
Abstract
We report five cases of multiple giant cell lesions in patients with typical Noonan syndrome. Such association has frequently been referred to as Noonan-like/multiple giant cell (NL/MGCL) syndrome before the molecular definition of Noonan syndrome. Two patients show mutations in PTPN11 (p.Tyr62Asp and p.Asn308Asp) and three in SOS1 (p.Arg552Ser and p.Arg552Thr). The latter are the first SOS1 mutations reported outside PTPN11 in NL/MGCL syndrome. MGCL lesions were observed in jaws (‘cherubism’) and joints (‘pigmented villonodular synovitis’). We show through those patients that both types of MGCL are not PTPN11-specific, but rather represent a low penetrant (or perhaps overlooked) complication of the dysregulated RAS/MAPK signaling pathway. We recommend discarding NL/MGCL syndrome from the nosology, as this presentation is neither gene-nor allele-specific of Noonan syndrome; these patients should be described as Noonan syndrome with MGCL (of the mandible, the long bone…). The term cherubism should be used only when multiple giant cell lesions occur without any other clinical and molecular evidence of Noonan syndrome, with or without mutations of the SH3BP2 gene.
Similar content being viewed by others

Introduction
Mutations in genes of the RAS/MAPK signaling pathway have been reported in several developmental disorders. In most cases, those mutations are expected to lead to overactivation of the pathway. Implications of germline mutations in the RAS/MAPK pathway in development and malignancies have been reviewed recently.1, 2, 3 Five core phenotypes are associated with constitutionally deregulated RAS/MAPK signaling: Noonan syndrome (NS), LEOPARD syndrome (LS), cardiofaciocutaneous syndrome (CFC), Costello syndrome (CS), and neurofibromatosis 1. They are collectively referred to as the neuro-CFC syndromes.4
The dominantly inherited NS is a well-defined syndrome characterized by typical facial dysmorphism, cardiac anomalies, growth retardation, and inconstant cognitive impairment. Birth prevalence is estimated between 1/1000 and 1/2500. Mutations in PTPN115 are reported in about 50% of cases, SOS16, 7 in 10–20%, RAF18 and KRAS,9 both in less than 5%, Rare patients with BRAF, MEK1, or MEK2 may exhibit an NS phenotype,10 although most of them have CFC syndrome.
Several ‘Noonan-like (NL) entities’ have been delineated. When NS and CFC genes were discovered, it became evident that those variants reflected intrinsic, non-allele-specific variability in the expression of those genes. The NL/multiple giant cell lesion (NL/MGCL) syndrome is one of those rare variants that were once considered as bona fide syndromes. MGCLs, also known as central giant cell lesions or giant cell granulomas, are non-neoplastic lesions characterized by a proliferation of granulation tissue containing many multinucleated giant cells (Langhans giant cells) embedded in a fibrous stroma. MGCLs are found in several disorders, among which two are related to NS, ‘cherubism’ and pigmented villonodular synovitis. MGCLs of the jaws are osteoclast-rich lesions. Most cases are sporadic, unilateral, and more common in women. By contrast, a bilateral, symmetric form is observed in a rare autosomal dominant disorder, called cherubism syndrome, with male preponderance. Cherubism syndrome is characterized by bilateral swelling of the jaws, full cheeks, hypertelorism, prognathism, malocclusion, and oligodontia. Enlargement of the cheeks first appears around the third or fourth year of life, usually stabilizes after puberty and abates by the third decade. Cystic lesions are symmetrical. On X-rays, they appear as multilocular cysts that expand the cortical plate and may displace tooth buds and erode roots. Local aggressiveness of the tumor is variable. Histologically, the lesions are indistinguishable from those of the sporadic, unilateral cases. They are similar to brown tumors of hyperparathyroidism. In the adult, the cystic lesions give place to sclerosis with progressive calcifications. Cherubism syndrome is fully penetrant in men and partially (50–70%) penetrant in women. A majority of cases have been associated with missense mutations affecting a 6-amino-acid sequence (aa 33–aa 38) upstream of the SH2 domain of the SH3BP2 protein. The corresponding gene SH3BP2 (Src homology 3-binding protein 2) maps to chromosome 4p16.3. Sporadic MGCLs of the jaw are not associated with the germline or somatic mutations of SH3BP2.11
Pigmented villonodular synovitis (PVS) is a benign proliferative disorder of the synovial tissue, which may affect the synovial-lined large joints (more commonly the knee and ankle) or tendon sheaths of small joints. It is more frequent in women. Patients usually present with painless swelling of the joints. PVS is sporadic and its pathogenesis is unknown. Histologically, PVS lesions are central giant cell lesions similar to those seen with cherubism. The presence of hemosiderin in giant cells and surrounding stroma gives the tissue a characteristic pigmentation. The lesions tend to be hyperplastic, hypervascular and hemorrhagic. PVS typically invades the surrounding tissues, including the subchondral bone, resulting in bone cyst formation.
NL/MGCL syndrome combines clinical signs of NS with multiple giant cell lesions similar to those observed in cherubism and PVS.12, 13, 14 Before the discovery of the NS-causing genes, nine patients have been described with NS and cherubism15, 16, 17, 18, 19, 20 and only one with NS and PVS.21 Nine patients with NS and cherubism have been reported with PTPN11 mutations.13, 14, 22, 23, 24, 25 These mutations are not specific (the same variation can be observed with or without MGCL lesions, even in a single pedigree).
To date, no mutations in SOS1, KRAS, RAF1, BRAF or MEK have been reported in patients with this phenotype and no PTPN11 alteration. Similarly, no mutation has been reported insofar in patients with NS and PVS. We report five cases of NS with MGCL, including three patients with SOS1 mutations.
Clinical cases reports
Clinical data of the five cases are summarized in Table 1.
Patient 1
The first patient had a birth length (BL) of 55 cm (>90th centile), a birth weight (BW) of 4288 g (>90th centile) and an occipitofrontal head circumference (OFC) of 35.5 cm. (50–75th centile) at term. Stenosis of the pulmonary valve was diagnosed in early infancy. We saw him first at the age of 8 years. He has typical features for NS. His karyotype was 46, XY, inv(9) (p11q13). An X-ray survey and CT scan showed bilateral radiolucent cysts of the mandible that were not operated. Eruption of the incisors was delayed. He had a normal stature and no learning difficulties.
Patient 2
This patient was born at 41 weeks of gestation (WG) with normal birth parameters: BL 48 cm (25th centile) and BW 3610 g (75–90th centile). Mitral valve prolapse was diagnosed at the age of 10 years. We saw him first at 11 years. He had typical NS, with cryptorchidism and decreased platelet aggregation function. Cerebral MRI showed moderate ventriculomegaly. Cysts of the mandible were present. At the age of 12 years, he had normal stature, dyslexia, and learning difficulties. His IQ was 93.
Patient 3
This patient was born at 39 WG. Hydramnios was present during pregnancy. BW was 3950 g (>90th centile), and BL 51 cm (90th centile). Stenosis of the pulmonary artery and atrial septal defect were diagnosed at birth. Clinical features were typical for NS. At 12 years, PVS of the left ankle was diagnosed and confirmed by bone scintigraphy, MRI, and biopsy. At the age of 15 years, his height was at −2 DS. He had delayed puberty, left cryptorchidism, and no learning disabilities. There was no involvement of the jaws.
Patient 4
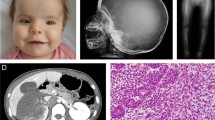
This boy was born at 41 WG, BW was 3850 g (75th centile), BL 51 cm (50th centile), and OFC 35.5 cm (50–75th centile). Stenosis of the pulmonary valve was diagnosed after birth. He carried a maternally inherited balanced translocation 46, XY, t(3;22) (p21q13)mat. At 5 years of age, he developed a cherubic appearance (Figure 1). Multiple radiolucent lesions in both ascending rami of the mandible were found (Figure 1). Partial bilateral resection was carried out at the age of 6 years. Pathological examination showed MGCL. Progressive bilateral enlargement of lesions and recurrence of the tumor in the ascending ramus of the right mandible at the age of 8 made further surgery mandatory. He had speech delay and learning disabilities. At 8 years of age, his DQ was 87, with specific visual constructional problems and discrepancy between verbal and performance scores. Final height was 162 cm (−1.9 DS).

Patient 4. (a) and (b) Facial appearance in adolescence after surgical correction of left eyelid ptosis. Characteristic gestalt of NS. The facial fullness with swollen cheeks and mild prognathism are the consequences of mandibular MGCL. (c) CT scan of the mandible with bilateral radiolucent lacunae corresponding to the extensive bilateral MGCL, more extensive on the left. (d) Partial orthopantogram of the left mandible showing the expansive lesions.
Patient 5
This boy was born at 38 WG, BW was 3320 g (50th centile), and BL 51 cm (75th centile). His father was 58 years of age at conception. Valvular and supravalvular pulmonary stenoses were diagnosed during gestation. NS was suspected at birth because of his facial features. A left unilocular mandibular radiolucency was coincidentally discovered at 12 years on an orthopantogram, and confirmed on a CT scan of the mandible (Figure 2). A histopathological examination of the lesion disclosed MGCL. His height was at −2 DS at 12 years. He had had no difficulties at school, but was depressive.

Patient 5. (a): Partial orthopantogram of the jaw showing cystic erosion of the mandible. (b) CT scan of the lower jaw showing bilateral cysts invading the mandibular rami.
Molecular investigations
Direct sequencing of the PTPN11 coding region (exons 1–15) was carried out in the five patients. Exons 16–23 of SOS1 were further sequenced in patients 1, 2, and 3 using standard protocols. On the basis of RefSeq NM_005633.2, the mutation c.1656G>C (p.Arg552Ser) was found in exon 11 of SOS1 in patients 1 and 3 (who are not related). This mutation has been reported earlier in common NS.26, 27 The mutation c. 1655G>C (p.Arg552Thr) was found in exon 11 of SOS1 in patient 2. This mutation has never been reported. On the basis of RefSeq NM_002834.3, mutations c.184T>G (p.Tyr62Asp) in exon 3 and c.922A>G (p.Asn308Asp) in exon 8 of PTPN11 were found in patients 4 and 5, respectively. These two mutations have been reported earlier in NS.26, 27 No PTPN11 mutations were found in patients 1, 2, and 3.
Discussion
To date, at least 23 patients with NL/MGCL phenotype have been reported under various names, most of them without molecular data.13, 14, 15, 17, 19, 20, 21, 22, 23, 24, 28, 29 Two were familial.20, 28 Taking these reports into account, 11 patients carrying a mutation of PTPN11 were found among 18 NL/MGCL-screened cases (61 %). In a series of eight patients with cherubism, Jafarov et al24 found three with a mutation in SH3BP2 and two with a mutation in PTPN11. One of the latter has clinical features of NL/MGCL syndrome; the other one was not described. None of them carried a mutation in both genes.
MGCL has been reported with other phenotypes linked to the RAS/MAPK pathway. A case of MGCL was reported with LS.30 Sarkozy et al31 reported a patient harboring a mutation in PTPN11 with a complex phenotype progressing throughout the years from NS toward LS and NL/MGCL. Interestingly, some patients with NF1 have been reported with MGCLs of various locations, including the jaws.32, 33, 34, 35, 36
Before the elucidation of the molecular basis of NS, some authors had suggested that the association of MGCL with an NS phenotype was an independent NL entity, the NL/MGCL syndrome. The mutations observed in patients with MGCL are not different from those observed in NS without any bone anomalies. A mutation can be observed with or without granulomas in the same family. NL/MGCL syndrome must be considered as a part of the NS spectrum, and not as an NL disorder. We strongly support the idea of discarding the use of NL/MGCL syndrome, Noonan-cherubism or other specific names to refer to this presentation of NS. NS with MGCL may be used to describe the affected patients. Cherubism should only be used for patients with sporadic or SH3BP2-related multiple giant cell lesions of the jaws, without any evidence of NS.
The pathophysiology of MGCL remains mysterious. SH3BP2 codes for the multidomain scaffolding protein, SH3BP2,37 a critical regulator of myeloid cell responses. Mutations in cherubism lead to a gain of function or act in a dominant-negative manner, leading to abnormal gain of myeloid cell function. Osteoclasts are multinucleated cells derived from the myeloid lineage. The major factors for osteoclast differentiation are RANKL (through its receptor RANK) and M-CSF; SH3BP2 functions downstream of both. It contributes to the activation of ERK, which is at the bottom of the MAPK-dependent pathway, leading to the production of TNFα and to the activation of the calcineurin/NFAT (nuclear factor of activated T cells) pathway and its downstream effector, NFATc1 (nuclear factor of activated T cells c1 isoform).38, 39 Although these observations give a frame to explain granuloma formation with RAS/MAPK-related genes on the one hand, and SH3BP2 on the other, whether this low penetrant manifestation results from the interference of modifier genes with the RAS/MAPK pathway or from an exogenous stimulus leading to focal development of granuloma in a genetically permissive environment remains unknown.
In summary, we present five patients with NL/MGCL. Three of them have SOS1 mutations (two patients with facial giant cell lesions and one patient with PVS of the ankle joint). The two others have PTPN11 mutations. NL/MGCL syndrome, previously regarded as an entity distinct from NS, is a phenotypic variation within the NS spectrum. MGCL appears to be a non-gene-specific and non-allele-specific manifestation of dysregulated RAS/MAPK pathway.
Note added in proof
Although this study was under reviewing process, two independent reports confirmed our observations. In this Journal, cherubism was reported in four patients with NS and SOS1 mutations, and in three patients with CFC and BRAF mutations.40 A SOS1 mutation41was reported in a single patient with NS and PVS. These reports confirm that MGCL is a rare complication of the deregulated RAS/MAPK pathway.
References
Tartaglia M, Martinelli S, Stella L et al: Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet 2006; 78: 279–290.
Gelb BD, Tartaglia M : Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet 2006; 15 (Spec No 2): R220–R226.
Schubbert S, Shannon K, Bollag G : Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7: 295–308.
Bentires-Alj M, Kontaridis MI, Neel BG : Stops along the RAS pathway in human genetic disease. Nat Med 2006; 12: 283–285.
Tartaglia M, Mehler EL, Goldberg R et al: Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 2001; 29: 465–468.
Roberts AE, Araki T, Swanson KD et al: Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet 2007; 39: 70–74.
Tartaglia M, Pennacchio LA, Zhao C et al: Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet 2007; 39: 75–79.
Razzaque MA, Nishizawa T, Komoike Y et al: Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 2007; 8: 1013–1017.
Schubbert S, Zenker M, Rowe SL et al: Germline KRAS mutations cause Noonan syndrome. Nat Genet 2006; 38: 331–336.
Nava C, Hanna N, Michot C et al: CFC and Noonan syndromes due to mutations in RAS/MAPK signaling pathway: genotype/phenotype relationships and overlap with Costello syndrome. J Med Genet 2007; 12: 763–771.
Idowu BD, Thomas G, Frow R et al: Mutations in SH3BP2, the cherubism gene, were not detected in central or peripheral giant cell tumours of the jaw. Br J Oral Maxillofac Surg 2008; 46: 229–230.
Cohen Jr MM, Gorlin RJ : Noonan-like/multiple giant cell lesion syndrome. Am J Med Genet 1991; 40: 159–166.
Lee JS, Tartaglia M, Gelb BD et al: Phenotypic and genotypic characterisation of Noonan-like/multiple giant cell lesion syndrome. J Med Genet 2005; 42: e11.
Wolvius EB, de LJ, Smeets EE et al: Noonan-like/multiple giant cell lesion syndrome: report of a case and review of the literature. J Oral Maxillofac Surg 2006; 64: 1289–1292.
Cancino CM, Gaiao L, Sant’Ana FM et al: Giant cell lesions with a Noonan-like phenotype: a case report. J Contemp Dent Pract 2007; 8: 67–73.
Lee SM, Cooper JC : Noonan syndrome with giant cell lesions. Int J Paediatr Dent 2005; 15: 140–145.
Addante RR, Breen GH : Cherubism in a patient with Noonan's syndrome. J Oral Maxillofac Surg 1996; 54: 210–213.
Betts NJ, Stewart JC, Fonseca RJ et al: Multiple central giant cell lesions with a Noonan-like phenotype. Oral Surg Oral Med Oral Pathol 1993; 76: 601–607.
Levine B, Skope L, Parker R : Cherubism in a patient with Noonan syndrome: report of a case. J Oral Maxillofac Surg 1991; 49: 1014–1018.
Dunlap C, Neville B, Vickers RA et al: The Noonan syndrome/cherubism association. Oral Surg Oral Med Oral Pathol 1989; 67: 698–705.
Minisola G, Porzio V, Ceralli F et al: Polyarticular pigmented villonodular synovitis associated with multiple congenital anomalies. A case of Noonan-like/multiple giant cell lesion syndrome. Clin Exp Rheumatol 1996; 14: 207–210.
Tartaglia M, Kalidas K, Shaw A et al: PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002; 70: 1555–1563.
Bertola DR, Pereira AC, Albano LM et al: PTPN11 gene analysis in 74 Brazilian patients with Noonan syndrome or Noonan-like phenotype. Genet Test 2006; 10: 186–191.
Jafarov T, Ferimazova N, Reichenberger E : Noonan-like syndrome mutations in PTPN11 in patients diagnosed with cherubism. Clin Genet 2005; 68: 190–191.
Bertola DR, Pereira AC, Passetti F et al: Neurofibromatosis-Noonan syndrome: molecular evidence of the concurrence of both disorders in a patient. Am J Med Genet A 2005; 136: 242–245.
Maheshwari M, Belmont J, Fernbach S et al: PTPN11 mutations in Noonan syndrome type I: detection of recurrent mutations in exons 3 and 13. Hum Mutat 2002; 20: 298–304.
Tartaglia M, Kalidas K, Shaw A et al: PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet 2002; 70: 1555–1563.
Bertola DR, Kim CA, Pereira AC et al: Are Noonan syndrome and Noonan-like/multiple giant cell lesion syndrome distinct entities? Am J Med Genet 2001; 98: 230–234.
Kirk JM, Betts PR, Butler GE et al: Short stature in Noonan syndrome: response to growth hormone therapy. Arch Dis Child 2001; 84: 440–443.
Wendt RG, Wolfe F, McQueen D et al: Polyarticular pigmented villonodular synovitis in children: evidence for a genetic contribution. J Rheumatol 1986; 13: 921–926.
Sarkozy A, Obregon MG, Conti E et al: A novel PTPN11 gene mutation bridges Noonan syndrome, multiple lentigines/LEOPARD syndrome and Noonan-like/multiple giant cell lesion syndrome. Eur J Hum Genet 2004; 12: 1069–1072.
Ruggieri M, Pavone V, Polizzi A et al: Unusual form of recurrent giant cell granuloma of the mandible and lower extremities in a patient with neurofibromatosis type 1. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999; 87: 67–72.
Edwards PC, Fantasia JE, Saini T et al: Clinically aggressive central giant cell granulomas in two patients with neurofibromatosis 1. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2006; 102: 765–772.
Krammer U, Wimmer K, Wiesbauer P et al: Neurofibromatosis 1: a novel NF1 mutation in an 11-year-old girl with a giant cell granuloma. J Child Neurol 2003; 18: 371–373.
Van Damme PA, Mooren RE : Differentiation of multiple giant cell lesions, Noonan-like syndrome, and (occult) hyperparathyroidism. Case report and review of the literature. Int J Oral Maxillofac Surg 1994; 23: 32–36.
Yazdizadeh M, Tapia JL, Baharvand M et al: A case of neurofibromatosis-Noonan syndrome with a central giant cell granuloma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2004; 98: 316–320.
Ueki Y, Tiziani V, Santanna C et al: Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet 2001; 28: 125–126.
Ueki Y, Lin CY, Senoo M : Increased myeloid cell responses to M-CSF and RANKL cause bone loss and inflammation in SH3BP2 ‘cherubism’ mice. Cell 2007; 128: 71–83.
Lietman SA, Yin L, Levine MA : SH3BP2 is an activator of NFAT activity and osteoclastogenesis. Biochem Biophys Res Commun 2008; 371: 644–648.
Neumann TE, Allanson J, Kavamura I et al: Multiple giant cell lesions in patients with Noonan syndrome and cardio-facio-cutaneous syndrome. Eur J Hum Genet 2008.
Mascheroni E, Digilio MC, Cortis E et al: Pigmented villonodular synovitis in a patient with Noonan syndrome and SOS1 gene mutation. Am J Med Genet A 2008; 146A: 2966–2967.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Beneteau, C., Cavé, H., Moncla, A. et al. SOS1 and PTPN11 mutations in five cases of Noonan syndrome with multiple giant cell lesions. Eur J Hum Genet 17, 1216–1221 (2009). https://doi.org/10.1038/ejhg.2009.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.44
Keywords
This article is cited by
-
Cutis verticis gyrata and Noonan syndrome: report of two cases with pathogenetic variant in SOS1 gene
Italian Journal of Pediatrics (2022)
-
Multifocal tenosynovial giant cell tumors in a child with Noonan syndrome
Pediatric Radiology (2017)
-
Mutations in RIT1 cause Noonan syndrome with possible juvenile myelomonocytic leukemia but are not involved in acute lymphoblastic leukemia
European Journal of Human Genetics (2016)
-
A new mutation in the C-SH2 domain of PTPN11 causes Noonan syndrome with multiple giant cell lesions
Journal of Human Genetics (2014)
-
Cherubism
Head and Neck Pathology (2014)
