
Abstract
Microarray-based comparative genomic hybridization analysis identified a 737-kb microdeletion of Xq11.1, including the cell division cycle 42 guanine nucleotide exchange factor (GEF)-9 gene (ARHGEF9), encoding collybistin, which has a pivotal role in formation of postsynaptic glycine and γ-aminobutyric acid receptor clusters, in a male patient with severe mental retardation and epilepsy. No overlapping deletion with this was identified in the database of genomic copy number variations. A cohort study of ARHGEF9 nucleotide sequence identified a nonsense mutation in another male patient with severe mental retardation and epilepsy. This mutation affects one of the three transcript variants of ARHGEF9, which was confirmed to be expressed in the brain by reverse transcription-PCR. Although this nonsense mutation was shared with the patient's mother, it was not observed in 100 normal individuals. Both male patients suffered epileptic seizures after 1 year of age. Brain magnetic resonance imaging revealed mild frontal atrophy in the first patient and right frontal polymicrogyria in the second patient. Three previously reported mutations of ARHGEF9 consisted of a missense mutation in a male patient with hyperekplexia and two chromosomal disruptions in two female patients. The common phenotypic effects of all ARHGEF9 mutations were mental retardation and epilepsy. Therefore, ARHGEF9 is likely to be responsible for syndromic X-linked mental retardation associated with epilepsy.
Similar content being viewed by others


Introduction
X-linked gene defects have long been considered to be important causes of mental retardation on the basis of the observation that mental retardation is significantly more common in men than in women.1 This bias is largely due to the involvement of X-linked genes that have roles in brain differentiation and function.2 Diagnosis of X-linked diseases has important implications for recurrence in the family.3 X-linked mental retardation (XLMR) is subdivided into syndromic XLMR and non-syndromic XLMR forms, depending on the complications in addition to mental retardation that are identified upon physical examination, laboratory investigation and brain imaging.1 Non-syndromic XLMR forms are characterized by ‘pure’ cognitive impairment, whereas many of the syndromic XLMR forms show additional complications, including epilepsy. Non-syndromic XLMR forms are more common than syndromic XLMR forms.4 This method of classification has benefited both clinicians seeking diagnoses and researchers seeking causative genes.1 Large cohorts of XLMR families have led us to the identification of more than 90 XLMR genes,5, 6 which are distributed through the entire X chromosome. Studies of XLMR show heterogeneous observations for both clinical and genetic aspects;7 therefore, it is suspected that many more genes are still uncharacterized, which would not be specific enough to guide mutation analysis for a specific XLMR gene.3
Recently, we identified a de novo microdeletion of Xq11.1, including the cell division cycle 42 guanine nucleotide exchange factor (GEF)-9 gene (ARHGEF9; (MIM 300429), in a patient with severe mental retardation and epilepsy. This identification led us to do a cohort study for nucleotide alterations of ARHGEF9 and identified a novel nonsense mutation in a male patient with XLMR and epilepsy.
Materials and methods
Subjects and controls
The indicated patient 1 was registered to our ongoing study of genomic copy number aberrations after obtaining informed consent from the patient's family, following the permission approval process of the ethical committee of the institution.
For the cohort study for ARHGEF9 mutation analyses, we recruited 23 male patients, whose genetic etiologies had not been revealed even by microarray-based comparative genomic hybridization analyses, and whose clinical phenotypes were similar to patient 1 with severe mental retardation and epilepsy. One hundred DNA samples were accumulated from 50 male and 50 female Japanese volunteers and used for normal controls.
Microarray-based comparative genomic hybridization analyses
The genomic copy numbers were analyzed using the Human Genome CGH Microarray 44 K (Agilent Technologies, Santa Clara, CA, USA), as described previously.8 In this study, genomic DNA was extracted from peripheral blood by using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany).
Fluorescent in situ hybridization analyses
The detected aberration was confirmed by fluorescent in situ hybridization, as described previously,8 with a bacterial artificial chromosome clone (RP11-943J20) as a probe, which was selected from the UCSC genome browser (http://www.genome.ucsc.edu).
Nucleotide mutation analyses for ARHGEF9
Genomic sequences of all exons of ARHGEF9 were analyzed by the standard PCR-direct sequencing method. The primers used for PCR and the big-dye sequencing reaction (Life Technologies, Foster City, CA, USA) are listed in Supplementary Table 1.
Expression study of ARHGEF9 transcript variants
According to the UCSC genome browser, ARHGEF9 has three transcript variants, including transcript variant 1 (NM_015185), transcript variant 2 (NM_001173479), and transcript variant 3 (NM_001173480) (Figure 1, Supplementary Table 2). Although ARHGEF9 had been confirmed to be highly expressed in the brain,9, 10 functional relevance of transcript variant 2 was analyzed in this study. To confirm the expression of transcript variant 2 of ARHGEF9 in the brain tissues, reverse transcription-PCR was performed using specific primers for transcript variants 2 and 3. Transcript variant 3 was also analyzed to compare with transcript variant 2. The sense primers, Primer 1a (5′-GTCAGCCCGCGACAACTCG-3′) and Primer 1b (5′-TGAGCTTCAACATGGCCTCG-3′), were specific for transcript variant 2 and 3, respectively (Figure 1). An anti-sense primer, Primer R (5′CATCCTCCTGGTTCACCCAG-3′), was on exon 3, which is common to both transcript variants. Skin fibroblasts and lymphocytes derived from healthy individuals were used for controls. Human Brain Total RNA (#636530) and Human Fetal Brain Total RNA (#636526) were purchased from Clontech Laboratories (Mountain View, CA, USA).

Schematic representation of the primary structure of cell division cycle 42 GEF-9 gene (ARHGEF9) and its transcript variants. ARHGEF9 is encoded in the anti-sense direction on the X chromosome and consisted of 11 different exons. There are three types of transcript variants, which share exons 2–10; the first exon is unique for each transcript variant. The location of the first methionine is indicated by ‘M’ on each transcript variant. The different use of exon 1b between transcript variant 1 and 3 is complicated, because the first methionine of transcript variant 1 is on the last part of exon 1b, which is spliced out in transcript variant 3 (Supplementary Figure 1). The location of the termination codon ‘stop’ of transcript variant 2 has not yet been determined. The locations of the primers used for expression analysis by reverse transcription-PCR are shown by arrows. The locations of the reported nucleotide mutations are indicated by black triangles.
Results
ARHGEF9 alterations
Microarray-based comparative genomic hybridization analysis showed a loss of genomic copy numbers at Xq11.1, with a length of 737-kb in patient 1 (Figures 2a and b). This was the only pathogenic aberration detected in patient 1. By fluorescent in situ hybridization analysis, the targeted signal was negative in patient 1 (Figure 2c), but it was hybridized to metaphase and interphase preparations from his parents; the results indicated de novo deletion in patient 1 (Figures 2d and e). His karyotype was indicated as 46,XY,del(X)(q11.1q11.1).ish del(RP11-943J20–).arr Xq11.1(62,321,746-63,058,548)x0.dn. The physical positions refer to March 2006 human reference sequence (NCBI36/hg18). The available web-based database, including the International Standards for Cytogenomic Arrays Consortium database (https://www.iscaconsortium.org/), DECIPHER (http://decipher.sanger.ac.uk/) and Database of Genomic Variants (http://projects.tcag.ca/variation/) were analyzed, and there was no data of the small deletion overlapping with this region.

A submicrodeletion identified in patient 1. Chromosome view by Agilent Genomic Workbench (Agilent) indicates a microdeletion of chrXq11.1 (a), which is expanded in Gene view (b). Fluorescent in situ hybridization analyses for patient 1 (c), his father (d) and mother (e). Patient 1 shows a green signal of RP11-75D20 on the X chromosome, but no red signal of RP11-943J20 covering ARHGEF9 (arrow) (c). His father and mother show one and two normal X chromosomes, respectively (d, e).
A nonsense mutation at the second codon of the transcript variant 2 of ARHGEF9 (c.4C>T, p.Q2X) was identified in patient 2 (Figure 3). Analysis of his parental samples showed that his mother had the same nonsense mutation. We could not get informed consent for segregation analysis in this family. This nonsense mutation was not found in 100 normal control DNA samples.

Sequencing analysis for exon 1a shows a C to T transition at the fourth nucleotide in patient 2, which indicates p.Q2X.
Expression study of ARHGEF9 transcript variants
The results of reverse transcription-PCR confirmed the expression of transcript variant 2 in the brain (Figure 4a). Direct sequencing of these PCR amplicons confirmed the specific splicing of transcript variants 2 and 3, and expression of transcript variant 1 was not detected (Figure 4b, Supplementary Figure 1).

Expression studies of transcript variants 2 and 3 by reverse transcription-PCR. (a) Although transcript variant 2 is not expressed in lymphocytes, it is highly expressed in both fetal and total brain samples. HDF, human dermal fibroblasts. (b) Nucleotide sequences of reverse transcription-PCR products are unique for transcript variant 2 and 3. All reverse transcription-PCR products amplified by primer 1b and primer R indicate transcript variant 3. nt, nucleotide position corresponds to build 18.
Clinical information of the patients
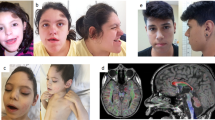
Patient 1 is a 5-year-old boy, second born to non-consanguineous healthy parents. He has a healthy 9-year-old elder brother. Patient 1 was born by vaginal delivery at 37 weeks of gestation, as a large-for-date baby, with a birth weight of 4254 g (>97 centile), birth length of 54 cm (>97 centile) and an occipito-frontal circumference of 36 cm (>97 centile). At the age of 2 years, he suffered complex partial seizures, which occurred once per month, but were well controlled by valproate. Electroencephalogram showed separate spikes at each temporal region. Brain magnetic resonance imaging revealed a relatively hypoplastic frontal lobe (Figures 5a and b). Ophthalmological and audiological examinations revealed no abnormalities. Conventional chromosomal G-banding analysis revealed a normal male karyotype of 46,XY. Developmental delay was noted at the age of 9 months; he began to sit at 12 months, roll over at 13 months and crawl at 20 months. At present, he shows overgrowth with a height of 118.3 cm (>97 centile), body weight of 20.8 kg (90–75 centile) and occipito-frontal circumference of 53 cm (90–97 centile). His head is trigonocephalic, but there are no other dysmorphic findings. He cannot stand without any support nor can he utter meaningful words. His toilet and diet habits are disorganized. Thus, his developmental quotient is estimated to be <15.

Brain magnetic resonance imaging of the patients. Patient 1 (a, b) shows relatively small frontal lobes, and patient 2 (c–f) shows right fronto-temporal polymicrogyria (*). (a) Spoiled gradient recalled (SPGR) image, (b) fluid attenuated IR (FLAIR) image, (c, e) T1-weighted image, (d, f) T2-weighted image.
Patient 2 is a boy (age, 5 years and 5 months). He was born with a birth weight of 3284 g (75–90 centile), length of 51 cm (90 centile) and occipito-frontal circumference of 32.5 cm (10–25 centile). He is the only child of healthy parents. His mother had a healthy elder brother. The maternal grandmother had a healthy brother and two female siblings. There are no other patients with mental retardation in this family (Supplementary Figure 2). He started to show psychomotor developmental delay in his early infantile period, with head control at 4 months, rolling over at 6 months, sitting at 12 months and walking alone at 24 months. He suffered convulsive seizures at the age of 20 months. His seizures were intractable for many anti-epileptic drugs. The electroencephalogram findings helped diagnose his epileptic status as continuous spike and wave during sleep. Brain magnetic resonance imaging findings indicated the presence of right frontal polymicrogyria (Figures 5c–f). At present, he shows normal stature with a height of 109 cm (50–75 centile), weight of 15.3 kg (10–25 centile) and occipito-frontal circumference of 52 cm (75–90 centile), without any dysmorphic findings. He shows severe developmental delay; he cannot speak any words and his walking is ataxic.
Discussion
In this study, a male patient (patient 1) with severe mental retardation and epilepsy showed a de novo nullisomy of Xq11.1 in which only one OMIM gene, ARHGEF9, was included. Such a small deletion including only ARHGEF9 has not been previously reported in the literature and is not found in the web-based database. ARHGEF9 encodes collybistin, a brain-specific GEF for the Rho GTPase cell division cycle 42, small Rho-like GTPases,11, 12, 13 and it has a pivotal role in the formation of postsynaptic glycine and γ-aminobutyric acid receptor clusters.7 Collybistin has been shown to be expressed in the brain and essential for the gephyrin-dependent clustering of a specific set of γ-aminobutyric acid receptors at inhibitory postsynaptic sites, and is also known to affect the plasticity and polarity of the eukaryotic actin cytoskeleton and to alter cell signaling transduction pathways.9, 10, 14, 15 Collybistin is required for both the initial localization and maintenance of gephyrin and gephyrin-dependent γ-aminobutyric acid receptors at inhibitory postsynaptic membrane specializations in the hippocampus.16
Previously, some other ARHGEF9 mutations have been reported. The first reported missense mutation, p.G55A in the SH3 domain, was found in a male patient with severe mental retardation, hyperekplexia, drug-resistant seizures and premature death.10 This missense mutation may have a dominant-negative effect and lead to a severe phenotype caused by somatic and dendritic trapping of gephyrin and receptors by collybistin aggregates.10 Compared with this, Xp11.1 nullisomy and the nonsense mutation identified in this study lead to loss-of-function of ARHGEF9.
The other previously reported mutations were ARHGEF9 disruptions caused by balanced chromosomal rearrangements discovered in two female patients with neurological phenotypes, including sensory hyperarousal, a disrupted sleep-wake cycle, late-onset epileptic seizures, increased anxiety, aggressive behavior and mental retardation.17, 18 Generally, X chromosome inactivation is randomly observed. However, patients with balanced translocation involving X chromosome often show non-random inactivation in normal X chromosome. Indeed, the reported female patients with X chromosome rearrangements showed non-random inactivation, which inactivated the normal X chromosome having a normal copy of ARHGEF9. Consequently, the patients showed low expression of ARHGEF9 and suffered mild mental retardation and sensory hyperarousal, but no hyperekplexia.17, 18 Although these female patients with X chromosome imbalances are specific cases, these findings suggested that ARHGEF9 is related to various phenotypes and would be a candidate gene for XLMR associated with epilepsy.17, 18
Previously Marco et al.17 analyzed the nucleotide sequences of ARHGEF9 in 477 mentally retarded males, and there was no pathogenic mutation. The three previously reported patients showed mental retardation and were associated with various types of epilepsies. Thus, we performed the cohort study focused on the male patients associated with epilepsy and identified a nonsense mutation, p.Q2X, in patient 2 who showed severe mental retardation and epilepsy. This nonsense mutation is located on exon 1a of ARHGEF9, which is used only in the transcript variant 2 of ARHGEF9 (Figure 1). As the other two transcript variants do not include this exon, this nonsense mutation does not influence the functions of those two variants. Thus, we studied the relevant importance by reverse transcription-PCR analysis for the transcript variants of ARHGEF9 in the brain RNA samples. The results confirmed high expression of the transcript variant 2 in the brain samples, including the adult and fetal brain samples (Figure 4). Thus, this nonsense mutation would lead to loss-of-function of the transcript variant 2. This nonsense mutation was not found in 100 normal controls, but was found to be inherited from the patient's healthy mother. Segregation pattern of this nonsense mutation in this family would be supposed to give us important knowledge; however, we could not get permission to analyze it. From these results, we think that this nonsense mutation of the transcript variant 2 identified in patient 2 would be responsible for the severe mental retardation and epilepsy seen in patient 2 with the X-linked recessive trait.
In this study, we identified loss-of-function alterations of ARHGEF9 in two male patients. The common characteristics of both patients were severe mental retardation, epilepsy and abnormal brain magnetic resonance imaging findings. Both patients suffered epilepsy after reaching 1 year of age. These clinical features are different from those of previously reported patients.10 The clinical features may be dependent on the differences in mutation types. The missense mutation identified in the patient with hyperekplexia might have a dominant negative effect on collybistin.10 The ARHGEF9 disruptions derived from X chromosome rearrangements in female patients with sensory hyperarousal would also have some other mechanisms.17, 18 The common features of all ARHGEF9 mutations were mental retardation and epilepsy. Therefore, ARHGEF9 is likely to be responsible for syndromic XLMR associated with epilepsy.
The microdeletion of Xq11.1 identified in patient 1 of this study included two unknown transcripts, other than ARHGEF9. One is the spindlin family, member 4 (SPIN4) and the other is non-coding RNA (Homo sapiens hypothetical LOC92249). Because patient 1 showed high stature, which was not seen in patient 2, this additional feature may be derived from the other transcripts included in the deletion region.
References
Ropers, H. H. & Hamel, B. C. X-linked mental retardation. Nat. Rev. Genet. 6, 46–57 (2005).
Ropers, H. H., Hoeltzenbein, M., Kalscheuer, V., Yntema, H., Hamel, B., Fryns, J. P. et al. Nonsyndromic X-linked mental retardation: where are the missing mutations? Trends Genet. 19, 316–320 (2003).
Stevenson, R. E. Advances in X-linked mental retardation. Curr. Opin. Pediatr. 17, 720–724 (2005).
Gecz, J., Shoubridge, C. & Corbett, M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 25, 308–316 (2009).
Chiurazzi, P., Schwartz, C. E., Gecz, J. & Neri, G. XLMR genes: update 2007. Eur. J. Hum. Genet. 16, 422–434 (2008).
Tarpey, P. S., Smith, R., Pleasance, E., Whibley, A., Edkins, S., Hardy, C. et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 41, 535–543 (2009).
Ropers, H. H. X-linked mental retardation: many genes for a complex disorder. Curr. Opin. Genet. Dev. 16, 260–269 (2006).
Shimojima, K., Komoike, Y., Tohyama, J., Takahashi, S., Paez, M. T., Nakagawa, E. et al. TULIP1 (RALGAPA1) haploinsufficiency with brain development delay. Genomics 94, 414–422 (2009).
Reid, T., Bathoorn, A., Ahmadian, M. R. & Collard, J. G. Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J. Biol. Chem. 274, 33587–33593 (1999).
Harvey, K., Duguid, I. C., Alldred, M. J., Beatty, S. E., Ward, H., Keep, N. H. et al. The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826 (2004).
Van Aelst, L. & D’Souza-Schorey, C. Rho GTPases and signaling networks. Genes Dev. 11, 2295–2322 (1997).
Kins, S., Betz, H. & Kirsch, J. Collybistin, a newly identified brain-specific GEF, induces submembrane clustering of gephyrin. Nat. Neurosci. 3, 22–29 (2000).
Grosskreutz, Y., Hermann, A., Kins, S., Fuhrmann, J. C., Betz, H. & Kneussel, M. Identification of a gephyrin-binding motif in the GDP/GTP exchange factor collybistin. Biol. Chem. 382, 1455–1462 (2001).
Jedlicka, P., Papadopoulos, T., Deller, T., Betz, H. & Schwarzacher, S. W. Increased network excitability and impaired induction of long-term potentiation in the dentate gyrus of collybistin-deficient mice in vivo. Mol. Cell Neurosci. 41, 94–100 (2009).
Hall, A. & Nobes, C. D. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 355, 965–970 (2000).
Papadopoulos, T., Eulenburg, V., Reddy-Alla, S., Mansuy, I. M., Li, Y. & Betz, H. Collybistin is required for both the formation and maintenance of GABAergic postsynapses in the hippocampus. Mol. Cell Neurosci. 39, 161–169 (2008).
Marco, E. J., Abidi, F. E., Bristow, J., Dean, W. B., Cotter, P., Jeremy, R. J. et al. ARHGEF9 disruption in a female patient is associated with X linked mental retardation and sensory hyperarousal. J. Med. Genet. 45, 100–105 (2008).
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E. et al. A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30, 61–68 (2009).
Acknowledgements
This work was partially supported by a grant from the Hayashi Memorial Foundation for Female Natural Scientists (KS) and a research grant, Scientific Research (c), 21591334, 2010 (TY), from the Japan Ministry of Education, Science, Sports and Culture.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Shimojima, K., Sugawara, M., Shichiji, M. et al. Loss-of-function mutation of collybistin is responsible for X-linked mental retardation associated with epilepsy. J Hum Genet 56, 561–565 (2011). https://doi.org/10.1038/jhg.2011.58
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.58
Keywords
This article is cited by
-
Human ARHGEF9 intellectual disability syndrome is phenocopied by a mutation that disrupts collybistin binding to the GABAA receptor α2 subunit
Molecular Psychiatry (2022)
-
The role of GABAergic signalling in neurodevelopmental disorders
Nature Reviews Neuroscience (2021)
-
De novo ARHGEF9 missense variants associated with neurodevelopmental disorder in females: expanding the genotypic and phenotypic spectrum of ARHGEF9 disease in females
neurogenetics (2021)
-
Developmental seizures and mortality result from reducing GABAA receptor α2-subunit interaction with collybistin
Nature Communications (2018)
-
Gephyrin: a key regulatory protein of inhibitory synapses and beyond
Histochemistry and Cell Biology (2018)

{kind=link}
{kind=link}