
Abstract
In fat and muscle cells, insulin-stimulated glucose uptake is mainly mediated by glucose transporter 4 (GLUT4), which translocates from intracellular compartments to the cell surface in response to insulin stimulation. AS160 is one of the substrates of Akt and plays important roles in insulin-regulated GLUT4 translocation. In this study, RuvB-like protein 2 (RUVBL2) is identified as a new AS160-binding protein using mammalian tandem affinity purification (TAP) combined with mass spectrometry. In 3T3-L1 adipocytes, RUVBL2 is highly expressed and is mainly distributed in the cytosol. Depletion of RUVBL2 in adipocytes inhibits insulin-stimulated GLUT4 translocation and glucose uptake through reducing insulin-stimulated AS160 phosphorylation. However, introduction of human RUVBL2 can reverse this inhibitory effect. These data suggest that RUVBL2 plays an important role in insulin-stimulated GLUT4 translocation through its interaction with AS160.
Similar content being viewed by others


Introduction
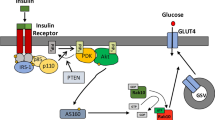
Insulin stimulates glucose uptake into adipocytes and skeletal muscle by inducing translocation of glucose transporter 4 (GLUT4) from intracellular sites to the plasma membrane (PM) 1, 2. This process involves sequential activation of PI3K and Akt after insulin binding to its cell surface receptor.
AS160, one of the substrates of Akt, plays critical roles in insulin-stimulated GLUT4 translocation. Under basal conditions, RNAi-mediated silencing of AS160 perturbs intracellular retention of GLUT4 and induces insulin-independent translocation of GLUT4 to the PM 3, 4. Insulin stimulation elicits phosphorylation of AS160 by Akt at six separate sites. Overexpression of an AS160 mutant, in which four of the six sites have been mutated to alanines (AS1604P), inhibits insulin-stimulated GLUT4 translocation. This inhibitory effect is dependent on the integrity of the Rab GTPase-activating protein (GAP) domain, because simultaneous disruption of the GAP domain in AS1604P (AS1604P, R/K) abolishes its inhibition of insulin-stimulated GLUT4 translocation 5, 6, 7. These data all support the hypothesis that AS160 negatively regulates the activity of Rab protein(s) involved in GLUT4 translocation. Recently, it has been suggested that Rab10 is the Rab protein downstream of AS160, since introduction of the constitutively active Rab10 (Rab10Q67L) increases the PM fraction of GLUT4 in the absence of insulin stimulation, and knockdown of Rab10 retards insulin-stimulated GLUT4 translocation 8, 9. These findings further support the existence of a negative regulatory mechanism between AS160 and Rab protein(s).
14-3–3 has been identified as a novel interaction partner of AS160, and their interaction is known to play an important role in insulin-stimulated GLUT4 translocation 10. This discovery provides new insights into AS160-mediated regulation and reveals that Rab protein(s) should not be the sole interaction partner of AS160.
To identify novel partners that interact with AS160 and are involved in GLUT4 trafficking, we performed tandem affinity purification (TAP) combined with mass spectrometry. We have identified a new AS160-binding protein called RuvB-like protein 2 (RUVBL2). Here, we present evidence that RUVBL2 is involved in insulin-stimulated GLUT4 translocation. These findings may provide novel insights into the precise functions of AS160.
Results
Identification of RUVBL2 as a new AS160-binding protein
To identify proteins that form complexes with AS160, a pCTAP-AS160-3×FLAG expression vector was generated in which streptavidin-binding peptide and calmodulin-binding peptide were tandemly linked to the C-terminus of AS160 with a 3×FLAG epitope in the middle (Figure 1A). For large-scale TAP, CHO cells that were stably transfected with pCTAP-AS160-3×FLAG or empty vector were prepared from 10 × 150-mm plates, which contained a total of 108 cells. Protein complexes associated with tagged AS160 were purified from cell lysates by TAP using streptavidin and calmodulin columns and then visualized by silver staining after SDS-PAGE. Protein bands specific to AS160 (Figure 1B) were excised and subjected to mass spectrometry. Two of these bands were successfully identified as RUVBL2 and 14-3-3. Information for one of the three peptides identified as RUVBL2 by MS/MS spectrum is shown in Figure 1C, while that for the remaining peptides and proteins identified was shown in Supplementary information, Table S1. That 14-3-3 was co-purified with AS160 further demonstrates the validity of TAP purification in this study 10.

Identification of RUVBL2 as an AS160-binding protein. (A) Schematic diagram of the modified pCTAP-A vector for TAP. The pCTAP-A vector includes streptavidin-binding peptide (SBP) and calmodulin-binding peptide (CBP), which is placed in tandem at the C-terminus of the bait protein. A 3×FLAG epitope was also inserted to facilitate the detection of the target protein expression. (B) Protein complexes containing tagged AS160 in cell lysates were purified first with streptavidin and then with calmodulin resin. The final eluates were visualized on a silver-stained SDS-PAGE gel. Four specific bands were excised and subjected to mass spectrometry. Three of them were identified as AS160, RUVBL2, and 14-3–3. The fourth one (indicated by the asterisk) was not identified successfully. (C) MS/MS of one of peptides identified as RUVBL2. (D) Small-scale TAP verified the interaction of RUVBL2 with AS160. CHO cells stably expressing FLAG-RUVBL2 were transfected with pCTAP-AS160-3×FLAG or with the empty vector as a control. Protein complexes were purified in the same way as for large-scale TAP and precipitated proteins were detected with anti-FLAG antibody. Bands corresponding to FLAG-RUVBL2 are displayed. (E) In vivo Co-IP confirmed the interaction between RUVBL2 and AS160. CHO cells were co-transfected with AS160 and FLAG-RUVBL2 or the empty vector. Using anti-FLAG resins to immunoprecipitate RUVBL2, we detected the presence of AS160 in precipitated protein complexes with anti-AS160 antibody. For small-scale TAP and Co-IP, results presented are representative of three independent experiments.
To confirm the interaction of AS160 with RUVBL2, small-scale TAP and Co-IP were performed in CHO cells. For small-scale TAP, CHO cells (a total of 107 cells) stably expressing FLAG-RUVBL2 were transfected with pCTAP-AS160-3×FLAG or the empty vector and subjected to the same purification procedures as those for large-scale TAP. Precipitated proteins were detected with anti-FLAG antibody. That RUVBL2 is specifically associated with AS160 was demonstrated clearly by the presence of a specific band after the two-step purification (Figure 1D). In the Co-IP experiment, AS160 was transiently transfected with or without FLAG-RUVBL2 (Figure 1E). Protein complexes were immunoprecipitated with agarose conjugated with anti-FLAG antibody and precipitated AS160 was detected with anti-AS160 antibody. As shown in Figure 1E, AS160 was immunoprecipitated with RUVBL2. Taken together, these data suggest that AS160 interacts with RUVBL2 in vivo.
RUVBL2 is highly expressed in 3T3-L1 adipocytes
Since the TAP experiment was carried out using CHO cells, we set out to profile the expression of RUVBL2 in 3T3-L1 cells. Cell lysates collected on different days during differentiation were subjected to western blotting for detection of RUVBL2 and AS160. As shown in Figure 2A, as differentiation progressed, RUVBL2 was increasingly upregulated (Supplementary information, Figure S1). However, the expression of AS160 reached a plateau at day 4 of differentiation and declined thereafter, consistent with a previous report 11.

The expression level of RUVBL2 correlates with insulin sensitivity of adipocytes. (A) RUVBL2 expression profile during 3T3-L1 adipocyte differentiation. 3T3-L1 adipocytes were harvested at the indicated times during differentiation, and cell lysates were subjected to immunoblotting for the detection of RUVBL2 and AS160. Actin was used as a loading control. (B) RUVBL2 expression is reduced in adipocytes of obese insulin-resistant diabetic KKAy mice. Epididymal fat tissues of obese insulin-resistant diabetic KKAy mice and C57BL/6J control mice were prepared and cell lysates were immunoblotted to detect the expression of RUVBL2 (C: Control; K: KKAy. Left C and K bands: 20 μg/lane. Right C and K bands: 10 μg/lane). Results presented are representative of three independent experiments.
RUVBL2 expression levels in epididymal fat tissues of obese insulin-resistant diabetic KKAy mice and C57BL/6J control mice were compared, and RUVBL2 expression was found to be markedly reduced in fat tissues of obese insulin-resistant diabetic KKAy mice (Figure 2B). That the expression level of RUVBL2 correlates with insulin sensitivity of adipocytes suggests that it may play some role(s) in insulin sensitivity.
RUVBL2 distributes to the cytosol during the differentiation of adipocytes
Immunofluorescence microscopy was used to determine subcellular distribution of RUVBL2, in both 3T3-L1 fibroblasts and adipocytes. Endogenous RUVBL2 was stained using an anti-RUVBL2 antibody, and DAPI was used to label the nucleus. RUVBL2 was localized predominantly in the nucleus in fibroblasts as demonstrated in Figure 3. After differentiation, RUVBL2 was much more uniformly distributed in adipocytes, with strong staining present in both the nucleus and the cytoplasm (Figure 3 and Supplementary information, Figure S2). The appearance of RUVBL2 in the cytosol in differentiated adipocytes suggests that RUVBL2 may function in both nucleus and cytoplasm.

Subcellular localization of RUVBL2 in 3T3-L1 fibroblasts and adipocytes. 3T3-L1 fibroblasts and adipocytes were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton. RUVBL2 was stained with anti-RUVBL2 antibody and FITC-conjugated goat anti-rabbit secondary antibody, and the cellular nucleus was stained with DAPI. Results presented are representative of three independent experiments.
Depletion of RUVBL2 in 3T3-L1 adipocytes attenuates insulin-stimulated GLUT4 translocation and glucose uptake
To explore the functions of RUVBL2 in adipocytes, where its expression is maintained at high levels and its distribution is mainly cytosolic, the effects of RUVBL2 knockdown on insulin-stimulated GLUT4-EGFP translocation were observed using total internal reflection fluorescence microscopy (TIRFM). As shown in Figure 4A, RUVBL2 levels were greatly reduced 3 days after the introduction of siRUVBL2. Meanwhile, depletion of RUVBL2 in adipocytes did not cause any change to the expression of proximal insulin-signaling molecules, including PI3K, Akt, and AS160, or to the differentiation status (Figure 4A and 4B).

Depletion of RUVBL2 in adipocytes does not alter the expression of proximal insulin-signaling molecules or the differentiation status. 3T3-L1 adipocytes were electroporated with scrambled RNA or siRUVBL2. At 3 days post transfection, adipocytes were collected for western blot or stained with Oil Red O. Results presented are representative of three independent experiments.
Quantification of insulin-stimulated GLUT4-EGFP translocation showed that depletion of RUVBL2 substantially inhibited GLUT4 translocation in adipocytes (Figure 5A and Supplementary information, Figure S3). Introduction of human RUVBL2, which was not targeted by the siRUVBL2 designed against the murine gene, into adipocytes restored insulin-stimulated GLUT4 translocation to the same level as controls (Figure 5A). Measurement of glucose uptake confirmed our TIRFM observations (Figure 5B and Supplementary information, Figure S4). These data all support the hypothesis that RUVBL2 plays roles in insulin-stimulated GLUT4 translocation.

Depletion of RUVBL2 in 3T3-L1 adipocytes inhibits insulin-stimulated GLUT4 translocation and glucose uptake. (A) Quantification of the time course of GLUT4-EGFP translocation to the PM in response to insulin stimulation. 3T3-L1 adipocytes were electroporated with GLUT4-EGFP, corresponding siRNAs, and human RUVBL2, as indicated. After 3 days, adipocytes were stimulated with 100 nM insulin and observed under TIRFM for 30 min. Fluorescence intensities of each image were normalized with the intensities of the images acquired immediately prior to insulin perfusion at 0 min. Data presented are mean value ± S.E.M. (Scrambled, n = 20 cells; siRUVBL2, n = 34 cells; siRUVBL2 + hRUVBL2, n = 30 cells). (B) Insulin-stimulated glucose uptake was attenuated by RUVBL2 knockdown. 3T3-L1 adipocytes were electroporated with scrambled RNA or siRUVBL2, and glucose uptake was determined 3 days after electroporation. All data were normalized with the readouts from the cells transfected with scrambled RNA and in basal condition. Error bars indicate the S.E.M. from three independent experiments. *P < 0.05.
RUVBL2 knockdown reduces insulin-stimulated phosphorylation of AS160 in 3T3-L1 adipocytes
Since RUVBL2 knockdown had an inhibitory effect on GLUT4 translocation, its impact on AS160 phosphorylation, an important regulatory point in the insulin-signaling pathway, was examined. We found that insulin-induced AS160 phosphorylation was reduced in RUVBL2 knockdown adipocytes (Figure 6A). Quantitative results showed that RUVBL2 knockdown significantly reduced insulin-stimulated AS160 phosphorylation to approximate 70% of that of control cells (Figure 6B). Thus RUVBL2 appears to exert its effect on GLUT4 translocation through regulation of insulin-induced AS160 phosphorylation.

RUVBL2 knockdown reduces insulin-stimulated AS160 phosphorylation in 3T3-L1 adipocytes. Adipocytes transfected with scrambled RNA (sc) or siRUVBL2 (si) were serum-starved for 2 h and then treated or not with insulin for 10 min. SDS lysates were prepared and immunoblotted with pAS160 antibody. (A) Representative western blot demonstrating AS160 phosphorylation in control and RUVBL2 knockdown adipocytes. Results presented are representative of three independent experiments. (B) The RUVBL2 knockdown effect on insulin-stimulated AS160 phosphorylation is statistically significant. Insulin-stimulated AS160 phosphorylation is normalized with the band-intensity of scramble RNA-transfected adipocytes with insulin stimulation. Data presented are mean ± S.E.M. from three independent experiments. *P < 0.05.
Discussion
AS160 was identified as one of the substrates of Akt and is known to be critically involved in insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes 4, 5, 6. AS160 exhibits GAP activity toward several Rab proteins including Rab2, 8, 10, and 14 12. Although the cognate Rab that is targeted by AS160 in vivo remains to be identified, it is acknowledged that the interaction between AS160 and certain Rab plays important roles in GLUT4 trafficking. It has been proposed that in the basal state, the GAP activity of AS160 maintains its target Rab in an inactive, GDP-bound form, consequently blocking translocation of GLUT4 to the PM 3, 6. Upon insulin stimulation, Akt phosphorylation of AS160 deactivates its GAP activity, transforming the target Rab into an active GTP-bound form, thereby relieving the inhibitory effect on GLUT4 trafficking 4, 6. Consistent with this idea, we have recently shown that AS160 participates in the docking of GLUT4 storage vesicles at the PM 13, 14.
The picture emerging from the literature is that large proteins like AS160 tend to have multiple interacting partners to fulfill its exquisite in vivo functions. Indeed, AS160 has been proposed as a hub that integrates signals from Akt, protein kinase C and AMP-activated protein kinase 15. It has been suggested that 14-3-3 functions as an AS160-binding protein and that this interaction plays an important role in the insulin-stimulated GLUT4 translocation 10. Calmodulin has also been identified as an AS160-binding protein and has been shown to regulate contraction – but not insulin – stimulated glucose uptake in skeletal muscle 16. In the current study, we searched for additional proteins that interact with AS160 using the TAP strategy combined with protein identification by mass spectrometry, and have identified a novel AS160-binding protein called RUVBL2. RUVBL2 is a member of a highly conserved protein family, is homologous to bacterial RuvB and possesses intrinsic ATPase activity that is stimulated by single-strand DNA 17. RUVBL2 has been shown to be involved in the regulation of gene expression 18, cell development 19, 20, and glucose metabolism 19. Consistent with a previously reported role in glucose metabolism, RUVBL2 was found to be highly expressed in adipocytes in this study (Figure 2A), and a large proportion of RUVBL2 distributes in the cytosol after differentiation (Figure 3). The reason why adipocytes need substantially elevated RUVBL2 and what the precise function of cytosolic RUVBL2 is are issues that are still unresolved. Here, we have demonstrated that depletion of RUVBL2 in adipocytes impairs insulin-stimulated GLUT4 translocation and glucose uptake, likely by decreasing insulin-induced AS160 phosphorylation. This suggests that RUVBL2's role is not restricted to the regulation of transcription in the nucleus, but also involves the regulation of insulin-stimulated GLUT4 trafficking in the cytosol. However, the mechanism of RUVBL2's effect on AS160 phosphorylation remains to be elucidated. In summary, we have demonstrated that RUVBL2 is a new interacting partner of AS160 and is involved in regulating insulin-stimulated GLUT4 translocation.
Materials and Methods
Plasmids construction and generation of stable cell line
The construction of GLUT4-EGFP has been described previously 21. The 3×FLAG epitope sequence synthesized by AuGCT Biotechnology (Beijing, China) was fused to the C-terminal of AS160. AS160-FLAG was cloned into pCTAP-A (Stratagene) to generate pCTAP-AS160-3×FLAG. pCTAP-AS160-3×FLAG was transfected into CHO cells with lipofectamine 2000 (Invitrogen). 48 h after transfection, cells were split into 4 × 10-cm plates using serial 10-fold dilutions (the final dilution was 1:10, 1:100, 1:1 000, and 1:10 000). The next day, the medium was replaced with F12 medium (Gibco) containing 500 μg/ml G418. The cells were incubated in this medium for approximately 15 days, with fresh selection medium change every 3 days. At the end of this period, single clones, which were well defined from their neighbors, could be picked out for large-scale culture. RUVBL2 cDNA was purchased from SANYING Biotechnology (Wuhan, China) and cloned into pcDNA3.1 (+) and pIRES2-DsRed expression vectors.
Antibodies and agents
Anti-FLAG M2 antibody (F1804) was purchased from Sigma. Anti-PI3K (#4292), anti-Akt (#9272), anti-AS160 (#2447), and anti-pAS160 (#9611) antibodies were from Cell Signaling Technologies, and anti-RUVBL2 antibody (ab36569) was purchased from Abcam. FITC-conjugated goat anti-rabbit antibody, HRP-conjugated goat anti-rabbit, and goat anti-mouse secondary antibodies were purchased from Jackson ImmunoResearch, Santa Cruz, and Sigma. DAPI was purchased from Invitrogen. Unless otherwise stated, all reagents were purchased from Sigma.
Cell culture and electroporation
Culture and electroporation of 3T3-L1 cells has been described previously 22. CHO cells stably transfected with pCTAP-AS160-3×FLAG were maintained in Ham's F12 medium supplemented with 10% fetal bovine serum and 2 mM L-Glutamine (Gibco). The siRNA targeting murine RUVBL2 (gi:142383697, nucleotides 732-752, 5′-GATGATCGAGTCCCTGACCAA-3′) was synthesized according to previous report 20. A scrambled sequence (5′-GAACTAGGACACGCTGTCGTA-3′), which has no sequence homology with any murine genes, was used as a negative control.
Animals
12-week-old male C57BL/6J mice and obese insulin-resistant diabetic KKAy mice were purchased from Laboratory Animal Center, Chinese Academy of Military Medical Science and treated according to animal care regulations. Epididymal fat tissues were dissected, ground to a powder in liquid nitrogen, and then lysed with RIPA solution for 20 min on ice.
Tandem affinity purification
For TAP experiment, CHO cells stably expressing AS160 or RUVBL2 were cultured on a large-scale (10 × 150-mm plates containing a total of 108 cells). Cells were lysed in lysis buffer provided in the Interplay TAP Mammalian Purification Kit (Stratagene) with the addition of a protease inhibitor mixture and PMSF (phenylmethyl sulfonylfluoride). TAP-tagged protein and its associated proteins in the soluble lysates were purified according to the protocol provided by the manufacturer. The proteins, which were co-purified with CTAP-AS160 were highly reproducible in several independent experiments.
Mass spectrometry
Silver-stained gel bands were excised and destained in 100 μl of 30 mM K3Fe(CN)6 and 100 mM Na2S2O3 for 30 min and then washed twice in distilled water for 15 min. The gel bands were dehydrated in 100% acetonitrile for 10 min and then dried in a Speedvac for approximately 15 min. The gel bands were reduced with 10 mM dithiotreitol in 100 mM NH4HCO3, at 56 °C for 45 min and alkylated by 40 mM iodacetamide in 100 mM NH4HCO3 in the dark for 30 min at room temperature. The gel pieces were washed with 40 μl of 100 mM NH4HCO3 for 5 min following addition of 40 μl acetonitrile and incubation for 15 min. After gel pieces were dried in Speedvac for 15 min, proteins were digested with sequence-grade modified trypsin (40 ng for each band) in 100 mM NH4HCO3 overnight at 37 °C. Tryptic digestion was stopped by adding formic acid to a 1% final concentration. Peptides were extracted thrice with 50 μl of 60% acetonitrile and the combined solutions were dried with Speedvac. Each tryptic peptide sample was dissolved in 20 μl of 1% formic acid and pressure-loaded onto a 10 cm of C18 capillary column (100 μm i.d.) packed with C18 materials (3 μm, 120A, Sunchrom), and analyzed by LTQ (Thermo). Nanospray ionization was accomplished with a spray voltage of 2.2 kV and heated capillary temperature of 230 °C. A cycle of one full-scan mass spectrum (400-1 600 m/z) was followed by six data-dependent tandem mass spectra. All tandem mass spectra were collected using normalized collision energy (a setting of 35%), an isolation window of 3 m/z, and 1 micro-scan. Application of mass spectrometer scan functions and HPLC solvent gradients were controlled by the XCalibur data system (Thermo Fisher). MS data were analyzed using SEQUEST against NCBI mouse protein database and results were filtered, sorted, and displayed using the Bioworks 3.2. Peptides with +1, +2, or +3 charge states were accepted if they were fully tryptic and had a cross correlation (Xcorr) of ≥1.90, >2.5, and >3.75, respectively.
Immunofluorescence microscopy
3T3-L1 fibroblasts and adipocytes were seeded on coverslips and left to recover for 24 h. Cells were then washed twice with PBS and fixed with 4% paraformaldehyde in PBS at 4 °C for 15 min. Fixed cells were washed thrice with PBS and then permeabilized with 0.2% Triton in PBS for 30 min. After blocking with PBS containing 5% goat serum for 1 h at room temperature, cells were incubated with 1:100 anti-RUVBL2 antibody diluted with PBS containing 5% goat serum at room temperature for 1 h. After washing thrice with PBS, cells were incubated with 1:200 FITC-conjugated anti-rabbit secondary antibody for another hour. After washout of the unbound secondary antibody, the nuclei were stained with DAPI. Images were acquired using Olympus confocal microscopy FV500.
2-Deoxyglucose uptake measurement
3T3-L1 adipocytes transfected with siRUVBL2 or scrambled RNA were serum starved for 2 h at 37 °C and then treated with or without insulin for 15 min. Cells were washed thrice with KRPH buffer (5 mM Na2HPO4, 20 mM Hepes, pH 7.4, 1 mM MgSO4, 1 mM CaCl2, 136 mM NaCl, 4.7 mM KCl, and 1% bovine serum albumin). Glucose transport was determined at 37 °C by incubation with 50 mM 2-deoxyglucose containing 0.5 mCi of 2-[3H] deoxyglucose. The reaction was stopped after 5 min by washing the cells thrice with ice-cold PBS. The cells were solubilized in 1% Triton X-100 at 37 °C for 30 min, and aliquots were subjected to scintillation counting. All the readouts were normalized using the mean value measured from the control cells in the same batch, and three independent experiments were conducted.
TIRFM imaging
The TIRFM setup was constructed based on through-the-lens configuration as described previously 21. Experiments were performed 3 days after transfection in KRBB solution (129 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES, 3 mM glucose, 2.5 mM CaCl2, 1.2 mM MgCl2, and 0.1% BSA. pH adjusted to 7.2). Prior to experiments, transfected adipocytes were serum starved for 2 h and transferred to a closed perfusion chamber. All experiments were performed at 30 °C. Insulin was applied at a final concentration of 100 nM throughout the study and perfused at 0 min.
For quantification of the time course of GLUT4-EGFP translocation, acquired images were processed as follows: first, the cell boundary was detected using the program developed in Matlab (The Math Works) 14. Then, mean fluorescence intensity within the cell boundary was measured. At last, mean values from different time points were normalized with the values from the images captured immediately prior to insulin perfusion at 0 min. Finally, intensity profiles measured from different cells are averaged, and the standard error of the mean (S.E.M.) is calculated.
Statistical analysis
Pooled data are represented as mean ± S.E.M. Statistical comparisons were made using student's t-test.
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Abbreviations
- GLUT4:
-
(glucose transporter 4)
- RUVBL2:
-
(RuvB-like protein 2)
- PI3K:
-
(phosphatidylinositide 3-phosphate kinase)
- AS160:
-
(Akt substrate of 160 kDa)
- TAP:
-
(tandem affinity purification)
- Co-IP:
-
(co-immunoprecipitation)
- CHO:
-
(Chinese hamster ovary)
- PBS:
-
(phosphate-buffered saline)
References
Bryant NJ, Govers R, James DE . Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 2002; 3:267–277.
Watson RT, Kanzaki M, Pessin JE . Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev 2004; 25:177–204.
Eguez L, Lee A, Chavez JA, et al. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab 2005; 2:263–272.
Larance M, Ramm G, Stöckli J, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem 2005; 280:37803–37813.
Kane S, Sano H, Liu SC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem 2002; 277:22115–22118.
Sano H, Kane S, Sano E, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 2003; 278:14599–14602.
Zeigerer A, McBrayer MK, McGraw TE . Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell 2004; 15:4406–4415.
Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE . Rab10 in insulin-stimulated GLUT4 translocation. Biochem J 2008; 411:89–95.
Sano H, Eguez L, Teruel MN, et al. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab 2007; 5:293–303.
Ramm G, Larance M, Guilhaus M, James DE . A role for 14–3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem 2006; 281:29174–29180.
Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE . Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem 2008; 283:9187–9195.
Miinea CP, Sano H, Kane S, et al. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J 2005; 391:87–93.
Jiang L, Fan J, Bai L, et al. Direct quantification of fusion rate reveals a distal role for AS160 in insulin-stimulated fusion of GLUT4 storage vesicles. J Biol Chem 2008; 283:8508–8516.
Bai L, Wang Y, Fan J, et al. Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab 2007; 5:47–57.
Taylor EB, An D, Kramer HF, et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem 2008; 283:9787–9796.
Kramer HF, Taylor EB, Witczak CA, Fujii N, Hirshman MF, Goodyear LJ . Calmodulin-binding domain of AS160 regulates contraction- but not insulin-stimulated glucose uptake in skeletal muscle. Diabetes 2007; 56:2854–2862.
Kanemaki M, Kurokawa Y, Matsu-ura T, et al. TIP49b, a new RuvB-like DNA helicase, is included in a complex together with another RuvB-like DNA helicase, TIP49a. J Biol Chem 1999; 274:22437–22444.
Gallant P . Control of transcription by Pontin and Reptin. Trends Cell Biol 2007; 17:187–192.
Mizuno K, Tokumasu A, Nakamura A, et al. Genes associated with the formation of germ cells from embryonic stem cells in cultures containing different glucose concentrations. Mol Reprod Dev 2006; 73:437–45.
Rousseau B, Ménard L, Haurie V, et al. Overexpression and role of the ATPase and putative DNA helicase RuvB-like 2 in human hepatocellular carcinoma. Hepatology 2007; 46:1108–1118.
Li CH, Bai L, Li DD, Xia S, Xu T . Dynamic tracking and mobility analysis of single GLUT4 storage vesicle in live 3T3-L1 cells. Cell Res 2004; 14:480–486.
Chen Y, Wang Y, Ji W, Xu P, Xu T . A pre-docking role for microtubules in insulin-stimulated glucose transporter 4 translocation. FEBS J 2008; 275:705–712.
Acknowledgements
We would like to thank our colleagues, Prof Hong Tang for providing pCTAP-A expression vector and Dr Li Zheng for valuable discussion. This work is supported by the National Natural Science Foundation of China (NO. 30630020), the National Basic Research Program of China (2004CB720000), and the CAS Project (KSCX1-YW-02-1).
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary information, Table S1
Peptides identified in TAP (PDF 32 kb)
Supplementary information, Figure S1
Western blots of 3T3-L1 cells with RUVBL2 antibodies from Abcam and Santa Cruz. RUVBL2 has a predicted molecular weight of 51 kDa. (PDF 31 kb)
Supplementary information, Figure S2
Knockdown of RUVBL2 greatly reduces immunofluorescence stained by RUVBL2 antibody from Abcam. (PDF 58 kb)
Supplementary information, Figure S3
GLUT4-EGFP expression and distribution in adipocytes quantified by epifluorescence microscopy and TIRFM. (PDF 31 kb)
Supplementary information, Figure S4
RUVBL2 knockdown in adipocytes has no effect on the expression of GLUT1 and GLUT4. (PDF 53 kb)
Rights and permissions
About this article
Cite this article
Xie, X., Chen, Y., Xue, P. et al. RUVBL2, a novel AS160-binding protein, regulates insulin-stimulated GLUT4 translocation. Cell Res 19, 1090–1097 (2009). https://doi.org/10.1038/cr.2009.68
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2009.68
Keywords
This article is cited by
-
Roles of TBC1D1 and TBC1D4 in insulin- and exercise-stimulated glucose transport of skeletal muscle
Diabetologia (2015)
-
The tandem affinity purification technology: an overview
Biotechnology Letters (2011)
