
Abstract
Transplantation of exogenous dopaminergic neuron (DA neurons) is a promising approach for treating Parkinson's disease (PD). However, a major stumbling block has been the lack of a reliable source of donor DA neurons. Here we show that a combination of five transcriptional factors Mash1, Ngn2, Sox2, Nurr1, and Pitx3 can directly and effectively reprogram human fibroblasts into DA neuron-like cells. The reprogrammed cells stained positive for various markers for DA neurons. They also showed characteristic DA uptake and production properties. Moreover, they exhibited DA neuron-specific electrophysiological profiles. Finally, they provided symptomatic relief in a rat PD model. Therefore, our directly reprogrammed DA neuron-like cells are a promising source of cell-replacement therapy for PD.
Similar content being viewed by others


Introduction
Parkinson's disease (PD) is a debilitating neurological disease affecting about half a million of Americans with around 50 000 new diagnoses each year. At present, there are a variety of clinical approaches being used to manage the symptoms of PD at different stages, including administration of levodopa (L-DOPA) 1, 2, or deep brain stimulation (DBS) where a brain pacemaker sending electrical pulses to specific parts of brain is surgically implanted 3. However, these treatments are mostly ineffective for advanced-stage PD patients.
A key feature of PD is progressive degeneration and death of DA neurons in the substantia niagra region of the midbrain. One promising approach to ameliorate the loss of DA neurons is cell-transplantation therapy 4, 5, 6, 7, 8. There have been numerous reports indicating that transplantation of embryonic stem (ES) cells or fetal tissues in experimental PD animal models or human patients showed positive results. In most cases, ES cells or fetal tissue were able to migrate and integrate into the substantia nigra area and provide symptomatic relief to either animal models or human patients of PD. Despite these promising results, transplantation of ES cells or fetus-derived material has encountered formidable technical, legal, and religious barriers.
An exciting new source of DA neurons is the directed differentiation of induced pluripotent stem cells (iPSCs) 9, 10, 11, 12, 13, 14. Because iPSCs can be derived from adult somatic tissues and differentiated into DA neurons with efficiencies similar to those of ES cells, they hold great promise as a replacement for ES cells. However, a major hurdle for this approach is the carcinogenic risk of iPSCs 15, 16. An alternative strategy to obtain DA neurons that may lower carcinogenic risk is the direct conversion approach where differentiated cells (e.g., fibroblast cells) are directly reprogrammed into DA neurons without going through the fully de-differentiated iPS cell route. Successful direct reprogramming of mouse and human fibroblasts has been shown for a variety of cells including neurons (e.g., general neurons 17, 18, DA neurons 19, 20, and motor neurons 21), cardiomyocytes 22, and hematopoietic cells 11. In this study, we report successful reprogramming of human fibroblasts into DA neuron-like cells with a novel set of transcriptional factors. These cells behaved like DA neurons morphologically and electrophysiologically, and could provide symptomatic relief in a rat model of PD.
Results
Five transcription factors that can directly reprogram primary fibroblasts into DA neurons
To reprogram human fibroblasts into DA neuron-like cells, we compiled a list of eight transcriptional factors that had been demonstrated to play critical roles in DA neurons from various publications (Supplementary information, Table S1). These transcription factors were cloned into the lentiviral vector pLEX. Vectors encoding the transcription factors were then packaged into replication-deficient lentiviruses following established procedures (see Materials and Methods section for details of lentivirus preparation).
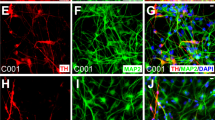
We used the widely published IMR90 human fibroblasts as our source cells. We initially transduced all eight transcription factors into IMR90 cells and placed them onto inactivated PA6 murine feeder fibroblasts 23, 24. About 10-20 days after lentiviral infection, we found that a significant fraction of the surviving cells became elongated and developed dendrite-like protrusions typical of neurons (Supplementary information, Figure S1). Furthermore, many of the neuron-like cells stained positive for markers of DA neurons. Subsequently, we carried out a series of experiments to derive a smaller set of transcriptional factors that are sufficient for the morphological transformation. Our efforts identified a combination of five transcription factors Mash1, Ngn2, Sox2, Nurr1, and Pitx3 that could efficiently transform IMR90 fibroblast cells into DA neuron-like cells following a protocol depicted in Figure 1A. Significant morphological transformation occurred 12-21 days after gene transduction. Interestingly, we found in our experiments that PA6 murine feeder cells were not required for the observed reprogramming. In fact, using the 5-transciption factor (5F) protocol, we could consistently reprogram IMR90 cells, in the absence of PA6 cells, into DA neuron-like cells that stained positive for the general neuron-specific marker Tuj1 and other markers more specifically expressed in DA neurons (Figure 1B, top panel), which included tyrosine hydroxylase (TH), dopa decarboxylase (DDC), and dopamine transporter (DAT) (see Supplementary information, Table S2 for list of protein markers used to characterize DA neurons). In addition, these cells stained negative for serotonin (a marker for serotogenic neurons) and ChAT (a marker for cholinergic neurons), consistent with the reprogrammed cells being DA neuron-like cells. We term these cells human-induced DA neuron cells (hiDA cells).

Successful conversion of human fibroblast into hiDA cells. (A) A schematic illustration of the protocol that we used to transform human fibroblasts into hiDA cells. (B) Top panel: Representative micrographs of hiDA cells stained with various DA neuron-specific (TH, DDC, DAT), general (Tuj1), and other types of neuron (serotonin and ChAT) antibodies. DAPI staining was used to identify individual cells. Lower panel: fraction of surviving cells (stained by DAPI) that stained positive for DDC. The scale bars represent 200 μm. The error bars represent SEM, n = 5. M: Mash1; MS: Mash1 + Sox2; MN: Mash1 + Ngn2; MSN: Mash1 + Sox2 + Ngn2; MSNN: Mash1 + Sox2 + Ngn2 + Nurr1; MSNNP: Mash1 + Sox2 + Ngn2 + Nurr1 + Pitx3. (C) Lack of proliferation in IMR90 fibroblast cells transduced with five factors. Top panel, growth curve of 5F-transduced (IMR90 + 5F) cells; Middle panel, fraction of cells labeled with BrdU when it was added at different times after transduction of 5F; Lower panel: photomicrographs of BrdU staining at day 0 and day 3 after gene transduction. The scale bars represent 200 μm. (D) Q-PCR analysis of key DA-neuron-specific gene expression (left panel). The right panel shows semi-quantitative PCR analysis of the gene. PCR products were electrophoresed in an agarose gel and stained with EtBr. Error bars represent SEM, n = 3.
Essential requirements of Mash1, Ngn2, and Sox2 for direct reprogramming of IMR90 cells into hiDA cells
Among the five factors, Mash1, Ngn2, Sox2 appeared to be essential, as leaving any one of them out of the 5F protocol abolished the reprogramming process (Supplementary information, Figure S2). Mash1 appeared to be of critical importance, as leaving it out abolished any positive staining for Tuj1 or DDC staining (Supplementary information, Figure S2). This is consistent with a recently published study showing that Mash1 is important in the conversion of mouse and human fibroblast into DA neuron-like cells 19. On the other hand, leaving one or both of the remaining two factors (Nurr1 and Pitx3) did not change the frequency of morphologically changed cells, but appeared to attenuate the number of dendrites developing out of the morphologically transformed cells (Supplementary information, Figure S3), indicating that these two factors mainly function to increase the maturity of the reprogrammed DA neurons.
Efficiency of reprogramming and lack of proliferation in hiDA cells
An important issue is the efficiency of reprogramming. There appeared to be a time-dependent increase in cells with dendrites (neuron-like cells) and those staining positive for general and DA neuron-specific markers (Supplementary information, Figure S4). At day 20 after transduction of the five transcription factor cocktail, approximately 40% of the attached cells remaining in the dish stained positive for DDC, a DA neuron-specific marker (Figure 1B, lower panel). The other combinations of transcriptional factors (those with less than 5) also showed clear expression of DDC (Figure 1B, lower panel) despite inducing cells with less dendrite and appeared to be less mature. Almost all DDC+ cells are also Tuj1+ and vice versa. In fact, when Tuj1+ cells were quantified, they exhibit an almost identical profile as DDC+ cells (Supplementary information, Figure S5). It is important to note that many of the cells appeared to have died in the conversion process. By day 20 after 5F transduction, only about 2% - 4% of initially plated IMR90 cells survived (Figure 1C, top panel). Therefore, the overall frequency of fibroblast to hiDA neuron conversion was around 1% - 2%. The decrease in cell numbers was consistent with reduced BrdU labeling. In fact, 3 days after 5F transduction, no cells were labeled positively by BrdU, indicating a total lack of cell proliferation 3 days after 5F transduction (Figure 1C, lower panel). Furthermore, there was clear evidence for cell death in the form of apoptosis, as demonstrated by TUNEL staining, which identifies fragmented DNA in the nuclei of cells experiencing programmed cell death (Supplementary information, Figure S6). The lack of cell proliferation provided solid evidence that the observed reprogramming did not go through de-differentiation and reversion to a stem or progenitor cell phenotype, which are usually associated with proliferation.
Expression levels of key DA neuron-specific genes
We next determined the expression levels of several DA neuron-specific genes through quantitative and semi-quantitative RT-PCR in the hiDA cells (Figure 1D and see Supplementary information, Table S3 for a list of the primers). Our results showed clear and strong expression of DA neuron-specific genes engrailed 1(EN1), TH, DAT, and vesicular monoamine transporter 2 (VMAT2). The expression levels of these genes increased tremendously (from 500-1 100-fold) in the hiDA cells when compared with control fibroblast cells. In fact, there was minimal expression of these genes in the IMR90 human fibroblasts. These data further confirmed the DA neuron-like identity of the hiDA cells. We also examined the endogenous expression of the five transcription factors used for reprogramming through semi-quantitative PCR using pairs of primers that each contains a primer specific for the endogenous gene (see Supplementary information, Table S4 for a list of the primers). Our results (Supplementary information, Figure S7) indicated that exogenous expression of the five transcription factors activated the expression of their endogenous counterparts significantly.
DA uptake and production in hiDA cells
The above results clearly show that our human fibroblast-derived, hiDA cells express key genes expressed in DA neurons. While very encouraging, these results did not confirm the functionality of the DA neuron-like cells. In order to provide evidence that our hiDA cells truly behave like DA neurons, we measured DA uptake in these cells because specific, DAT-mediated dopamine uptake is one of the key characteristics of DA neurons. Our results indicated clearly that uptake of [3H]-labeled dopamine in the reprogrammed putative DA-neuron cells was significantly higher than that of parental IMR90 fibroblasts (Figure 2A). Supplementary information, Table S5 shows additional details of uptake measurements. One interesting fact was that only those cells transduced with the five factors for more than 17 days showed specific DA uptake (Supplementary information, Table S5), indicating that the hiDA cells only exhibit dopamine uptake when they become more mature.

Dopamine release and uptake of fibroblast-derived DA neuron-like cells. (A) Dopamine uptake in putative DA-neuron cells. Cells in 12-well plates were incubated for 10 min with 50 nM [3H]Dopamin in the absence and presence of 0.1 mM cocaine. Cells were then washed in ice-cold KRH buffer, lysed, and radioactivity was determined by a scintillation counter. Specific uptake of dopamine was calculated as the difference between measurement with and without 0.1 mM cocaine. Error bars represent SEM, n = 3. (B) Dopamine production from putative DA-neuron cells. Left panel are representative HPLC chromatograms showing the peaks that were detected for 10 nM DHBA and DA standard solutions (a), DHBA internal standard and endogenous DA in neurons (b), and only DHBA internal standard in fibroblasts (c). Samples were injected at time 0, and retention times of the standards were used to identify DHBA and DA in the tissue samples. Right panel shows quantitative results of HPLC chromatography experiments measuring DA from DA-neuron like cells. Error bars represent SEM, n = 5.
We also measured tissue levels of endogenous DA in hiDA cells using high-performance liquid chromatography (HPLC). Our experiments indicated that our hiDA cells produced a significant amount of DA, in contrast to parental fibroblasts, which produced no DA (Figure 2B). This is consistent with the fact that DA synthesis is a key property of DA neurons.
DA neuron-like electrophysiology of hiDA cells
Further functional proof of the neuron-like identity of the hiDA cells came from whole-cell electrophysiological recordings to measure voltage-gated sodium ion currents (Figure 3 and Supplementary information, Figure S8). When voltage was clamped at different holding potentials with depolarizing voltage steps (starting from −100 mV with successive 10 mV steps), hiDA cells showed a rapidly inactivating inward current (Figure 3A top and middle panel) at each level, followed by a non-inactivating outward current, characteristic of typical neuronal voltage-gated sodium and potassium channels, respectively. Furthermore, when tetrodotoxin (TTX, at 1 μM), a selective sodium channel blocker, was administered, the fast inward currents (activated at voltages below ENa+ = 83.5 mV) were completely blocked (Figure 3A, lower panel), indicating that the observed currents were exclusively mediated through voltage-gated sodium channels. Furthermore, when current was clamped, prolonged depolarizing current injections induced typical neuron-like action potentials driven by sodium channels (Figure 3B, top panel). Moreover, these action potentials were again completely inhibited by TTX in the same cells (Figure 3B, lower panel). Overall, 39 cells were recorded, of which 25 cells fired and 14 did not (Supplementary information, Table S6). These data provide compelling evidence that our hiDA cells do possess neuron-like sodium and potassium channels.

Electrophysiological profiles of putative hiDA neurons. (A) Whole-cell membrane currents from a representative DIV12 cell held at an initial holding potential of −70 mV was first hyperpolarized to −100 mV before successive 10 mV steps were delivered (average of 5 repetitions at a rate of 1 Hz). Fast, rapidly inactivating inward current, detailed in inset, was followed by a non-inactivating outward current. Fast inward currents were quantified from another cell (DIV11), isolated with cesium-based electrode solution to demonstrate expected ENa+ of +83.5 mV and were completely blocked by TTX (1 μM). Symbols ± SEM bars represent the mean of y-axis currents (pA) in the presence and absence of TTX. (B) Current clamp recording in which current injections maintained a starting voltage of −70 mV, followed by a hyperpolarizing current step (−40 pA) and successive 30 pA steps. In response, a typical DIV13 cell demonstrated fast, rapidly inactivating voltage spikes upon depolarizing current injections, which were also sensitive to TTX (1 μM). These are typical of DA neuron action potentials. (C) Overlay of two consecutive whole-cell recording events shown in a representative DIV21 cell before and after application of the D2-receptor selective antagonist (raclopride, 1 μM). Action potentials resulted after a hyperpolarizing step (−40 pA) in the presence of the antagonist. (D) In the every cell assessed, the membrane potential was significantly depolarized in the presence of raclopride (5 cells, mean ± SEM (mV) *P < 0.05, **P < 0.001, paired Student's t-test) compared to baseline control. The input resistance similarly increased in the presence of raclopride for each cell, suggesting D2-autoreceptor antagonism (5 cells, mean ± SEM (MΩ) *P < 0.05, **P < 0.001, paired Student's t-test, input resistance baseline control versus raclopride for each cell).
To establish whether hiDA cells exhibited DA release, whole-cell recordings were done in combination with the competitive DA receptor (D2R)-selective antagonist, raclopride 25. The D2-autoreceptor, a receptor localized on DA neurons, is well known for its involvement in negative feedback regulation of DA neuronal firing 26, and in the presence of raclopride, greater action potential firing results were observed 27. Indeed, hiDA cells that previously did not fire in response to repolarization following a hyperpolarized current step showed robust action potential firing upon antagonist application (Figure 3C, 40% of cells tested), consistent with DA release from the hiDA cells.
Furthermore, it has been established previously that activation of D2-autoreceptors decreases the firing and causes subsequent membrane hyperpolarization of recorded DA neurons 26, 28 and this effect is most likely mediated through GIRKs (G protein-coupled inward rectifying potassium channels) 26, 29. Consequently, raclopride depolarizes the membrane potential of dopamine neurons 27 that should also increase the input resistance 26. Indeed, these changes were observed in the presence of raclopride for these hiDA cells (Figure 3D). To our knowledge, this is the first demonstration of functional dopamine release with D2-autoreceptor expression in directly reprogrammed human DA cells.
Relief of PD symptoms by transplantation of hiDA cells in a rat model of PD
In order to evaluate the functional relevance of our reprogrammed DA neuron-like cells in vivo, we injected the reprogrammed cells into the middle of the striatum of an established rat model for PD (Sprague-Dawley rats lesioned with 6-hydroxydopamine (6-OHDA) and show amphetamine-induced ipsilateral rotations). Our data indicate that compared with controls, which were 6-OHDA lesioned rats injected with saline, those injected with hiDA cells showed a significant stabilization of their rotational behaviors while those injected with saline showed progressively worse rotational behaviors (Figure 4A). When individual rats were examined, four of eight rats injected with cells showed attenuated rotational behavior at 8 weeks post injection (Figure 4B).

In vivo cell transplantation studies with reprogrammed DA neuron-like cells. (A) About 3 × 105 reprogrammed cells (12 days post infection) were injected into the middle of striatum of rats to the side with 6-OHDA-induced lesions with a Hamilton syringe. Right before and at different times after cell transplant, rats were evaluated for amphetamine-induced rotational behavior with a rotometer. Error bars represent SEM, n = 8. (B) Rotational behaviors of four 6-OHDA-lesioned rats injected with hiDA cells. (C) Brain tissues from the two rats that showed consistent correction of amphetamine-induced rotational behaviors were examined for evidence of transplanted cells through immunoflurescenece. HN: staining for human nuclear protein, which is an indicator of injected human cells. Scale bars represent 200 μm.
To show evidence of long-term engraftment, we sacrificed two rats that showed improvement at 16 weeks after hiDA cellular transplantation and examined their brain tissues by immunofluorescence staining. Our results indicate clear evidence for the presence of human cells, as demonstrated by positive staining for a human nuclear protein (HN, Figure 4C). These cells also showed clear staining for TH, DDC, and DAT, indicating that they retained their DA neuron-like properties in vivo for up to 16 weeks post transplantation.
Discussion
The advent of iPSC renewed hopes for cell-replacement therapy for PD. It is now possible to derive an unlimited number of pluripotent stem cell from either skin or other somatic tissues. Indeed, it has been shown that much like ES cells, iPS cells could be differentiated into DA neuron-like cells and these cells could provide symptomatic relieve in rodent models of PD (iPS cells 12 and ES cells 30). In addition, because iPS cells are derived from the patients' own cells, they should not elicit an immunogenic response when transplanted back into the patients' brain, as is likely with transplantation of donor fetal tissues. Despite these significant advantages, there exists a significant hurdle before patient-derived iPS cells could be evaluated in human patients. Because of the use of genes such as Oct4 and Myc, which are known to promote carcinogenesis, there is a clear risk that there may exist a small number of cells that may become oncogenic during the iPSC induction process 12, 31. Therefore, for any iPSC-derived cell-replacement treatment to be evaluated in humans, technological advances have to be made to reduce the carcinogenic risks of iPSCs significantly.
Direct-reprogramming approaches avoid much of the carcinogenic risks of the iPSC approach because differentiated cells (including both the starting and converted cells here) are less likely to form tumors when transplanted in vivo. However, in its current form most direct-reprogramming approaches still use lentiviral vectors, which carry inherent risks of genome integration and inactivation of key tumor suppressor genes that may lead to tumor formation, albeit at much lower risks than the iPSC-associated methods. In the regard, promising alternative gene transduction approaches, such as RNA-based approach, have shown that lentiviral vectors could be replaced all together 32, 33, thereby reducing much of the remaining risks for carcinogenesis in direct conversion of differentiated cells into DA neurons or other differentiated cell lineages.
Recently, there has been a wave of publications that report successful direct reprogramming of fibroblasts into neurons. The earliest of these is the successful conversion of mouse fibroblasts into induced neurons (iNs) using three transcription factors Ascl1 (Mash1), Brn2, and Myt1l by Vierbuchen et al. 17. The same group also showed that the above three factors could convert human fibroblasts into neurons when combined with an additional transcription factor NeuroD1 18. These cells showed morphological and electrophysiological behaviors that are typical of neurons. However, the exact identities of the converted neurons were not clear and they clearly lacked signatures of DA neurons. Using two of the above three transcription factors (MYT1L and BRN2) but with the addition of a microRNA (miR-124), Ambasudhan et al. 34 showed that it is possible to convert human fibroblast cells into functional neurons. Again, these neurons, despite being functional, are not clearly defined in their subtypes. Similarly, Yoo et al. 35 showed that it is possible to convert human fibroblasts into neurons with microRNAs miR-9/9*, miR-124 in combination with NeuroD2. Qiang et al. 36 showed that it is possible to directly convert fibroblasts from patients with Alzheimer's diseases into functional glutmatergenic forebrain neurons with transcription factors Ascl1, Brn2, Zic1, and Myt1l. Several of these protocols appear to confirm the importance of Ascl1 (Mash1) in the reprogramming of fibroblasts into neurons, which was also proved important in our study.
More recently, two reports showed successful direct conversion of DA neuron-like cells in mouse 19 and human fibroblasts 19, 20. Of these, Caiazzo et al. 19 used transcription factors Ascl1 (Mash1), Nurr1, and Lmx1a. In the second report, Ascl1, Brn2, and Mytl were used in combination with Lmx1a and FoxA1 20. Both reports showed DA neuron-like cellular morphology and electrophysiology. However, our hiDA cells appear to be more morphologically mature when compared with those reported in those two reports. Furthermore, compared with our study, only limited in vivo evaluation, in the form of injection and examination of brain slices shortly afterwards (4 days), was done in the report from Caiazzo et al. 19, and no in vivo characterization was carried out in the report of Pfisterer et al. 20. No functional evaluation was carried out in rats with PD symptoms. In comparison, our study involves in vivo functional studies in PD rats over an extended period of time.
In summary, results from this study suggest that our 5F cocktail can successfully reprogram human fibroblast cells into DA neuron-like cells. These cells behave like DA neuron cells both morphologically and functionally. Aside from adding to an increasing collection of successful and exciting direct cellular-reprogramming stories 17, 18, 19, 22, 34, 37, 38, the main implication for our discovery is a potentially safe strategy to derive DA neuron-like cells for treatment of PD, a devastating neurological disease with no universally satisfactory treatment strategy currently available. Because these cells are of human origin and could be obtained with robust efficiency, we believe that they provide a promising source of DA neurons for evaluation as potential treatments for PD.
Materials and Methods
Cell culture
Early passage human fibroblasts IMR90 (from fetal lung), were obtained from the Coriell Institute for Medical Research (Camden, NJ, USA). They were maintained in standard medium (MEM + non-essential amino acid + 10% bovine serum) and sub-cultured every 3-5 days as necessary. To induce DA neuron-like cells, we used DMEM/F12 medium (Fisher) with B27 supplement (Invitrogen), 25 ng/ml Sonic Hedgehog (SHH, R&D), 25 ng/ml FGF-8 (R&D), and non-essential amino acid (Fisher).
Plasmid cloning and lentivirus production
From published literature, we selected a list of eight transcriptional factors that were shown to carry out critical roles in DA neurons (Supplementary information, Table S1). We then obtained cDNAs encoding the factors from Open Biosystems (Huntsville, AL, USA). Brief descriptions of each of the selected transcription factors were included in Supplementary information, Table S1. The ORF of the transcription factors were transferred into the pLEX lentiviral vectors (Open Biosystems, Huntsville, AL, USA) by combined use of PCR and subcloning.
Lentiviral packaging plasmids, psPAX2 and pMD2.G, were obtained from Addgene (Cambridge, MA, USA). These plasmids were originally deposited to Addgene by Dr Didier Trono of EPFL (Ecole Ploytechnique Fédérale de Lausanne, Switzerland).
To produce lentiviral vectors encoding various transcription factors, lentiviral plasmids encoding the individual transcription factors were transduced into 293T cells together with second-generation packaging plasmids from the Trono laboratory (psPAX2, pMD2.G) 39 following a published, calcium phosphate-based Trono lab protocol (http://tronolab.epfl.ch/page58122.html). Supernatants were collected 48 h after transfection. The supernatants, which contain active recombinant lentiviral vectors, were then used in subsequent gene transduction experiments.
Lentivirus transduction
Fibroblasts at the exponential phase of growth were infected with a cocktail of lentiviral vector supernatants with polybrene (10 μg/ml). At 24 h after infection, infected fibroblast cells were trypsinized and replated into a 10-cm Petri dish at a density of 3 × 106 cells or into a 12-well plate at 2.5 × 105 cells/well or into a 24-well plate at 1.5 × 105 cells with polybrene free medium. 24 h later, the medium was changed to DMEM/F12 medium (1:1) containing B27 supplement (Invitrogen), 25 ng/ml SHH (R&D), and 25 ng/ml FGF8 (R&D). Medium was changed every 48 h subsequently. Under some circumstances, putative dopamine neuron-like cells were also digested with Accutase (Gibco) for transferring to a new Petri dish or culturing plates.
Immunofluorescence staining
For immunofluorescence staining, cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 10 min, washed twice with PBS and then blocked for 45 min at room temperature with PBS containing 1% bovine serum albumin and 0.1% Triton X-100. Primary antibodies were used at dilutions recommended by the manufacturer. The antibodies (anti-human in all cases) used were mouse anti-Tuj1 (Covance, 1:800), rabbit anti-TH (Pel-Freez, 1:1 000), rat anti-DAT (Chemicon, 1:5 000), rabbit anti-DDC (Chemicon, 1:500), rat anti-Serotonin (Chemicon, 1:200), goat anti-ChAT (Chemicon, 1:100), mouse anti-HN (Millipore, 1:200). Secondary antibodies labeled with either Alexa488 or Alexa555 were obtained from Invitrogen (Carlsbad, CA, USA). In all cases, cellular nuclei were stained with DAPI in VectorShield mounting medium (Vector Laboratories, Burlingame, CA, USA).
Calculation of transduction efficiency
To calculate the efficiency of neuronal induction, the average number of neuron-specific marker-positive cells present in DAPI-positive cells of 10 randomly selected ×20 visual fields was used to determine the fraction of total number of neurons present. The total numbers of cells were determined using cells cultured in parallel wells or dishes.
Real-time PCR and PCR
Total RNA was extracted using RNAeasy Mini Kit (Qiagen) according to the manufacturer's instructions. RNA was then subjected to complementary DNA synthesis with random hexamer primers using Superscript III reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed using SYBR Green PCR Master Mix (Qiagen). A full description of primer sequence can be found in Supplementary information, Tables S3 and S4.
Quantification of tissue dopamine (DA) levels
We used a previously published method to quantify tissue DA levels 40. Media was removed from the wells and replaced with 100 μl of 0.1 M HClO4 solution containing 7.9 mM sodium metabisulfite and 1.3 mM EDTA, as well as 10 nM dihydroxybenzylamine (DHBA) as an internal standard. The cells were removed by scrapping, and the wells were washed with an additional 100 μl HClO4 solution. Samples were either pooled from all wells for assay, or assayed individually. Cellular suspensions (200 μl) were sonicated and centrifuged for 15 min at 13 000× g and 4 °C. Samples were filtered (0.22 μm), and 50 μl aliquots were subjected to HPLC analysis. The HPLC consisted of an ESA Coulochem III EC detector with a model 5011A analytical cell (guard cell = 300 mV, E1 = −100 mV, E2 = + 220 mV) (ESA Analytical, Chelmsford, MA, USA) and a Brownlee Velosep RP-18 3-μm column (Perkin Elmer, Chelton, CT, USA). The mobile phase consisted of 150 mM NaH2PO4, 4.76 mM citric acid, 50 μM EDTA, 25 mM SDS, 20% methanol and 17% acetonitrile (pH 5.6). Flow rate was 0.6 ml/min. All chemicals were purchased from Sigma-Aldrich, St Louis, MO, USA.
[3H]DA uptake assay
A previously published method 41 was used. Medium was removed from cells that were grown on a 12-well plate and replaced with KRH buffer (120 mM NaCl, 10 mM HEPES, 10 mM glucose, 4.7 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, and 1.2 mM KH2PO4, pH 7.4) supplemented with 0.01 mM pargyline, ascorbic acid and catechol. Specific uptake of 50 nM [3H]DA was then measured for 10 min at 37 °C as the difference between uptake in the absence and presence of 0.1 mM cocaine (∼3 wells/condition/cell type; assay volume = 1 ml). The assay was terminated by quickly washing the cells three times with 1 ml of ice-cold KRH buffer. Cells were then lysed with 0.5 ml 5% trichloroacetic acid for 60 min with gentle shaking before a 0.1 ml aliquot was added to 4 ml of scintillation cocktail. Radioactivity was determined by liquid scintillation spectrometry.
Whole-cell electrophysiological recordings
Whole-cell, patch-clamped recordings were conducted on visually identified cells at DIV11-21 using infrared differential interference contrast optics as previously described 42. Borosilicate glass recording electrodes (Harvard Instruments, Holliston, MA, USA) were pulled to a resistance of 5-9 MΩ using a Sutter P-97 electrode puller (Sutter Instruments, Novato, CA, USA). For voltage-clamped recordings, the internal patch solution comprised 135 mM K-gluconate (interleaved with 135 mM CsMeSO4), 10 mM HEPES, 10 mM EGTA, 0.4 mM Na+-GTP, 4 mM Na+-ATP, 1.5 mM NaCl2, 1 mM MgCl2, 5 mM glucose, pH adjusted to 7.35 with 1 M KOH (or CsOH), with an osmolarity of 316 mOsm. Current-clamped recordings were obtained with the above internal patch solution consisting of 135 mM K-gluconate. The external recording solution was comprised of 140 mM NaCl2, 6 mM KCl, 10 mM HEPES, 15 mM D-glucose, 1.5 mM CaCl2, 1 mM MgCl2, pH adjusted to 7.34 with NaOH at 312 mOsm. TTX (1 μM) was added in some experiments to the extracellular solution to block voltage-gated sodium channels. All salts and drugs were purchased from Sigma Aldrich (St Louis, MO, USA). Membrane potentials were maintained at −70 mV, and voltage steps were delivered from a hyperpolarized potential of −100 mV to −110 mV and subsequently depolarized with 10 mV steps up to either 10 mV or 80 mV at a rate of 1 Hz for up to seven repetitions. Action potentials were recorded by current injection to maintain an initial holding potential of approximately −70 mV and then a hyperpolarizing current step was delivered (−40 pA), with subsequent 30 pA depolarizing steps up to 100 pA at a rate of 1 Hz up to seven repetitions. In a subset of experiments, successive traces were recorded every 10 s in order to monitor the membrane-holding potential and the input resistance up to 5 min. At the start of each trace, the membrane potential was hyperpolarized (−40 pA) from a starting potential near rest (baseline current adjusted but maintained during the experiment to achieve a membrane potential between −60 and −80 mV). Alterations in membrane potential and input resistance in the presence of the D2 receptor antagonist raclopride (1 μM) were also recorded up to 5 min. Additionally, series resistance and membrane capacitance cancellations were performed for all cells larger than Rinput = 1 GΩ. For voltage-clamp recordings in the presence of cesium, no resting membrane potential was recorded. Responses were amplified and filtered (Axopatch 200B, MDS, Sunnyvale, CA, USA) as previously described 42.
Electrophysiological analysis
Responses were measured and analyzed using the software pClamp version 8.2 (MDS, Sunnyvale, CA, USA). Voltage-clamped traces were averaged at each holding potential across repetitions and initial baseline was consistently set between 56.5 ms and 106.5 ms at −70 mV. For I-V (current-voltage) plots, amplitudes were measured by averaging at the peaks of both inward and outward current. Current amplitudes were subsequently plotted with respect to the voltage at which currents were obtained. Amplitudes originating from the same voltage step were then averaged, and error bars represent the mean amplitude ± SEM for a given voltage. To assess D2-autoreceptor antagonism, membrane-holding potential was averaged by selecting multiple points over a time course of 40 s up to 2 min for a control baseline. The same was done in the presence of raclopride, and the baseline difference was calculated per cell. Input resistance measurements were based on dV/dI per baseline of each cell. Membrane potential and input resistances before and during drug (raclopride, 1 μM) application were compared for each cell with paired Student's t-test. Passive membrane properties were additionally assessed in which n = number of recordings from individual cells and were expressed as mean ± SEM. Mann-Whitney Rank Sum tests were performed for two group comparisons. Statistical significance was set at P ≤ 0.05 (Sigmaplot, Systat, Point Richmond, CA, USA).
Transplantation of cells into a rat model of Parkinson's disease
We followed a previously established cellular transplant protocol 43. Sprague-Dawley rats (3 months of age from Taconic) were unilaterally lesioned in the medial forebrain bundle (stereotaxic coordinates from Bregma were AP − 4.3, ML + 1.2, DV − 8.3) with 6-hydroxydopamine (2 μg/μl infused at a rate of 1 μl/min over 4 min). The rats were evaluated for completeness of the lesion by their rotational behavior in response to D-amphetamine. The animals were placed in a rotometer (San Diego Instruments) and the rotational behavior in response to D-amphetamine (5 mg/kg) was recorded quantitatively for 18 consecutive 5-min periods (90 min total).
Reprogrammed DA cells were transplanted into the middle of the striatum using stereotaxic surgery (coordinates from Bregma AP + 0.5, ML + 2.5, DV − 5.0). Cells were implanted using a Hamilton syringe (3 μl of concentration = 105 cells/μl at a rate of 1 μl/min). At 4 weeks, 8 weeks, and 12 weeks after implantation, the animals were tested for rotational behavior in response to D-amphetamine as previously described.
Animals were sacrificed at the conclusion of the study by barbiturate overdose and perfused with 4% paraformaldehyde. Brains were removed and post-fixed in 4% paraformaldehyde overnight and subsequently immersed in 30% sucrose. Fixed tissue was sectioned in the coronal plane on a freezing microtome at 30 μm thickness and subsequently processed for immunofluorescence and immunohistochemistry. Staining of the tissue sections for tyrosine hydroxylase, dopamine transporter, and human nuclei antibodies was performed as markers of the implanted cells.
References
Bezard E, Brotchie JM, Gross CE . Pathophysiology of levodopa-induced dyskinesia: potential for new therapies. Nat Rev Neurosci 2001; 2:577–588.
Jenner P . Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci 2008; 9:665–677.
Brodsky MA, Nutt JG . Parkinson disease: deep brain stimulation versus best medical therapy for PD. Nat Rev Neurol 2010; 6:530–532.
Sladek JR Jr, Redmond DE Jr, Collier TJ, et al. Transplantation of fetal dopamine neurons in primate brain reverses MPTP induced parkinsonism. Prog Brain Res 1987; 71:309–323.
Lindvall O, Brundin P, Widner H, et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson's disease. Science 1990; 247:574–577.
Spencer DD, Robbins RJ, Naftolin F, et al. Unilateral transplantation of human fetal mesencephalic tissue into the caudate nucleus of patients with Parkinson's disease. N Engl J Med 1992; 327:1541–1548.
Widner H, Tetrud J, Rehncrona S, et al. Bilateral fetal mesencephalic grafting in two patients with parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N Engl J Med 1992; 327:1556–1563.
Deacon T, Schumacher J, Dinsmore J, et al. Histological evidence of fetal pig neural cell survival after transplantation into a patient with Parkinson's disease. Nat Med 1997; 3:350–353.
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131:861–872.
Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663–676.
Szabo E, Rampalli S, Risueno RM, et al. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 2010; 468:521–526.
Wernig M, Zhao JP, Pruszak J, et al. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson's disease. Proc Natl Acad Sci USA 2008; 105:5856–5861.
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L . Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 2009; 27:275–280.
Sanchez-Danes A, Consiglio A, Richaud Y, et al. Efficient generation of A9 midbrain dopaminergic neurons by lentiviral delivery of Lmx1a in human embryonic stem cells and iPS cells. Hum Gene Ther 2011 Aug 30; doi:10.1089/hum.2011.054.
Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 2008; 26:101–106.
Miura K, Okada Y, Aoi T, et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol 2009; 27:743–745.
Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M . Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010; 463:1035–1041.
Pang ZP, Yang N, Vierbuchen T, et al. Induction of human neuronal cells by defined transcription factors. Nature 2011; 476:220–223.
Caiazzo M, Dell'anno MT, Dvoretskova E, et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011; 476:224–227.
Pfisterer U, Kirkeby A, Torper O, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci USA 2011; 108:10343–10348.
Son EY, Ichida JK, Wainger BJ, et al. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 2011; 9:205–218.
Ieda M, Fu JD, Delgado-Olguin P, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010; 142:375–386.
Kawasaki H, Mizuseki K, Nishikawa S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 2000; 28:31–40.
Sasai Y . Generation of dopaminergic neurons from embryonic stem cells. J Neurol 2002; 249 Suppl 2:II41–II44.
Goldstein M . Dopaminergic mechanisms in self-inflicting biting behavior. Psychopharmacol Bull 1989; 25:349–352.
Lacey MG, Mercuri NB, North RA . Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol 1987; 392:397–416.
Prasad BM, Amara SG . The dopamine transporter in mesencephalic cultures is refractory to physiological changes in membrane voltage. J Neurosci 2001; 21:7561–7567.
Rayport S, Sulzer D, Shi WX, et al. Identified postnatal mesolimbic dopamine neurons in culture: morphology and electrophysiology. J Neurosci 1992; 12:4264–4280.
Beckstead MJ, Grandy DK, Wickman K, Williams JT . Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 2004; 42:939–946.
Kim JH, Auerbach JM, Rodriguez-Gomez JA, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson's disease. Nature 2002; 418:50–56.
Nakagawa M, Takizawa N, Narita M, Ichisaka T, Yamanaka S . Promotion of direct reprogramming by transformation-deficient Myc. Proc Natl Acad Sci USA 2010; 107:14152–14157.
Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010; 7:618–630.
Miyoshi N, Ishii H, Nagano H, et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011; 8:633–638.
Ambasudhan R, Talantova M, Coleman R, et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 2011; 9:113–118.
Yoo AS, Sun AX, Li L, et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011; 476:228–231.
Qiang L, Fujita R, Yamashita T, et al. Directed conversion of Alzheimer's disease patient skin fibroblasts into functional neurons. Cell 2011; 146:359–371.
Huang P, He Z, Ji S, et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011; 475:386–389.
Sekiya S, Suzuki A . Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 2011; 475:390–393.
Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D . Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 1997; 15:871–875.
Nelson AM, Larson GA, Zahniser NR . Low or high cocaine responding rats differ in striatal extracellular dopamine levels and dopamine transporter number. J Pharmacol Exp Ther 2009; 331:985–997.
Sorkina T, Richards TL, Rao A, Zahniser NR, Sorkin A . Negative regulation of dopamine transporter endocytosis by membrane-proximal N-terminal residues. J Neurosci 2009; 29:1361–1374.
Stubblefield EA, Benke TA . Distinct AMPA-type glutamatergic synapses in developing rat CA1 hippocampus. J Neurophysiol 2010; 104:1899–1912.
Collier TJ, Gallagher MJ, Sladek CD . Cryopreservation and storage of embryonic rat mesencephalic dopamine neurons for one year: comparison to fresh tissue in culture and neural grafts. Brain Res 1993; 623:249–256.
Acknowledgements
We thank Zhilin Song (University of Colorado Denver, USA) for insightful discussions. This study was supported in part by grants from the US National Cancer Institute (CA131408, CA136748 and CA155270) and US National Aeronautics and Space Administration Ground-based Space Radiation Biology Research Program (NNX09AH19G) (to C-Y Li). It was also supported by US NIH grants (DA015050 and DA004216) (to NR Zahniser), and NIH grant NS065353 (to JR Sladek). Qian Huang was supported by the grant from the National Basic Research Project of China (2010CB529902).
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Table S1
A select list of transcription factors important mDA neurons (PDF 112 kb)
Supplementary information, Table S2
A summary of various neuron-specific proteins used to identify DA-neurons. (PDF 109 kb)
Supplementary information, Table S3
Primers used for quantitative and semi-quantitative PCR (Figure 1D). (PDF 117 kb)
Supplementary information, Table S4
Primers used for semi-quantitative PCR of exogenous and endodgenous genes. (PDF 108 kb)
Supplementary information, Table S5
Data of [3H]DA uptake in putative DA neuron-like cells and in IMR90 fibroblasts. (PDF 108 kb)
Supplementary information, Tabe S6
A summary of electrophysiological recordings of Na+ currents of putative neurons. (PDF 142 kb)
Supplementary information, Figure S1
Morphological changes in IMR90 cells (left panel) after tranduction of 8-factors for 12 days (right panel). (PDF 650 kb)
Supplementary information, Figure S2
Top panel: Requirement for individual transcription factors in reprogramming of IMR90 cells into DA neuron-like cells. (PDF 184 kb)
Supplementary information, Figure S3
Increased dendrite network in 5F vs 3F tranduced IMR90 cell at 18 days post gene transduction. (PDF 107 kb)
Supplementary information, Figure S4
Top panel: Time dependent increase in dendrite-like structures observed in 5F transduced IMR90 cells (100x). (PDF 282 kb)
Supplementary information, Figure S5
Fraction of surviving cells that stained positive for Tuj1 cell IMR90 cells transduced with various combinations of transcription factors. (PDF 34 kb)
Supplementary information, Figure S6
Apoptotic cell death as measured by TUNEL staining in IMR90 cells transduced with 5F. (PDF 69 kb)
Supplementary information, Figure S7
Semi-quantitative PCR analysis of the expression of five factors in parent IMR90(as denoted by a “-” sign”) as well 5F-transduced IMR90 cells. (PDF 151 kb)
Supplementary information, Figure S8
Patch clamp recording. (PDF 531 kb)
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0
About this article
Cite this article
Liu, X., Li, F., Stubblefield, E. et al. Direct reprogramming of human fibroblasts into dopaminergic neuron-like cells. Cell Res 22, 321–332 (2012). https://doi.org/10.1038/cr.2011.181
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2011.181
Keywords
This article is cited by
-
The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders
Translational Neurodegeneration (2022)
-
Induced Dopaminergic Neurons for Parkinson’s Disease Therapy: Targeting the Striatum or Midbrain/Substantia Nigra Pars Compacta?
Neuroscience Bulletin (2022)
-
Neurog2 directly converts astrocytes into functional neurons in midbrain and spinal cord
Cell Death & Disease (2021)
-
Recent advances on drug development and emerging therapeutic agents for Alzheimer’s disease
Molecular Biology Reports (2021)
-
Direct conversion of human fibroblasts into dopaminergic neuron-like cells using small molecules and protein factors
Military Medical Research (2020)
