracellular niche required for pathogen growth.19 The process of pyroptosis is not bactericidal per se, and the released pathogens are killed by the cytotoxic mechanisms of neutrophils recruited to the inflamed tissue.18 However, cell death associated with IL-1β release does not occur only in response to infection with NLRC4-activating pathogens. Stimulation of LPS-primed peritoneal macrophages with the NLRP3-activating stimulus ATP (via the P2x7 receptor20) or allospecific cytotoxic T-lymphocytes induces cell death in addition to IL-1β processing and release.21 Brief (30-min) incubation with ATP causes LPS-primed murine peritoneal macrophages to ‘round up’ and bleb, which is closely followed by the release of the cytolytic marker lactate dehydrogenase.22 ATP-induced death of LPS-treated mouse peritoneal macrophages is caspase-1-dependent and completely independent of IL-1 secretion.20 NLRP3-inflammasome-dependent pyroptosis is also activated by infection of macrophages with Staphylococcus aureus.23 Caspase-1 activation via the AIM2 inflammasome also results in pyroptotic cell death,24 as does activation of the NLRP1 inflammasome.25 Thus, pyroptotic cell death can be activated by many diverse stimuli and by multiple inflammasomes, and is independent of IL-1β.18, 20 Interestingly, caspase-1-dependent cleavage of pro-IL-1β appears to be an exclusively mammalian trait, as other vertebrate pro-IL-1β sequences lack a caspase-1 cleavage site.26 Thus, in these organisms pro-IL-1β must be cleaved by additional proteases, and there is also evidence for alternative processing in mammals.27, 28, 29, 30 Does caspase-1 therefore have a conserved role in cell death independent of IL-1β?
Caspase-1 substrates
The specificity of caspase-1 for cleavage of pro-IL-1β is suggested to be due to the labile nature of its activity, while it appears a rather promiscuous enzyme based on substrate cleavage profiles.31 Several independent proteomic-based approaches to identify caspase-1 substrates have identified numerous proteins, the cleavage of which could result in rapid cell death.31, 32, 33, 34 Identified caspase-1 substrates from these and other studies35, 36, 37, 38, 39 (there are 121 in total, see Supplementary Table 1) suggest a diverse substrate specificity, the cleavage of which could result in the rapid dismantling of the cell, characteristic of pyroptosis. The activation of classical apoptotic caspases, and associated regulators, in addition to cytoskeletal proteins and other proteins essential for cell sustaining processes will give rise to the pyroptotic phenotype of a rapid oncolytic-like cell death with features of apoptosis. Some of these substrates and their ontologies are summarised in Figure 1. The full list of identified caspase-1 substrates and their UniProt identification are supplied in Supplementary Table 1.

Is IL-1 just the tip of the ICEberg? Caspase-1 has a broad range of substrate specificity that extends far beyond inflammation. Depicted here is an iceberg of caspase-1 substrates in which IL-1- and inflammation-related protein substrates are situated at the tip of the iceberg. The uppercase ICE in ICEberg relates to the former name for caspase-1, ICE. Also shown are some selected substrates that may contribute to the phenotype of a pyroptotic cell death. In addition to inflammation, substrates related to the ontologies of cell death, cytoskeleton and metabolism are shown. The full list of substrates is provided as Supplementary information (Supplementary Table 1)
Caspase-1 and Cell Death in Non-mammalian Vertebrates
Chicken pro-IL-1β does not contain a caspase-1 cleavage site,26 although chickens do express caspase-1.40 Cell death induced by trophic factor withdrawl in primary cultured chick dorsal root ganglia (DRG) neurones is inhibited when crmA (cytokine response modifier A gene from cowpox virus that encodes an inhibitor of caspase-141) or bcl-2 are overexpressed,42 and in chick motoneurones treated with the caspase-1 inhibitor YVAD.43 Chicken DRG neurones that over express a dominant-negative caspase-1 enzyme are protected from cell death induced by trophic factor withdrawal.44 The lack of a caspase-1-cleavage site on chicken pro-IL-1β26 suggests that this protective effect of caspase-1 inhibition must be independent of pro-IL-1β processing.
Caspase-1 orthologues have also been identified in fish. In zebrafish (Danio rerio) two orthologues of caspase-1 have been identified, caspy1 and caspy2, as have ASC- and NLR-like molecules.45, 46 Caspy activity is activated by oligimerisation of the zebrafish orthologue of ASC and induces apoptosis when expressed in mammalian 293 T cells.45 YVAD also protects zebrafish embryos from camptothecin-induced cell death.47 Seabream (Sparus aurata L) express caspase-1, and its activity is also effectively blocked by YVAD.48 Classical DAMPs that activate caspase-1 in mammalian cells such as ATP and mono sodium urate do not induce activation of caspase-1 in seabream macrophages.49 However, infection of seabream macrophages with S. typhimurium induces a caspase-1-dependent pyroptotic cell death, and caspase-1-independent processing and secretion of IL-1β.49 These data suggest that the association between caspase-1 activation and IL-1β developed later in evolution, thus further suggesting a conserved role for caspase-1 in cell death (Figure 2).

Conservation of caspase-1-dependent cell death and IL-1β processing. The development of caspase-1 as an inflammatory, in addition to a cell death protease occurred in mammals. Highlighted here are conserved caspase-1 cleavage sites in mammalian vertebrate pro-IL-1β that are not observed in non-mammalian pro-IL-1β sequences. The schematic diagram shows the link between caspase-1, cell death, and inflammation in mammals, and that the link is not present between caspase-1 and inflammation in non-mammalian vertebrates. DANGER represents any source of stress that could drive an inflammatory response such as tissue injury, disease, or infection
Caspase-1 and Cell Death in Mammals
In mammals, caspase-1 cannot be considered a typical regulator of apoptosis. Caspase-1 KO mice develop normally and KO cells undergo apoptosis in response to typical apoptotic stimuli.50, 51 However, there are examples where ectopic overexpression of caspase-1 and its substrates can cause cell death. These effects are due either to the effects of IL-1β, or the direct cell-death-inducing effects of caspase-1, and caspase-1-dependent cleavage of pro-IL-1β may induce apoptosis differently to exogenously administered mature IL-1β. For example, apoptosis in COS cells induced by co-expression of caspase-1 and pro-IL-1β is inhibited by IL-1 receptor antagonist (IL-1Ra). However, the addition of exogenous mature IL-1β before hypoxia is anti-apoptotic via the downregulation of IL-1RI.52 As with the zebrafish caspy described above, overexpression of murine caspase-1 in the rat fibroblast cell line Rat-1 results in cell death and this can be blocked by co-expression of crmA, and of the anti-apoptotic bcl-2.48 There are, however, many examples where endogenous caspase-1 is involved directly in cell death in disease and tissue injury.
Brain injury/neuronal cell death
Acute brain injuries such as stroke, trauma, and haemorrhage, and chronic neurodegenerative diseases, including Alzheimer’s and Parkinson’s diseases, are devastating conditions with pathologies that are exacerbated by inflammation and IL-1.2, 53 Transgenic mice overexpressing a dominant-negative caspase-1 under the control of a neurone-specific promoter exhibit reduced ischaemic brain injury compared with wild-type mice.44 Caspase-1 KO mice also have reduced infarcts compared with wild type after experimental stroke induced by occlusion of the middle cerebral artery (MCAo),54, 55 and intracerebroventricular (i.c.v.) administration of the caspase-1 inhibitor Ac-YVAD-cmk is neuroprotective in this model.54, 56 Following stroke in mice (permanent MCAo) neuronal caspase-1 is activated rapidly (within 30 min), preceding the activation of caspase-3.57 Caspase-1 is also reported to be expressed in neurones in the mouse brain after thromboembolic stroke (as are inflammasome components) but is not expressed by microglia until 24 h post stroke.58 After spinal cord injury in rats, caspase-1 expression is induced rapidly in neurones.59 In addition to the activation of caspase-1 in macrophages (in response to various DAMPs60, 61, 62), lysosomal destabilisation and cathepsin B are also required for caspase-1 activation in neurones after stroke.63 A selective caspase-1 inhibitor is protective after MCAo in rats when injected i.c.v. up to 3 h following reperfusion, but not after 6 h,64 even though IL-1β, protein or message, is below detection limits in the brain parenchyma at this time,65 and IL-1β KO mice are not protected.66 The major IL-1 form present early after stroke is IL-1α,65 which is consistent with the temporal profile of IL-1 family member expression in other paradigms of sterile inflammation.67, 68 Although i.c.v. injection of an anti-IL-1β antibody (given at reperfusion) reduces ischaemic brain injury,69 it may be neutralising low levels of IL-1β in the cerebrospinal fluid rather than parenchymal IL-1β processed by caspase-1. IL-18, another IL-1 family member and the only other cytokine activated by caspase-1 is suggested to have no role in ischaemic brain injury in adult animals.70 In contrast, in rodent neonatal hypoxia/ischaemia, protection against ischaemic brain injury was observed in IL-18-deficient mice.71 In cultured mouse cortical neurones oxygen glucose deprivation (OGD) induces a caspase-1-dependent cell death where caspase-1 activation is apical to the cleavage of BH3-interacting domain death agonist (Bid) and the mitochondrial-dependent activation of caspase-3.72 Furthermore, OGD-induced neuronal cell death in rat organotypic hippocampal slices is blocked by the caspase-1 inhibitor Ac-YVAD-cmk, and this is neither reversed by the addition of IL-1β to the culture, nor is IL-1Ra protective in the absence of Ac-YVAD-cmk.73 In vivo IL-1Ra is protective only when administered within 3 h after MCAo in rats,74 suggesting that its effects may be via the inhibition of IL-1α, and that caspase-1 inhibition in acute brain injury may be targeting cell death directly and independently of IL-1.
In rat pheochromocytoma (PC12) cells, apoptosis induced by the downregulation of Cu2+/Zn2+ superoxide dismutase (SOD1), or by trophic factor/nerve growth factor deprivation, can be prevented by caspase-1 inhibition. Cell death induced by SOD1 suppression is prevented by interventions against IL-1, but a neutralising IL-1β antibody does not protect against trophic factor withdrawal-induced cell death.75 Murine caspase-1 KO DRG neurones are also protected from trophic factor withdrawal-induced death.44 Caspase-1-dependent Bid cleavage drives neurodegeneration in a mouse model of amyotrophic lateral sclerosis induced by transgenic expression of mutant SOD1.76 Inhibition of caspase-1 or caspase-3 also delays mortality in mouse models of amyotrophic lateral sclerosis (mSOD1(G93A) or SOD(G93R) mice).77, 78 This effect is generally thought to be dependent on IL-1β.79, 80 However, chronic expression of IL-1β mRNA in the spinal cord or the absence of IL-1β in SOD1G37R mice does not modify disease progression and motorneuron death.81 In neuroAIDS gp120, a membrane glycoprotein of HIV-1, elevates cytochrome C immunoreactivity, which is blocked by the caspase-1 inhibitor Ac-YVAD-(acyloxy)mk). Ac-YVAD-(acyloxy)mk) did not affect IL-1β levels or gp120-induced cleavage of pro-IL-1β.82 It has been suggested that gp120-induced processing of pro-IL-1β and neuronal apoptosis are matrix metalloproteinase-dependent.83 These data indicate that both caspase-1 and its substrates can be involved in neuronal injury independently of each other. Some examples of the direct cell death-related activities of caspase-1 independent of IL-1 are presented in Table 1.
Other tissue injury
Similarly to the effects of inhibiting caspase-1 or its pro-inflammatory substrates in the brain, there is evidence for the involvement of these pathways in many other models of tissue injury and disease. Caspase-1 is strongly implicated in TNF-α or Fas-ligand-induced hepatocyte apoptosis,84, 85 and a caspase-1 inhibitor completely inhibits Fas-induced mortality in vivo.86 Liver injury induced by major trauma is also caspase-1-dependent, independently of its effects on IL-1β and IL-18 processing.87 There is evidence that IL-18 can also be secreted by macrophages via a caspase-1-independent, Fas/Fas-ligand-mediated manner and contribute to acute liver injury in mice.88 Endotoxemic acute renal failure is attenuated in caspase-1 KO mice, and in these mice blockade of IL-1 by IL-1Ra or neutralisation of IL-18 is not protective.89 In renal ischaemia/reperfusion models, caspase-1 inhibition may be beneficial owing to an inhibition of pro-IL-18 processing,90 whereas no protection is observed in IL-1RI KO mice, or after IL-1Ra administration.91 However, inhibition of IL-18 has no effect against caspase-1-dependent hypoxia-induced death in proximal renal tubules in vitro,92 suggesting that caspase-1 also has a role in renal injury that is independent of IL-1 and IL-18 processing. In renal ischaemia/reperfusion injury NLRP3 KO mice are protected independently of NLRP3’s inflammasome function.93 Inhibition or deletion of caspase-1 also improves outcome after myocardial infarction,94, 95, 96 and this protection may also occur independently of IL-1β and IL-18. Caspase-1 KO mice are protected from ischaemia/reperfusion-induced cardiomyocyte apoptosis, whereas mice with cardiomyocyte-specific overexpression of caspase-1 develop heart failure, in the absence of IL-1β or IL-18.97 Ischaemia/reperfusion injury in transgenic mice overexpressing caspase-1 results in myocardial infarcts that are 50% larger than their non-transgenic littermates and this is suggested to depend upon cross talk with caspase-3.98 Caspase-1 ablation protected photoreceptors in a model of autosomal dominant retinitis pigmentosa, although no protective effect was observed in IL-1R1 KO mice.99 Caspase-1 KO mice are resistant to sepsis induced lethality,50 yet there is no protection in IL-1β KO mice.100 Caspase-1 KO mice are also completely protected from septic shock induced by administration of live Escherichia coli while IL-1β and IL-1β/IL-18 double KO mice suffered the same mortality as the wild-type controls, but were protected by administration of a caspase-1 inhibitor.101 Histological analysis revealed high levels of apoptosis within the B-cell population in the spleen of septic mice that was not present in the caspase-1 KO mice,101 again suggesting that the effects of caspase-1 on cell death are independent of the cleavage of its classical substrates pro-IL-1β and pro-IL-18.
Caspase-1 in humans
IL-1-driven pathology defines a new emerging family of diseases classified as autoinflammatory.102 Within this group of diseases there is some where a link between caspase-1 and IL-1β processing is clearly established, while it remains to be seen in others. Currently, evidence implicating caspase-1 in direct cell death processes in human disease is lacking, although the animal studies described above provide evidence that a role for caspase-1 in cell death processes in humans should not be entirely unexpected. A group of diseases where a role for caspase-1-dependent IL-1β-driven responses is unequivocal are the cryopyrin-associated periodic syndromes (CAPS). These diseases share many features including recurrent bouts of fever, elevated acute phase proteins, fatigue and hearing loss.102 CAPS are caused by gain of function mutations in NLRP3 leading to increased inflammasome formation, caspase-1 activation, and subsequently increased release of IL-1β.103 Disease symptoms resolve following treatment with anti-IL-1β therapies.102
Studies describing clinical use of anti-IL-1 therapies focus almost exclusively on the use of biologicals such as IL-1Ra (anakinra) or anti-IL-1β antibodies such as canakinumab.1 Clinical data on drugs that target caspase-1 are much more limited. Pralnacasan is an orally available caspase-1 inhibitor, that when trialled in phase II for rheumatoid arthritis showed amelioration of disease symptoms, but not for osteoarthritis.104 Trials using pralnacasan were subsequently suspended when a preclinical study in dogs reported liver abnormalities when treated long term with high doses.104 Pralnacasan is currently in phase II trials for the treatment of drug resistant epilepsy. Currently, we can only speculate based upon the animal studies described above, that caspase-1 will also have a direct role in cell death processes in human disease.
Cross Talk Between Inflammatory and Apoptotic Caspases
As discussed above, there is evidence supporting a significant interaction between the pathways regulating inflammatory, and apoptotic caspases (Figure 3). For example, the anti-apoptotic protein bcl-2, which is classically associated with an inhibition of the intrinsic pathway of apoptosis, can also inhibit caspase-1-dependent cell death.39, 42 More recently bcl-2 has subsequently been discovered to bind to, and suppress activation of the NLRP1 inflammasome, inhibiting release of IL-1β in response to the NLRP1-activating ligand muramyl dipeptide,105 and also blocks the activation of the NLRP3 inflammasome in response to apoptotic stimuli in mouse macrophages.106 Members of the inhibitors of apoptosis (IAP) family suppress apoptotic caspases through their E3 ubiquitin ligase activity,107 but have now been shown to regulate the activity of caspase-1 and IL-1β release.108, 109 X-linked IAP protein, a caspase-1 substrate (Figure 1), is reported to be a component of the NLRP1 inflammasome formed after spinal cord injury and after thromboembolic stroke in rodents.58, 59 In several of the models of neuronal cell death described above, caspase-1 is apical to the activation of caspase-3.57, 72 From the proteomic work summarised in Figure 1 we know that caspase-1 can function directly upon caspase-3 and Bid,31 and in several disease models described above caspase-1 is reported to activate Bid.72, 76 Cleavage of Bid is central to the intrinsic cell death pathway of apoptosis, with the truncated form of Bid triggering the process that leads to mitochondrial outer membrane permeabilisation, cytochrome C release, apoptosome formation, and the subsequent activation of caspases 9 and 3/7.110 Caspase-8 is typically associated with Bid cleavage, although the data discussed in this review suggest that caspase-1 could also contribute to this intrinsic cell death pathway. This cross talk between caspase-1 and apoptotic pathways has also been reported in reverse. For example, caspase-8 can cleave pro-IL-1β, at the same site as caspase-1, in response to Toll-like receptor stimulation,111 or following treatment of LPS-primed macrophages with an IAP antagonist.109 In addition, the pro-apoptotic drug staurosporine induces NLRP3-dependent activation of caspase-1 and IL-1β secretion from LPS-primed macrophages via release of oxidised mitochondrial DNA.106 Inflammatory and apoptotic caspase cross talk also occurs during pyroptotic cell death in macrophages, where caspase-7 is activated downstream of NLRP3- and NLRC4-inflammasome-dependent caspase-1 activation.112, 113 Caspase-7 is also activated downstream of caspase-1 in response to LPS in the absence of cell death.114 These data suggest that there is significant overlap between inflammatory and apoptotic caspases and that the signalling processes controlling their regulation are not exclusive to inflammation or to apoptosis.

Inflammatory and apoptotic caspase cross talk. Shown is a summary of some of the interactions between inflammatory and apoptotic caspases. In particular, parallels and overlap between the formation of the inflammasome and the apoptosome are shown. Bold arrows are established links. The dashed arrows highlight possible interactions as suggested by the reviewed literature
Summary
Sterile inflammation is the inflammatory response to injury and disease in the absence of infection and is driven by DAMPs; endogenous host molecules modified during disease, or intracellular proteins released after necrosis.5 The same indiscriminate weapons used during inflammation by recruited leucocytes to kill pathogens (e.g., reactive oxygen species and proteases) kill surrounding healthy cells and thus sterile inflammation exacerbates disease and injury.5, 115 Thus, considering the evidence discussed above for the direct role of caspase-1 in cell death processes after sterile insults, an inhibition of caspase-1 would be anti-inflammatory by preserving cell viability and therefore limiting the release of DAMPs, consequently resulting in less inflammation. In this way caspase-1 could drive an IL-1-dependent inflammation across all vertebrate classes. In mammals, caspase-1-dependent processing of pro-IL-1β can occur although there are additional pathways of pro-IL-1β processing that have been described in disease.27, 28, 29, 30 Thus, a challenge when devising future studies and interpreting current literature will be to dissociate the effects of caspase-1 on cell death and on the processing of IL-1β as it is possible that caspase-1-dependent cell death could lead to an IL-1-dependent inflammatory response, independent of caspase-1-processing of pro-IL-1β. The conserved caspase-1-dependent cell death in non-mammalian vertebrates and the cross talk between inflammatory and apoptotic caspases discussed above, suggest that the involvement of caspase-1 in cell death pathways is much greater than is considered currently.
Abbreviations
- AIM2:
-
absent in melanoma 2
- ASC:
-
apoptosis-associated speck-like protein containing a CARD
- Bid:
-
BH3-interacting domain death agonist
- CAPS:
-
cryopyrin-associated periodic syndrome
- CARD:
-
caspase activation and recruitment domain
- DAMPs:
-
damage-associated molecular patterns
- DRG:
-
dorsal root ganglion
- IAP:
-
inhibitor of apoptosis
- ICE:
-
interleukin (IL)-1-converting enzyme
- IL-1Ra:
-
IL-1 receptor antagonist
- MCAo:
-
middle cerebral artery occlusion
- NLR:
-
NOD-like receptor
- NLRC4:
-
NLR family CARD domain-containing protein 4
- NLRP3:
-
NLR family, pyrin domain (PYD) containing 3
- OGD:
-
oxygen glucose deprivation
- PAMPs:
-
pathogen-associated molecular patterns
- PRRs:
-
pattern recognition receptors
- SOD:
-
superoxide dismutase
References
Dinarello CA . Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011; 117: 3720–3732.
Brough D, Tyrrell PJ, Allan SM . Regulation of interleukin-1 in acute brain injury. Trends Pharmacol Sci 2011; 32: 617–622.
Lopez-Castejon G, Brough D . Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev 2011; 22: 189–195.
Takeuchi O, Akira S . Pattern recognition receptors and inflammation. Cell 2010; 140: 805–820.
Chen GY, Nunez G . Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010; 10: 826–837.
Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci USA 1989; 86: 5227–5231.
Howard AD, Kostura MJ, Thornberry N, Ding GJ, Limjuco G, Weidner J et al. IL-1-converting enzyme requires aspartic acid residues for processing of the IL-1 beta precursor at two distinct sites and does not cleave 31-kDa IL-1 alpha. J Immunol 1991; 147: 2964–2969.
Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR . The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 1993; 75: 641–652.
Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992; 356: 768–774.
Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW et al. Human ICE/CED-3 protease nomenclature. Cell 1996; 87: 171.
Siegel RM . Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol 2006; 6: 308–317.
Bergsbaken T, Fink SL, Cookson BT . Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009; 7: 99–109.
Schroder K, Tschopp J . The inflammasomes. Cell 2010; 140: 821–832.
Strowig T, Henao-Mejia J, Elinav E, Flavell R . Inflammasomes in health and disease. Nature 2012; 481: 278–286.
Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol 2010; 11: 63–69.
Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012; 36: 464–476.
Monack DM, Detweiler CS, Falkow S . Salmonella pathogenicity island 2-dependent macrophage death is mediated in part by the host cysteine protease caspase-1. Cell Microbiol 2001; 3: 825–837.
Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 2010; 11: 1136–1142.
Brodsky IE, Medzhitov R . Pyroptosis: macrophage suicide exposes hidden invaders. Curr Biol 2011; 21: R72–R75.
Le Feuvre RA, Brough D, Iwakura Y, Takeda K, Rothwell NJ . Priming of macrophages with lipopolysaccharide potentiates P2 × 7-mediated cell death via a caspase-1-dependent mechanism, independently of cytokine production. J Biol Chem 2002; 277: 3210–3218.
Hogquist KA, Nett MA, Unanue ER, Chaplin DD . Interleukin 1 is processed and released during apoptosis. Proc Natl Acad Sci USA 1991; 88: 8485–8489.
Perregaux D, Gabel CA . Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 1994; 269: 15195–15203.
Munoz-Planillo R, Franchi L, Miller LS, Nunez G . A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol 2009; 183: 3942–3948.
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES . AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458: 509–513.
Fink SL, Bergsbaken T, Cookson BT . Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA 2008; 105: 4312–4317.
Bird S, Zou J, Wang T, Munday B, Cunningham C, Secombes CJ . Evolution of interleukin-1beta. Cytokine Growth Factor Rev 2002; 13: 483–502.
Fantuzzi G, Ku G, Harding MW, Livingston DJ, Sipe JD, Kuida K et al. Response to local inflammation of IL-1 beta-converting enzyme-deficient mice. J Immunol 1997; 158: 1818–1824.
Joosten LA, Netea MG, Fantuzzi G, Koenders MI, Helsen MM, Sparrer H et al. Inflammatory arthritis in caspase 1 gene-deficient mice: contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum 2009; 60: 3651–3662.
Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M . Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum 2009; 60: 3642–3650.
Stehlik C . Multiple interleukin-1beta-converting enzymes contribute to inflammatory arthritis. Arthritis Rheum 2009; 60: 3524–3530.
Walsh JG, Logue SE, Luthi AU, Martin SJ . Caspase-1 promiscuity is counterbalanced by rapid inactivation of processed enzyme. J Biol Chem 2011; 286: 32513–32524.
Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 2008; 7: 2350–2363.
Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M . The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 2007; 282: 36321–36329.
Agard NJ, Maltby D, Wells JA . Inflammatory stimuli regulate caspase substrate profiles. Mol Cell Proteomics 2010; 9: 880–893.
Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 1997; 275: 206–209.
Cayrol C, Girard JP . The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA 2009; 106: 9021–9026.
Kahns S, Kalai M, Jakobsen LD, Clark BF, Vandenabeele P, Jensen PH . Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem 2003; 278: 23376–23380.
Miggin SM, Palsson-McDermott E, Dunne A, Jefferies C, Pinteaux E, Banahan K et al. NF-kappaB activation by the Toll-IL-1 receptor domain protein MyD88 adapter-like is regulated by caspase-1. Proc Natl Acad Sci USA 2007; 104: 3372–3377.
Weigert A, Cremer S, Schmidt MV, von Knethen A, Angioni C, Geisslinger G et al. Cleavage of sphingosine kinase 2 by caspase-1 provokes its release from apoptotic cells. Blood 2010; 115: 3531–3540.
Johnson AL, Bridgham JT, Munks M, Witty JP . Characterization of the chicken interleukin-1beta converting enzyme (caspase-1) cDNA and expression of caspase-1 mRNA in the hen. Gene 1998; 219: 55–62.
Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS et al. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell 1992; 69: 597–604.
Gagliardini V, Fernandez PA, Lee RK, Drexler HC, Rotello RJ, Fishman MC et al. Prevention of vertebrate neuronal death by the crmA gene. Science 1994; 263: 826–828.
Milligan CE, Prevette D, Yaginuma H, Homma S, Cardwell C, Fritz LC et al. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron 1995; 15: 385–393.
Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G et al. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med 1997; 185: 933–940.
Masumoto J, Zhou W, Chen FF, Su F, Kuwada JY, Hidaka E et al. Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development. J Biol Chem 2003; 278: 4268–4276.
Stein C, Caccamo M, Laird G, Leptin M . Conservation and divergence of gene families encoding components of innate immune response systems in zebrafish. Genome Biol 2007; 8: R251.
Ikegami R, Hunter P, Yager TD . Developmental activation of the capability to undergo checkpoint-induced apoptosis in the early zebrafish embryo. Dev Biol 1999; 209: 409–433.
Lopez-Castejon G, Sepulcre MP, Mulero I, Pelegrin P, Meseguer J, Mulero V . Molecular and functional characterization of gilthead seabream Sparus aurata caspase-1: the first identification of an inflammatory caspase in fish. Mol Immunol 2008; 45: 49–57.
Angosto D, Lopez-Castejon G, Lopez-Munoz A, Sepulcre MP, Arizcun M, Meseguer J et al. Evolution of inflammasome functions in vertebrates: Inflammasome and caspase-1 trigger fish macrophage cell death but are dispensable for the processing of IL-1β. Innate Immun 2012 e-pub ahead of print 28 March 2012.
Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995; 80: 401–411.
Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 1995; 267: 2000–2003.
Friedlander RM, Gagliardini V, Rotello RJ, Yuan J . Functional role of interleukin 1 beta (IL-1 beta) in IL-1 beta-converting enzyme-mediated apoptosis. J Exp Med 1996; 184: 717–724.
Allan SM, Tyrrell PJ, Rothwell NJ . Interleukin-1 and neuronal injury. Nat Rev Immunol 2005; 5: 629–640.
Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z et al. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA 1997; 94: 2007–2012.
Schielke GP, Yang GY, Shivers BD, Betz AL . Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J Cereb Blood Flow Metab 1998; 18: 180–185.
Rabuffetti M, Sciorati C, Tarozzo G, Clementi E, Manfredi AA, Beltramo M . Inhibition of caspase-1-like activity by Ac-Tyr-Val-Ala-Asp-chloromethyl ketone induces long-lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J Neurosci 2000; 20: 4398–4404.
Benchoua A, Guegan C, Couriaud C, Hosseini H, Sampaio N, Morin D et al. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci 2001; 21: 7127–7134.
Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD . Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab 2009; 29: 534–544.
de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW . A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 2008; 28: 3404–3414.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464: 1357–1361.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 2008; 9: 857–865.
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9: 847–856.
Benchoua A, Braudeau J, Reis A, Couriaud C, Onteniente B . Activation of proinflammatory caspases by cathepsin B in focal cerebral ischemia. J Cereb Blood Flow Metab 2004; 24: 1272–1279.
Ross J, Brough D, Gibson RM, Loddick SA, Rothwell NJ . A selective, non-peptide caspase-1 inhibitor, VRT-018858, markedly reduces brain damage induced by transient ischemia in the rat. Neuropharmacol 2007; 53: 638–642.
Luheshi NM, Kovacs KJ, Lopez-Castejon G, Brough D, Denes A . Interleukin-1alpha expression precedes IL-1beta after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J Neuroinflamm 2011; 8: 186.
Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ . Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci 2001; 21: 5528–5534.
Kono H, Karmarkar D, Iwakura Y, Rock KL . Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol 2010; 184: 4470–4478.
Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E et al. IL-1{alpha} and IL-1{beta} recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 2011; 187: 4835–4843.
Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K . Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995; 26: 676–680.
Wheeler RD, Boutin H, Touzani O, Luheshi GN, Takeda K, Rothwell NJ . No role for interleukin-18 in acute murine stroke-induced brain injury. J Cereb Blood Flow Metab 2003; 23: 531–535.
Hedtjarn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H . Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci 2002; 22: 5910–5919.
Zhang WH, Wang X, Narayanan M, Zhang Y, Huo C, Reed JC et al. Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci USA 2003; 100: 16012–16017.
Ray AM, Owen DE, Evans ML, Davis JB, Benham CD . Caspase inhibitors are functionally neuroprotective against oxygen glucose deprivation induced CA1 death in rat organotypic hippocampal slices. Brain Res 2000; 867: 62–69.
Mulcahy NJ, Ross J, Rothwell NJ, Loddick SA . Delayed administration of interleukin-1 receptor antagonist protects against transient cerebral ischaemia in the rat. Br J Pharmacol 2003; 140: 471–476.
Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML . The contrasting roles of ICE family proteases and interleukin-1beta in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci USA 1996; 93: 5635–5640.
Guegan C, Vila M, Teismann P, Chen C, Onteniente B, Li M et al. Instrumental activation of bid by caspase-1 in a transgenic mouse model of ALS. Mol Cell Neurosci 2002; 20: 553–562.
Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan J . Inhibition of ICE slows ALS in mice. Nature 1997; 388: 31.
Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 2000; 288: 335–339.
Pasinelli P, Borchelt DR, Houseweart MK, Cleveland DW, Brown R . Caspase-1 is activated in neural cells and tissue with amyotrophic lateral sclerosis-associated mutations in copper-zinc superoxide dismutase. Proc Natl Acad Sci USA 1998; 95: 15763–15768.
Meissner F, Molawi K, Zychlinsky A . Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc Natl Acad Sci USA 2010; 107: 13046–13050.
Nguyen MD, Julien JP, Rivest S . Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol 2001; 50: 630–639.
Corasaniti MT, Russo R, Amantea D, Gliozzi M, Siviglia E, Stringaro AR et al. Neuroprotection by the caspase-1 inhibitor Ac-YVAD-(acyloxy)mk in experimental neuroAIDS is independent from IL-1beta generation. Cell Death Differ 2005; 12: 999–1001.
Russo R, Siviglia E, Gliozzi M, Amantea D, Paoletti A, Berliocchi L et al. Evidence implicating matrix metalloproteinases in the mechanism underlying accumulation of IL-1beta and neuronal apoptosis in the neocortex of HIV/gp120-exposed rats. Int Rev Neurobiol 2007; 82: 407–421.
Enari M, Hug H, Nagata S . Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature 1995; 375: 78–81.
Los M, Van de Craen M, Penning LC, Schenk H, Westendorp M, Baeuerle PA et al. Requirement of an ICE/CED-3 protease for Fas/APO-1-mediated apoptosis. Nature 1995; 375: 81–83.
Rouquet N, Pages JC, Molina T, Briand P, Joulin V . ICE inhibitor YVADcmk is a potent therapeutic agent against in vivo liver apoptosis. Curr Biol 1996; 6: 1192–1195.
Menzel CL, Sun Q, Loughran PA, Pape HC, Billiar TR, Scott MJ . Caspase-1 is hepatoprotective during trauma and hemorrhagic shock by reducing liver injury and inflammation. Mol Med 2011; 17: 1031–1038.
Tsutsui H, Kayagaki N, Kuida K, Nakano H, Hayashi N, Takeda K et al. Caspase-1-independent Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 1999; 11: 359–367.
Wang W, Faubel S, Ljubanovic D, Mitra A, Falk SA, Kim J et al. Endotoxemic acute renal failure is attenuated in caspase-1-deficient mice. Am J Physiol Renal Physiol 2005; 288: F997–1004.
Melnikov VY, Ecder T, Fantuzzi G, Siegmund B, Lucia MS, Dinarello CA et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest 2001; 107: 1145–1152.
Haq M, Norman J, Saba SR, Ramirez G, Rabb H . Role of IL-1 in renal ischemic reperfusion injury. J Am Soc Nephrol 1998; 9: 614–619.
Edelstein CL, Hoke TS, Somerset H, Fang W, Klein CL, Dinarello CA et al. Proximal tubules from caspase-1-deficient mice are protected against hypoxia-induced membrane injury. Nephrol Dial Transplant 2007; 22: 1052–1061.
Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF et al. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol 2010; 185: 6277–6285.
Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA . Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci USA 2001; 98: 2871–2876.
Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R et al. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol 2003; 35: 685–694.
Holly TA, Drincic A, Byun Y, Nakamura S, Harris K, Klocke FJ et al. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J Mol Cell Cardiol 1999; 31: 1709–1715.
Merkle S, Frantz S, Schon MP, Bauersachs J, Buitrago M, Frost RJ et al. A role for caspase-1 in heart failure. Circ Res 2007; 100: 645–653.
Syed FM, Hahn HS, Odley A, Guo Y, Vallejo JG, Lynch RA et al. Proapoptotic effects of caspase-1/interleukin-converting enzyme dominate in myocardial ischemia. Circ Res 2005; 96: 1103–1109.
Samardzija M, Wenzel A, Thiersch M, Frigg R, Reme C, Grimm C . Caspase-1 ablation protects photoreceptors in a model of autosomal dominant retinitis pigmentosa. Invest Ophth Vis Sci 2006; 47: 5181–5190.
Fantuzzi G, Zheng H, Faggioni R, Benigni F, Ghezzi P, Sipe JD et al. Effect of endotoxin in IL-1 beta-deficient mice. J Immunol 1996; 157: 291–296.
Sarkar A, Hall MW, Exline M, Hart J, Knatz N, Gatson NT et al. Caspase-1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin-1beta and interleukin-18. Am J Resp Crit Care 2006; 174: 1003–1010.
Dinarello CA . Blocking interleukin-1beta in acute and chronic autoinflammatory diseases. J Intern Med 2011; 269: 16–28.
Hoffman HM, Brydges SD . Genetic and molecular basis of inflammasome-mediated disease. J Biol Chem 2011; 286: 10889–10896.
Kersse K, Declercq W, Vandenabeele P . Encyclopedia of Molecular Pharmacology Vol. 1 2nd edn Springer-Verlag: Basel, 2008.
Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C et al. Bcl-2 and Bcl-X(L) regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 2007; 129: 45–56.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012; 36: 401–414.
Vucic D, Dixit VM, Wertz IE . Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev 2011; 12: 439–452.
Labbe K, McIntire CR, Doiron K, Leblanc PM, Saleh M . Cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase-1 activation by the inflammasome. Immunity 2011; 35: 897–907.
Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012; 36: 215–227.
Kantari C, Walczak H . Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochimica et Biophysica Acta 2011; 1813: 558–563.
Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S et al. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med 2008; 205: 1967–1973.
Malireddi RK, Ippagunta S, Lamkanfi M, Kanneganti TD . Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J Immunol 2010; 185: 3127–3130.
Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog 2009; 5: e1000361.
Erener S, Petrilli V, Kassner I, Minotti R, Castillo R, Santoro R et al. Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-kappaB target genes. Mol Cell 2012; 46: 200–211.
Rock KL, Latz E, Ontiveros F, Kono H . The sterile inflammatory response. Annu Rev Immunol 2010; 28: 321–342.
Acknowledgements
We thank Professor Nancy Rothwell and Drs Sylvie Girard and Catherine Lawrence for providing critical feedback on the draft manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Ciliberto
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Denes, A., Lopez-Castejon, G. & Brough, D. Caspase-1: is IL-1 just the tip of the ICEberg?. Cell Death Dis 3, e338 (2012). https://doi.org/10.1038/cddis.2012.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.86
Keywords
This article is cited by
-
Leukocyte differential gene expression prognostic value for high versus low seizure frequency in temporal lobe epilepsy
BMC Neurology (2024)
-
Sodium tanshinone IIA sulfonate ameliorates neointima by protecting endothelial progenitor cells in diabetic mice
BMC Cardiovascular Disorders (2023)
-
Extracellular tau stimulates phagocytosis of living neurons by activated microglia via Toll-like 4 receptor–NLRP3 inflammasome–caspase-1 signalling axis
Scientific Reports (2023)
-
Role of some inflammasomes in rheumatoid arthritis patients in Egypt
Molecular Biology Reports (2023)
-
The NLRP3 inflammasome: molecular activation and regulation in spermatogenesis and male infertility; a systematic review
Basic and Clinical Andrology (2022)
