
Abstract
The genetic structure of human populations is important in population genetics, forensics and medicine. Using genome-wide scans and individuals with all four grandparents born in the same settlement, we here demonstrate remarkable geographical structure across 8–30 km in three different parts of rural Europe. After excluding close kin and inbreeding, village of origin could still be predicted correctly on the basis of genetic data for 89–100% of individuals.
Similar content being viewed by others

Introduction
High-density genome-wide scans have revealed a considerable degree of geographical structure among populations across the globe.1 Even within Europe, the continent with the lowest among-population genetic diversity, populations separated by <500 km, such as the English and Irish,2 Italians from Lombardy and Tuscany,1 Finns from neighbouring regions3 and Estonians from different counties,4 can be differentiated. However, it remains to be seen whether the populations of villages a few kilometers apart can be distinguished.
Methods
Data are from Illumina Human Hap300 genome-wide scans (Illumina, San Diego, CA, USA). We made use of only the subset of each present-day population with all four grandparents from one location and this was reduced further when exclusions were made on the basis of kinship and inbreeding. First-, second- and third-degree relatives were removed, using genomic sharing estimates based on identity-by-state with a cutoff of 0.1 (in R). We also used more stringent thresholds until no more individuals remained in each subgroup. Principal component analysis (PCA) was performed using Eigensoft5 and model-based clustering using Frappe.6
We used PCA plus linear discriminant analysis (LDA) to predict subpopulation membership using the genetic data.7 We used all single-nucleotide polymorphisms (SNPs) except for those on the X chromosome and regions of high linkage disequilibrium identified in Table 1 of Price et al.8 Principal component (PC) scores were obtained according to described methods,5 substituting each SNP with the residuals produced by linear regression on the three previous SNPs. A double cross-validation procedure was used to correct for overfitting of the validation set, using the ratio of PC scores between the validation and training samples to calculate a scaling factor. A second validation cycle trained an LDA step, which was used to classify a separate outgroup of individuals, corrected with the scaling factor. The predicted classes (using the first three PCs) were compared with the known geographical groups to calculate an error rate. Written informed consent was obtained from all subjects.
Results
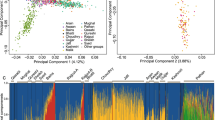
Using 300 000 SNPs and only unrelated, non-inbred individuals with all four grandparents from the same valley, village or isle, we here show the genomic differentiation across 8–30 km in three disparate areas of rural Europe, using genetic information alone (Figure 1). PCA of genomic sharing and model-based clustering (not shown) both allow separation of individuals with grandparents from each of three small Scottish isles, three alpine valleys in the north of Italy and two villages on one small island in Croatia. We used a supervised classification approach to predict subpopulation membership. Highly reliable levels of prediction were achieved with 100, 96 and 89% of individuals correctly classified on the basis of their genetic data for Italy, the Scottish Isles and Croatia, respectively. In each area, when individuals with grandparents from more than one village were included in PCA, they were scattered among the clusters, consistent with mixed origins (not shown).

Fine-scale genetic structure in rural European populations illustrated using principal component analysis. Individuals are a subset of participants from the EUROSPAN project, with all four grandparents from the same isle (Scotland; n=36), village (Croatia, n=157) or valley (Italy, n=57). Individuals with ancestry in different settlements are coloured red, blue and/or green. (a) Italy, (b) Scotland and (c) Croatia.
Discussion
It is interesting to consider the time depth of this differentiation. By removing first-, second- and third-degree relatives, we controlled for structure arising from mating behaviour in the past ∼120 years, and thus focused on the patterning arising earlier than this. The signal of structure is stronger when we include close relatives, but also persists when we use more stringent thresholds for relatedness (not shown), indicating that the patterns arise from ancient shared ancestry within the villages compared to their neighbouring subpopulations. Inbreeding and more-distant shared parental ancestry will also contribute to among-population differences. We used the genomic measure FROH9 to remove all individuals with total shared parental ancestry equivalent to one second-cousin pedigree loop (FROH >0.015), whereas including inbred subjects led to more obvious structuring (not shown). Thus, the observed structure in each of the populations arises partly from recent relatedness and shared parental ancestry and partly from deeper patterns of kinship within the subpopulations, overlaid with mating among them and now with immigrants.
To explore how many markers are required to recover these fine scale patterns of structure, we ranked SNPs by FST among villages and repeated the PCA for the most differentiated subsets of 30 000, 10 000, 3000 and 300 SNPs in each population. In all three populations, 10 000 or more high FST SNPs recovered an essentially identical picture to that using the full data set, and even 3000 SNPs preserved considerable separation between the villages (not shown). Using only the most discriminating 300 SNPs, little structure could be observed between the two Croatian villages; however, in Scotland and Italy one of the three settlements included in each location remained completely differentiated from the other two (not shown). We note that these results are only indicative of the minimum number of SNPs required to separate these populations, as by necessity SNPs have been selected intrinsically on the basis of FST within the same data set, rather than extrinsically from other data.
The slightly lower differentiation of the Croatian villages is not surprising given the fact that they are physically the closest of those considered here, being 8 km apart, with only low hills separating them. In contrast, the settlements in the Scottish Isles and Italy are separated by 15–30 km of sea in the former case, and of 3000 m mountains in the latter, although there are deep connecting valleys.
Such fine-scale differentiation is consistent with the highly non-random nature of human mate choice over the millennia. The average distance between the birthplaces of spouses in rural parts of Finland, the Po valley in northern Italy and the isles of Scotland in the nineteenth century was ∼1.5–3 km.10 Such close endogamy was probably the norm in rural Europe due to lack of transport or economic opportunities. The breakdown of these isolates has since dramatically altered the population structure.11
The exquisite structure preserved in the genomes of people with all grandparents from the same settlement demonstrates that very detailed genetic and geographical ancestry information can be obtained by genome-wide SNP analyses. This provides novel opportunities, under certain circumstances, to predict the micro-geographical origin of an individual. Genetic association studies that include rural populations must also model this genetic structure, but it is not a barrier to gene discovery.12 When whole-genome sequences become widely available, the ability to use many more variants, including rarer ones, to identify short shared genomic segments will perhaps allow routine identification of regional ancestries, given a suitably large and carefully collected reference sample.
References
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Novembre J, Johnson T, Bryc K et al: Genes mirror geography within Europe. Nature 2008; 456: 98–101.
Sabatti C, Service SK, Hartikainen AL et al: Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet 2009; 41: 35–46.
Nelis M, Esko T, Mägi R et al: Genetic structure of Europeans: a view from the North-East. PLoS One 2009; 4: e5742.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: e190.
Tang H, Peng J, Wang P, Risch NJ : Estimation of individual admixture: analytical and study design considerations. Genet Epidemiol 2005; 28: 289–301.
Egeland T, Bøvelstad HM, Storvik GO, Salas A : Inferring the most likely geographical origin of mtDNA sequence profiles. Ann Hum Genet 2004; 68: 461–471.
Price AL, Weale ME, Patterson N et al: Long-range LD can confound genome scans in admixed populations. Am J Hum Genet 2008; 83: 132–135.
McQuillan R, Leutenegger AL, Abdel-Rahman R et al: Runs of homozygosity in European populations. Am J Hum Genet 2008; 83: 359–372.
Cavalli-Sforza LL, Menozzi P, Piazza A : The History and Geography of Human Genes. Princeton: Princeton University Press, 1994.
Rudan I, Carothers AD, Polasek O et al: Quantifying the increase in average human heterozygosity due to urbanisation. Eur J Hum Genet 2008; 16: 1097–1102.
Vitart V, Rudan I, Hayward C et al: SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet 2008; 40: 437–442.
Acknowledgements
We thank the study volunteers in each population. EUROSPAN (European Special Populations Research Network) was supported by European Union FP6 Grant number 018947 (LSHG-CT-2006-01947). For the MICROS study, we thank the primary-care practitioners and the Department of Laboratory Medicine, Hospital of Silandro. The study was supported by the Ministry of Health and Department of Educational Assistance, University and Research of the Autonomous Province of Bolzano and the South Tyrolean Sparkasse Foundation. ORCADES was supported by the Scottish Government Chief Scientist Office and the Royal Society. DNA extractions were performed at the Wellcome Trust Clinical Research Facility (WTCRF) in Edinburgh. We acknowledge the invaluable contributions of L Anderson and the research nurses and the administrative team in Edinburgh. The Croatian study was supported through grants from the Medical Research Council, UK, and the Ministry of Science, Education and Sport of the Republic of Croatia (number 108-1080315-0302). We thank Professor P Rudan and the staff of the Institute for Anthropological Research in Zagreb; genotyping of the Croatian samples was carried out at the WTCRF, Edinburgh. CO’D was funded by a postdoctoral fellowship from the Irish Research Council for Science Engineering and Technology and AC from Science Foundation Ireland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
O'Dushlaine, C., McQuillan, R., Weale, M. et al. Genes predict village of origin in rural Europe. Eur J Hum Genet 18, 1269–1270 (2010). https://doi.org/10.1038/ejhg.2010.92
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.92
Keywords
This article is cited by
-
Understanding 6th-century barbarian social organization and migration through paleogenomics
Nature Communications (2018)
-
The Italian genome reflects the history of Europe and the Mediterranean basin
European Journal of Human Genetics (2016)
-
Fine-scale human genetic structure in Western France
European Journal of Human Genetics (2015)
-
Geographic population structure analysis of worldwide human populations infers their biogeographical origins
Nature Communications (2014)
-
Genetic variation in the Sorbs of eastern Germany in the context of broader European genetic diversity
European Journal of Human Genetics (2011)
