
Abstract
In facioscapulohumeral muscular dystrophy (FSHD) the majority of patients carry a D4Z4 macrosatellite repeat contraction in the subtelomere of chromosome 4q. Several disease mechanisms have been proposed to explain how repeat contraction causes muscular dystrophy. All proposed mechanisms foresee a change from a closed to a more open chromatin structure followed by loss of control over expression of genes in or proximal to D4Z4. Initially, a distance and residual repeat size-dependent upregulation of the candidate genes FRG2, FRG1 and ANT1 was observed, but most successive expression studies failed to support transcriptional upregulation of 4qter genes. Moreover, chromatin studies do not provide evidence for a cis-spreading mechanism operating at 4qter in FSHD. In part, this inconsistency may be explained by differences in the techniques used, and the use of RNA samples obtained from different muscle groups. The aim of this study is to comprehensively and uniformly study the expression of the FSHD candidate genes FRG1, FRG2, CRYM, ANT1, ALP, PITX1 and LRP2BP at the RNA and protein level in identically processed primary myoblasts, myotubes and quadriceps muscle. Expression was compared between samples obtained from FSHD patients and normal controls with samples from myotonic dystrophy type 1 patients as disease controls. No consistent changes in RNA or protein expression levels were observed between the samples. The one exception was a selective increase in FRG2 mRNA expression in FSHD myotubes. This study provides further evidence that there is no demonstrable consistent, large magnitude, overexpression of any of the FSHD candidate genes.
Similar content being viewed by others


Introduction
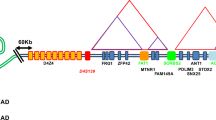
Facioscapulohumeral muscular dystrophy (FSHD) was the first genetic disease to be mapped by microsatellite markers,1, 2, 3, 4, 5 and although its genetic mutation was discovered soon after,6, 7 the exact pathogenic mechanism remains elusive. To a large extent this uncertainty is because of the unusual genetic mutation, a macrosatellite repeat contraction, but also genetic heterogeneity of the disease.8 Clinically, FSHD is characterized by progressive weakness and wasting starting in the facial, shoulder and upper arm muscles. The disease manifestations also include non-muscular features such as sensorineural deafness and retinal vasculopathy.9, 10, 11 More than 50% of patients present with asymmetric muscle weakness. Typically, noticeable muscle weakness starts in the second decade of life, although early onset cases have also been reported. Significant inter- and intrafamilial variability in disease onset, progression and presentation is common with 20% of gene carriers remaining asymptomatic and an equal proportion of patients eventually becoming wheelchair-dependent.12, 13, 14 A macrosatellite repeat contraction in the subtelomere of chromosome 4q is causal to the disease in >97% of patients.6, 7 This polymorphic D4Z4 macrosatellite repeat consists of 11–100 units, each 3.3 kb in size, in the control population whereas patients with FSHD have one array of 1–10 units. There is no linear relationship between residual repeat size and disease severity, although patients with the smallest number of repeat units (1–3 units) are generally more severely affected.12, 13, 14 Contraction of D4Z4 is necessary but not sufficient to cause FSHD.15 At least 9 different haplotypes of 4qter have been identified based on small sequence variations proximal and distal to the repeat. Contractions in only one of these haplotypes (4qA161) have been associated with FSHD.16 The cause for this association is not known but it is speculated that 4qA161 contains a combination of sequence polymorphisms that are essential for disease development. Genetic heterogeneity is also reported for FSHD. Some patients have a partial deletion of the D4Z4 repeat that extends into proximal non-repeated sequences.17, 18 These so-called proximal deletion patients are clinically indistinguishable from FSHD. Moreover, some patients with phenotypic FSHD do not show a contraction of D4Z4 but share epigenetic changes at the FSHD locus with patients with a contraction suggesting that this disease mechanism is also mediated through D4Z4.19, 20 Several disease mechanisms have been proposed to explain how a repeat contraction causes muscular dystrophy.20, 21, 22, 23, 24 Most mechanisms predict a chromatin change from a closed to a more open chromatin structure followed by loss of control over gene expression of genes in or proximal to the repeat. Several studies support this model including the observation of partial loss of DNA methylation at D4Z4 in FSHD.20 Loss of control is proposed to occur through cis-looping or cis-spreading mechanisms but the evidence for these models is controversial. Initially a distance- and residual repeat size-dependent upregulation of the candidate genes FRG2, FRG1 and ANT1 was observed,22 but subsequent studies have both disputed23, 25, 26 as well as supported27, 28 the transcriptional dysregulation of 4qter genes. In addition, chromatin studies have failed to provide evidence for a cis-spreading mechanism operating at 4qter in FSHD.23 In part, this discrepancy may be explained by differences in techniques used and differences in the muscle groups studied. The conflicting or unconfirmed data have generated a number of divergent disease mechanisms that are being actively pursued in FSHD.
The aim of this study is to try to address in a systematic, uniform and comprehensive manner, the status of expression of proposed FSHD candidate genes. To this end, we analyzed gene expression of the candidate 4qter genes FRG1, FRG2, ANT1, ALP (PDLIM3) and LRP2BP, as well as the recently published FSHD candidate genes PITX1 and CRYM,29, 30 at both the mRNA and protein level in primary myoblast and myotube cultures as well as quadriceps muscle. A study of transcripts emanating from D4Z4, including DUX4, in the same set of biomaterial are extensively described elsewhere.31 We compared expression between samples derived from FSHD patients and normal controls with samples from myotonic dystrophy type 1 (DM1) patients as disease controls. All RNA and protein sources were identically processed and examined to minimize technical variation.
Materials and methods
Most protocols described in this paper and other protocols used by researchers of the Fields Center for FSHD and Neuromuscular Research are described in detail at the Fields Center website (http://www.urmc.rochester.edu/fields-center/).
Study subjects
All subjects with FSHD satisfied research criteria for the clinical diagnosis for FSHD and were genetically confirmed. Similarly, DM1 samples originated from genetically confirmed patients. Normal control samples originated from healthy individuals recruited for this study, with a normal neuromuscular examination and no family history of neuromuscular diseases. The study protocol was approved by the respective institutional research subjects review board.
Muscle biopsies
Biopsy material was obtained from the Vastus Lateralis muscle by the needle muscle biopsy method from which three types of samples were obtained, one for RNA and protein extraction, one for myoblast isolation and one for histological analysis (for a detailed description see Supplementary information S1 or the Fields Center website). Trichrome and hematoxylin and eosin stained sections were used for pathological grading of FSHD muscle biopsy samples. Pathological grading criteria are outlined in the legend of Table 2.
RNA and protein isolation from primary myoblast and myotubes
Myoblasts were isolated from the muscle biopsy samples by preplating and the quality of these cultures was determined by desmin staining (for a detailed description see Supplementary information S2 and S3 or the Fields Center website). For RNA and protein isolation, proliferating myoblasts of four control, four FSHD and three DM1 patients were grown in 9 cm diameter Petri dishes till 40–50% confluency to minimize spontaneous differentiation. Myotubes were obtained by growing the myoblasts till 70% confluency, followed by induction of differentiation by growing them on differentiation media (DMEM (+glucose, +L-glutamin, +pyruvate), 2% horse serum) for 6 days.
Total RNA was isolated from myoblast and myotube cultures using the nucleospin RNA II kit (Macherey Nagel, Düren, Germany) according to the instructions of the manufacturer. The RNA concentration of the samples was determined on the ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and the quality was analyzed with a RNA 6000 nanochip on an Agilent 2100 BioAnalyzer (Agilent Technologies Netherlands BV, Amstelveen, The Netherlands). A total protein extract was obtained by first washing the cells with 10 ml PBS. Thereafter, myoblast and myotubes were incubated for 5 min on ice in respectively 250 and 500 μl RIPA buffer (20 mM triethanolamine; 140 mM NaCl; 0.1% DOC (w/v); 0.1% SDS (w/v); 0.1% Triton X-100 (v/v); protease inhibitors Complete (Roche, Mannheim, Germany)). Cells were scraped and the lysate was pulled through a 29G U-100 insulin needle five times. Non-soluble material was spun down (20 000 g/10 min/4°C) and the protein content of the supernatant was determined with a BCA protein quantification assay (Pierce, Rockford, USA).
RNA isolation from biopsy material
Ten to 50 mg of frozen muscle biopsy was homogenized in 1 ml of trizol reagent (Life technologies, Gaithersburg, MD, USA) and incubated for 5 min at room temperature. Thereafter, 100 μl 1-bromo, 3-chloro propane was added, vortexed for 10 s and incubated at room temperature for 10 min. Next, the mixture was centrifuged at 13000 g for 15 min at 4°C. The upper aqueous phase was transferred to a clean reaction vial, vortexed shortly with 0.5 ml isopropanol, and incubated for 10 min at room temperature. The precipitated RNA was centrifuged at 13000 g for 8 min at 4°C.
The pellet was washed with 1 ml 75% ethanol. Thereafter, the pellet was air-dried, dissolved in 20 μl water and incubated for 10 min at 60°C. A quantity of 1.5 μg RNA per sample was DNAse I treated according to the instructions of the manufacturer (Applied Biosystems, Foster City, CA, USA).
Quantitative RNA expression analysis
For the myoblast cultures cDNA was made from 2 μg purified total RNA using the Revert Aid H Minus first strand cDNA synthesis kit using random hexamer primers (Fermentas, St Leon-Rot, Germany) according to the instructions of the manufacturer. The cDNA was subsequently treated with 0.5U RNase H for 20 min at 37°C, purified with the Nucleospin Extract II kit (Macherey Nagel) and the cDNA concentration was determined on the ND-1000 spectrophotometer (Thermo Scientific). Five or 20 ng cDNA was used in a real time RT-PCR experiment using SYBR green master mix on a MyiQ (Biorad Laboratories, Veenendaal, The Netherlands) running an initial denaturation step at 95°C for 3 min, followed by 35 cycles of 10 s at 95°C and 45 s at 60°C. All samples were run in duplo. The melting curve of the PCR product was obtained by running an initial denaturation step at 95°C, followed by a 1 min incubation at 65°C and thereafter 0.5°C increase of the temperature every 10 s till 95°C. All primer sets (Table 1) were designed using Primer3 and spanned at least one intron. PCR products were analyzed for specificity by melting curve analysis and on a 2% agarose gel, which showed respectively a single peak and a single band running at the expected size for all PCR products. Furthermore, PCR products were sequence verified. The results of the real time RT-PCR were analyzed and quantified using iQ5 optical system software version 2.0 (Biorad Laboratories, Veenendaal, The Netherlands). All expression levels were calculated using GAPDH as constitutively expressed standard for cDNA input, and the relative steady-state RNA levels of the target gene were calculated by the method of Pfaffl.32
Expression analysis of FRG1 was also performed by using an internal standard. This analysis was performed as described earlier.33
Western blot analysis
For protein expression analysis in biopsy muscle samples, homogenates of the muscle biopsy samples were made with an ultratorax using RIPA buffer. Insoluble material was boiled for 10 min in 4xSB. Equivalent amounts of both fractions were pooled before analysis on western blot.
For protein expression analysis of the biopsy material as well as the primary myoblasts and myotubes, equal amounts of each sample was run on a 12% poly-acrylamide gel. Separated proteins were transferred to a PVDF membrane (Millipore, Amsterdam, The Netherlands). The membranes were stained with ponceau red (0.1% (w/v) ponceau red; 0.5% (v/v) HAc) to verify equal transfer of protein and thereafter, blocked with 3% mPBS for 1 h. Next, membranes were incubated for 1 h with primary antibody diluted in blockbuffer and subsequently washed five times with PBST. Primary antibodies that were used in this study are a goat anti-ALP antibody (Abcam), a rabbit anti-LRP2BP antibody (Abcam), a rabbit anti-histone 2B antibody (Abcam), a rabbit anti-FRG1 antibody,34 a mouse anti-myosin MF20 antibody (University Iowa), a mouse anti-ANT1 antibody (Calbiochem, Santa cruz biotechnology), a mouse anti-CRYM antibody (Abnova), a mouse anti-PITX1 antibody (Abnova) and a mouse anti-tubulin antibody (Sigma). Bound antibodies were detected by incubation for 1 h with IRDye 800CW conjugated secondary antibody diluted 1:5000 in blockbuffer. Next, the membranes were washed four times with PBST and twice with PBS and signals were detected on the Odyssey (LI-COR Biosciences GmbH, Bad Homburg, Germany).
Results
Genetic analysis and quality assessment of the primary myoblast cultures
Before expression analysis of the target genes, the quality of the primary myoblast and myotube cultures was assessed. First, the D4Z4 repeat length of the primary cells was determined with pulsed field gel electrophoresis using DNA marker p13E-11.2, 6 These studies confirmed the presence of a contracted repeat on the disease allele in FSHD myoblasts, whereas FSHD was excluded in the control and DM1 myoblasts (Table 2). Second, the purity of the myoblast isolations was assessed by IF staining of desmin and by morphological appearance. The desmin staining showed that all cultures contained 85–99% cells that expressed desmin (data not shown). We did not observe morphological differences between the myoblasts and myotubes of controls and FSHD patients (Supplemental information S4). However, the DM1 myoblasts did not appear to have a morphology that resembled control myoblasts. Furthermore, the proliferation rate of these cells was very low and they did not appear to differentiate efficiently into multi-nucleated myotubes.
RNA and protein expression analysis of the FSHD candidate genes in primary myoblasts
The relative steady-state levels of the candidate genes FRG1, PITX1, CRYM, FRG2, ALP, LRP2BP and ANT1 was determined with quantitative RT-PCR using primers that spanned at least one intron (Table 1). For FRG1, 4q-specific primers were used that annealed in the 5′ UTR of FRG1.
Of these genes, we could not reliably quantify CRYM expression in our samples. The Ct values were either very high or could not be determined at all. For the genes FRG1 (Figure 1a), ALP, LRP2BP and ANT1 (Supplementary information S5) we could not detect a significant difference in the relative steady-state RNA levels in both the proliferating myoblast as well as the differentiated myotubes. However, there seemed to be a trend, although not significant, towards upregulation of PITX1 in the myotubes, while no difference was observed in the proliferating myoblasts (Figure 1b). Interestingly, we could confirm the deregulated expression of FRG2 in differentiated myotubes of FSHD patients (Figure 1c). No reliable expression of this gene could be observed in the proliferating myoblasts of all patient groups, whereas expression could be observed in the FSHD samples of the differentiated myotubes only. Sequencing of the PCR products revealed both chromosome 4 and 10 copies of FRG2.

RNA expression analysis in primary myoblasts and myotubes of four healthy controls (Control A–D), four FSHD patients (FSHD A–D) and three DM1 patients (DM1 A–C) as disease controls. The relative abundance compared with the average expression level of the control cultures are depicted for FRG1 (a) and PITX1 (b). FRG2 RNA expression (c) could only be detected in myotube cultures of FSHD patients and not in controls.
In addition, we analyzed the protein expression levels of FRG1, PITX1, CRYM, ALP, LRP2BP and ANT1 in the primary myoblast and myotube cultures. FRG2 expression levels could not be analyzed, as we could not obtain a FRG2-specific antibody. Equal protein amounts of each lysate were blotted on PVDF membrane. The blots were first probed with an anti-tubulin antibody as loading control and an anti-myosin antibody to determine the differentiation status of the cells at the time of lysis. Both FSHD and control myoblasts as well as one DM1 cell line did reveal a clear upregulation of myosin upon differentiation (Figure 2). For two out of three DM1 cell lines, the protein yield was too low to give a decisive answer. In both the myoblast as the myotube samples, we could not reliably detect CRYM, ANT1 and PITX1 protein levels. Either, we could not detect a band (CRYM, PITX1) or multiple bands were visible (ANT1). For FRG1, ALP and LRP2BP no clear differences in protein expression levels were observed between the FSHD and control groups (Figure 2). In general, FRG1 expression levels compared with histone 2B and tubulin levels seemed to decrease upon differentiation, whereas ALP and LRP2BP showed an upregulation during differentiation.

Protein expression analysis in primary myoblasts and myotubes of four healthy controls (Control A–D), four FSHD patients (FSHD A–D) and three DM1 patients (DM1 A–C) as disease controls. Equal protein loading for each sample was verified by probing the western blots with antibodies against tubulin and histone 2B. Both FSHD and control myoblast reveal comparable expression of myosin upon differentiation. No clear differences in expression of the FSHD candidate genes FRG1, LRP2BP and ALP is observed. Overall, FRG1 protein expression decreases upon differentiation, whereas LRP2BP and ALP expression levels seem to increase.
RNA and protein expression analysis of the FSHD candidate genes in muscle biopsies
Expression analysis of the FSHD candidate genes was also performed on quadriceps muscle of 10 FSHD and 10 control individuals. All FSHD muscle samples used showed pathological changes. With quantitative RT-PCR, no significant difference in relative steady-state expression of the genes FRG1, ANT1, ALP, PITX1 or LRP2BP could be detected between the two groups. There seemed to be a trend toward downregulation of PITX1, although there was a high degree of variability in expression within the control and FSHD group (Figure 3). For FRG1, we also performed a competitive quantitative RT-PCR.33 This confirmed that there was no difference in FRG1 expression between FSHD samples and controls (Figure 3). For CRYM, a gene that has been proposed to be upregulated in FSHD on the protein level, very low expression levels were detected, except for three control samples (Controls 2, 3 and 5) that showed a very high expression level compared with all other samples (Figure 3). This result was confirmed by using an additional primer set for CRYM (Supplementary information S6) and by direct sequencing of the RT-PCR products. The only transcript that could not be detected in the biopsy samples was FRG2.

RNA expression analysis in biopsy material isolated from 10 healthy controls (Control 1–10) and 10 FSHD patients (FSHD 1–10). The relative expression in each sample compared with the average of the controls are depicted. No difference in expression is observed for the FSHD candidate genes FRG1, ALP, LRP2BP and ANT1, while a high degree of variability in expression was observed for PITX1 and CRYM.
On the protein level we could detect FRG1, LRP2BP, ALP and CRYM, but not ANT1 or PITX1. For FRG1, ALP and LRP2BP no clear differences were observed between or within the two groups (Figure 4), although some samples showed expression level differences that did not appear to be FSHD related. For CRYM, however, clear differences could be observed that matched the results of the RNA steady-state expression analysis. Three control samples (Controls 2, 3 and 5) showed relatively high expression levels compared with all other control and FSHD samples (Figure 4). This, together with the quantitative RT-PCR data, indicates a very high degree of variability in CRYM expression, which is not FSHD related.

Protein expression analysis of biopsy material isolated from 10 healthy controls (Control 1–10) and 10 FSHD patients (FSHD 1–10) that were also analyzed on steady-state RNA expression levels. Equal loading of each sample was determined by ponceau and mouse anti-tubulin staining of the blots. The expression analysis of the proteins FRG1, LRP2BP, ALP and CRYM did not reveal any clear differences between FSHD and controls. However, CRYM showed a high degree of variability in expression that did not seem to be disease related, but confirmed the steady-state RNA expression analysis.
Discussion
A number of candidate genes have been proposed in FSHD based on selective upregulation in FSHD-derived tissue or cell lines.22, 27, 28, 29, 30 Yet many of these findings have been difficult to confirm resulting in divergent hypotheses regarding the underlying pathophysiology of FSHD. In this study, utilizing a uniform and systematic approach, the expression levels of the FSHD candidate genes FRG1, PITX1, CRYM, FRG2, ALP, LRP2BP and ANT1 were determined in primary myoblasts and myotubes as well as in biopsy material of affected quadriceps muscle. We did not observe consistent and significant deregulation of the FSHD candidate genes we examined in all samples tested, either at the RNA, or at the protein level. Only FRG2 showed increased expression levels in FSHD samples and then only in myotubes at the RNA level. Although the FRG2 expression level was very low in both myoblasts and muscle biopsy samples, expression was consistently observed in myotubes from FSHD patients and not in controls. This observation corroborates an earlier report showing the transcriptional upregulation of chromosome 4 and 10 copies of FRG2 in FSHD myotubes only.28 Whether FRG2 is involved in FSHD pathogenesis remains doubtful, as muscle-specific overexpression of FRG2 in mice does not result in a muscular dystrophy phenotype.27 Moreover, individuals with proximal deletion encompassing FRG2 have phenotypically typical FSHD.17
For all other genes tested, we did not observe expression level differences, which is in line with23, 25, 26 but also in contrast to22, 27, 29, 30 data described before. Most of these studies were performed with limited sample size and sometimes included samples isolated from different muscle groups. Perhaps the gene of greatest relevance given the conflicting data is FRG1 as it is still considered one of the leading candidate genes in FSHD as muscle-specific overexpression, at high levels, in mice causes a muscular dystrophy phenotype.27 In our studies, no difference in FRG1 expression was observed between FSHD and controls either at the RNA or protein level. Moreover, for RNA expression studies in muscle we confirmed our results by testing expression using two separate experiments with different reference methods, utilizing the same samples. In addition, there was no relationship between expression levels and deletion size or between expression levels and the pathological grading of the muscle in FSHD-derived samples as for all other genes tested. As FRG1 is a nuclear protein, it is important to emphasize that for the protein studies the FRG1 expression levels were compared with both total protein levels using tubulin or ponceau staining as well as nuclear protein levels using histone 2B. In addition, we found no clear upregulation of PITX1, a gene that has recently been reported to undergo DUX4-dependent upregulation.29 Only in the myotubes a trend toward upregulation was observed. It should be noted, however, that the reported deregulation of PITX1 was found in muscle samples of the biceps and deltoid muscle, whereas quadriceps muscle was used in our study.
The quadriceps muscle was chosen in this study because it is easily accessible for needle biopsy and because it is relatively spared early in FSHD. Nevertheless, quantitative muscle strength testing35 shows more involvement of this muscle group than is suspected by bedside examination. Indeed, all the FSHD biopsy samples in our study showed mild-to-moderate pathological changes (Table 2). Therefore, possible observed changes in this muscle are more likely to be early changes in FSHD pathophysiology and not secondary changes caused by muscle wasting or damage, which could explain some of the conflicting data regarding myoblast phenotypes from affected and unaffected muscle.25, 36, 37
The importance of sample size in verifying selective deregulation of FSHD candidate genes is illustrated by the high inter sample variability in RNA and protein expression for several target genes. Expression level differences determined with a limited number of samples can result in unreliable results. In biopsy material this can be caused by variability in the presence of non-muscle cells (eg, blood cells, endothelial cells and so on). Of note is that CRYM showed high RNA and protein expression level differences in the muscle biopsy samples, which did not seem to be disease related. Recently, Reed et al30 reported that CRYM protein levels are upregulated in FSHD deltoid muscle. In this study, three FSHD samples were compared with two controls. In our study, we cannot confirm a FSHD-specific upregulation of CRYM in FSHD quadriceps muscle biopsies (n=10). In contrast, some control samples showed high CRYM RNA and protein expression levels, while most had barely detectable expression levels. These results clearly show the heterogeneity of biological samples in studying RNA and protein expression levels and emphasize the need to examine large sample sizes and especially the need for adequate normal control samples to understand the intrinsic variability in expression of a particular gene in normal tissue.
This study clearly shows that none of the FSHD candidate genes, except for FRG2, are upregulated at the levels described earlier in some studies.22, 27, 28, 29, 30 We cannot exclude low levels of deregulation that is beyond the sensitivity of our methods or exclude the possibility that one or more of the candidate genes show a transient spatiotemporal deregulation under conditions we have not tested. Our study emphasizes the need for large collections of identically processed samples to investigate FSHD-specific transcriptional changes to overcome sample heterogeneity. As humans are genetically and phenotypically heterogeneous in nature, a more promising approach to investigate the role of the established candidate genes for FSHD might be to elucidate their cellular function and relate that function to FSHD pathology.
A paper by Masny and et al,38 utilizing a different approach to study a potential cis effect in FSHD reached a similar conclusion. The authors studied native transcripts emanating from chromosome 4 by RNA–DNA FISH to quantitatively establish the expression level of the nascent transcript from each allele. Masny and co-workers found no difference in expression level between the normal and contracted allele, providing further evidence against a cis effect of transcriptional deregulation of 4qter genes in FSHD.
Recent evidence offers an alternative hypothesis to pathophysiology of FSHD. As all patients with FSHD share a change in the chromatin structure of D4Z4 on chromosome 4qA161,16, 19, 20 it might well be that a shift in balance of the complex transcriptional activity recently discovered to emanate from the repeat29, 31 is causally related to the pathology.
References
Wijmenga C, Frants RR, Brouwer OF, Moerer P, Weber JL, Padberg GW : Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 1990; 336: 651–653.
Wijmenga C, Sandkuijl LA, Moerer P et al: Genetic linkage map of facioscapulohumeral muscular dystrophy and five polymorphic loci on chromosome 4q35-qter. Am J Hum Genet 1992; 51: 411–415.
Weiffenbach B, Bagley R, Falls K et al: Linkage analyses of five chromosome 4 markers localizes the facioscapulohumeral muscular dystrophy (FSHD) gene to distal 4q35. Am J Hum Genet 1992; 51: 416–423.
Sarfarazi M, Wijmenga C, Upadhyaya M et al: Regional mapping of facioscapulohumeral muscular dystrophy gene on 4q35: combined analysis of an international consortium. Am J Hum Genet 1992; 51: 396–403.
Mills KA, Buetow KH, Xu Y et al: Genetic and physical mapping on chromosome 4 narrows the localization of the gene for facioscapulohumeral muscular dystrophy (FSHD). Am J Hum Genet 1992; 51: 432–439.
Wijmenga C, Hewitt JE, Sandkuijl LA et al: Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 1992; 2: 26–30.
van Deutekom JC, Wijmenga C, van Tienhoven EA et al: FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet 1993; 2: 2037–2042.
Gilbert JR, Stajich JM, Wall S et al: Evidence for heterogeneity in facioscapulohumeral muscular dystrophy (FSHD). Am J Hum Genet 1993; 53: 401–408.
Padberg GW, Brouwer OF, de Keizer RJ et al: On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve 1995; 2: S73–S80.
Fitzsimons RB, Gurwin EB, Bird AC : Retinal vascular abnormalities in facioscapulohumeral muscular dystrophy A general association with genetic and therapeutic implications. Brain 1987; 110 (Part 3): 631–648.
van der Maarel SM, Frants RR, Padberg GW : Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta 2007; 1772: 186–194.
Lunt PW, Jardine PE, Koch MC et al: Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD). Hum Mol Genet 1995; 4: 951–958.
Tawil R, Forrester J, Griggs RC et al: Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. The FSH-DY Group. Ann Neurol 1996; 39: 744–748.
Ricci E, Galluzzi G, Deidda G et al: Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann Neurol 1999; 45: 751–757.
Lemmers RJ, Wohlgemuth M, Frants RR, Padberg GW, Morava E, van der Maarel SM : Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am J Hum Genet 2004; 75: 1124–1130.
Lemmers RJ, Wohlgemuth M, van der Gaag KJ et al: Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet 2007; 81: 884–894.
Lemmers RJ, Osborn M, Haaf T et al: D4F104S1 deletion in facioscapulohumeral muscular dystrophy: phenotype, size, and detection. Neurology 2003; 61: 178–183.
Deak KL, Lemmers RJ, Stajich JM et al: Genotype-phenotype study in an FSHD family with a proximal deletion encompassing p13E-11 and D4Z4. Neurology 2007; 68: 578–582.
de Greef JC, Wohlgemuth M, Chan OA et al: Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology 2007; 69: 1018–1026.
van Overveld PG, Lemmers RJ, Sandkuijl LA et al.: Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet 2003; 35: 315–317.
Petrov A, Allinne J, Pirozhkova I, Laoudj D, Lipinski M, Vassetzky YS : A nuclear matrix attachment site in the 4q35 locus has an enhancer-blocking activity in vivo: implications for the facio-scapulo-humeral dystrophy. Genome Res 2008; 18: 39–45.
Gabellini D, Green MR, Tupler R : Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 2002; 110: 339–348.
Jiang G, Yang F, van Overveld PG, Vedanarayanan V, van der Maarel S, Ehrlich M : Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum Mol Genet 2003; 12: 2909–2921.
Yang F, Shao C, Vedanarayanan V, Ehrlich M : Cytogenetic and immuno-FISH analysis of the 4q subtelomeric region, which is associated with facioscapulohumeral muscular dystrophy. Chromosoma 2004; 112: 350–359.
Winokur ST, Chen YW, Masny PS et al: Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet 2003; 12: 2895–2907.
Bouju S, Pietu G, Le Cunff M et al: Exclusion of muscle specific actinin-associated LIM protein (ALP) gene from 4q35 facioscapulohumeral muscular dystrophy (FSHD) candidate genes. Neuromuscul Disord 1999; 9: 3–10.
Gabellini D, D'Antona G, Moggio M et al: Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature 2006; 439: 973–977.
Rijkers T, Deidda G, van Koningsbruggen S et al: FRG2, an FSHD candidate gene, is transcriptionally upregulated in differentiating primary myoblast cultures of FSHD patients. J Med Genet 2004; 41: 826–836.
Dixit M, Ansseau E, Tassin A et al: DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA 2007; 104: 18157–18162.
Reed PW, Corse AM, Porter NC, Flanigan KM, Bloch RJ : Abnormal expression of mu-crystallin in facioscapulohumeral muscular dystrophy. Exp Neurol 2007; 205: 583–586.
Snider L, Asawachaicharn A, Tyler AE et al: RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet 2009; 18: 2414–2430.
Pfaffl MW : A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29: e45.
Welle S, Bhatt K, Thornton CA : High-abundance mRNAs in human muscle: comparison between young and old. J Appl Physiol 2000; 89: 297–304.
van Koningsbruggen S, Straasheijm KR, Sterrenburg E et al: FRG1P-mediated aggregation of proteins involved in pre-mRNA processing. Chromosoma 2007; 116: 53–64.
The FSH-DY Group: A prospective quantitative study of the natural history of facioscapulohumeral muscular dystrophy (FSHD) implications for therapeutic trials. Neurology 1997; 48: 38–46.
Vilquin JT, Marolleau JP, Sacconi S et al: Normal growth and regenerating ability of myoblasts from unaffected muscles of facioscapulohumeral muscular dystrophy patients. Gene Therapy 2005; 12: 1651–1662.
Barro M, Carnac G, Flavier S, Mercier J, Vassetzky Y, Laoudj-Chenivesse D : Myoblasts from affected and non affected FSHD muscles exhibit morphological differentiation defects. J Cell Mol Med 2008 (e-pub ahead of print 24 May 2008).
Masny PS, Chan OYA, de Greef JC et al: Analysis of allele-specific RNA transcription in FSHD by RNA-DNA FISH in single myonuclei. Eur J Hum Genet 2009; doi:10.1038/ejhg.2009.183.
Acknowledgements
We thank the patients and their relatives for their ongoing support to our studies. This study was financially supported by the Fields Center for FSHD and Neuromuscular Research and by a grant from the Dutch Organization of Scientific Research (NWO 016.056.338). This publication was made possible by Grant number UL1 RR 024160 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Information on NCRR is available at http://www.ncrr.nih.gov/. Information on re-engineering the Clinical Research Enterprise can be obtained from http://nihroadmap.nih.gov/clinicalresearch/overview-translational.asp. This publication was made passible by Grant Number UL1 RR 024160 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsiblity of the authors and do not necessarily represent the official view of NCRR or NIH. Information on available at http://www.ncrr.nih.gov/. Information on Reengineering the Clinical Research Enterprise can be obtained from http://nihroadmap.nih.gov/clinicalresearch/overview-translational.asp.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Klooster, R., Straasheijm, K., Shah, B. et al. Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur J Hum Genet 17, 1615–1624 (2009). https://doi.org/10.1038/ejhg.2009.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.62
Keywords
This article is cited by
-
Facioscapulohumeral dystrophy: the path to consensus on pathophysiology
Skeletal Muscle (2014)
-
DUX4 promotes transcription of FRG2 by directly activating its promoter in facioscapulohumeral muscular dystrophy
Skeletal Muscle (2014)
-
Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy
Nature Structural & Molecular Biology (2013)
-
A unique library of myogenic cells from facioscapulohumeral muscular dystrophy subjects and unaffected relatives: family, disease and cell function
European Journal of Human Genetics (2012)
-
Deciphering transcription dysregulation in FSH muscular dystrophy
Journal of Human Genetics (2012)
