
Abstract
In a multi-institutional collaborative project, 1473 patients with myeloproliferative neoplasms (MPN) were screened for isocitrate dehydrogenase 1 (IDH1)/IDH2 mutations: 594 essential thrombocythemia (ET), 421 polycythemia vera (PV), 312 primary myelofibrosis (PMF), 95 post-PV/ET MF and 51 blast-phase MPN. A total of 38 IDH mutations (18 IDH1-R132, 19 IDH2-R140 and 1 IDH2-R172) were detected: 5 (0.8%) ET, 8 (1.9%) PV, 13 (4.2%) PMF, 1 (1%) post-PV/ET MF and 11 (21.6%) blast-phase MPN (P<0.01). Mutant IDH was documented in the presence or absence of JAK2, MPL and TET2 mutations, with similar mutational frequencies. However, IDH-mutated patients were more likely to be nullizygous for JAK2 46/1 haplotype, especially in PMF (P=0.04), and less likely to display complex karyotype, in blast-phase disease (P<0.01). In chronic-phase PMF, JAK2 46/1 haplotype nullizygosity (P<0.01; hazard ratio (HR) 2.9, 95% confidence interval (CI) 1.7–5.2), but not IDH mutational status (P=0.55; HR 1.3, 95% CI 0.5–3.4), had an adverse effect on survival. This was confirmed by multivariable analysis. In contrast, in both blast-phase PMF (P=0.04) and blast-phase MPN (P=0.01), the presence of an IDH mutation predicted worse survival. The current study clarifies disease- and stage-specific IDH mutation incidence and prognostic relevance in MPN and provides additional evidence for the biological effect of distinct JAK2 haplotypes.
Similar content being viewed by others

Introduction
Despite the seminal discovery of JAK2 or MPL mutations in the majority of patients with BCR-ABL1-negative myeloproliferative neoplasms (MPN),1, 2, 3, 4 it is becoming increasingly evident that these mutations do not signify either disease-initiating or leukemia-promoting events.5, 6 It is therefore important to keep looking for additional molecular alterations to clarify the genetic underpinnings of both chronic- and blast-phase MPN. In the last 2 years, mutations involving TET2, ASXL1 and CBL have been described in some patients with BCR-ABL1-negative MPN, including polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF).7 The precise pathogenetic contribution of these mutations and their clinical relevance are currently under investigation. The glioma-associated8 isocitrate dehydrogenase 1 (IDH1) and IDH2 mutations are the latest to be added to the ‘MPN mutations list’.9
IDH1, located on chromosome 2q33.3, and IDH2, located on chromosome 15q26.1, encode enzymes that catalyze oxidative decarboxylation of isocitrate to α-ketoglutarate. IDH1 (cytoplasm and peroxisome) and IDH2 (mitochondria) use NADP+ as a co-factor to generate NADPH, which is important in the production of intracellular glutathione. Intact IDH activity is therefore necessary for cellular protection from oxidative stress. Mutant IDH has decreased affinity to isocitrate, but displays neomorphic catalytic activity toward α-ketoglutarate, the net result being decreased supply of α-ketoglutarate and accumulation of 2-hydroxyglutarate.10, 11, 12, 13 It is currently believed that these intracellular changes facilitate oncogenic pathways including activation of HIF-1α.10
IDH1 and IDH2 mutations were first described in low-grade gliomas/secondary glioblastomas8 and subsequently in acute myeloid leukemia (AML),14 with respective mutational frequencies of ∼70 and 8%. We recently screened 200 patients with either chronic- or blast-phase MPN for IDH mutations, and identified 9 patients with either IDH1 (n=5) or IDH2 (n=4) mutations.9 Mutational frequencies were ∼21% for blast-phase MPN and ∼4% for PMF. In the current study, we expanded our study cohort to include 1473 patients recruited from three MPN centers of excellence, with the intent to accurately describe the prevalence of IDH mutations in chronic-, fibrotic- and blast-phase PV, ET and PMF. In addition, IDH-mutated patients were analyzed for their cytogenetic and molecular (that is, JAK2, MPL and TET2 mutation and JAK2 haplotype status) phenotype, as well as their prognostic relevance.
Materials and methods
This study was approved by the Mayo Clinic institutional review board. All patients provided authorization for use of their medical records for research purposes, and the research was carried out according to the principles of the Declaration of Helsinki. Patient samples were obtained from the Mayo Clinic, Harvard Medical Institute and University of Florence. Mutational analyses were performed on DNA derived from either bone marrow or peripheral blood granulocytes. JAK2 46/1 haplotype analysis on patient samples accrued from Harvard was performed on germline DNA. Diagnoses of MPN, post-PV/ET MF and AML, in patient samples accrued from the Mayo Clinic and the University of Florence, were according to the World Health Organization and International Working Group criteria.7, 15 Diagnoses in patients accrued from Harvard were self-reported during an internet-based collection of samples, as previously detailed.16
DNA from either bone marrow (Mayo Clinic samples) or granulocytes (samples from Harvard and the University of Florence) was extracted using conventional methods. MPL, JAK2 and TET2 mutation and JAK2 haplotype analyses were performed according to previously published methods.4, 17, 18, 19 With regard to IDH mutation analysis, Harvard patient samples were analyzed using the following primers for IDH1, which cover amino acid residues 41–138: sense, 5′-TGTGTTGAGATGGACGCCTA-3′ and anti-sense, 5′-GGTGTACTCAGAGCCTTCGC-3′. Sequencing of IDH2 used primers that covered amino acid residues 125–226: sense, 5′-CTGCCTCTTTGTGGCCTAAG-3′ and anti-sense, 5′-ATTCTGGTTGAAAGATGGCG-3′. Sequence analysis was performed using Mutation Surveyor (SoftGenetics, State College, PA, USA) and all mutations were validated by repeat PCR and sequencing on unamplified DNA from the archival sample.
Mayo Clinic and University of Florence patient samples were screened for IDH1 and IDH2 mutations by direct sequencing and/or high-resolution melting assay. Direct sequencing for IDH1 exon 4 mutations was carried out using the following primer sequences: sense, 5′-CGGTCTTCAGAGAAGCCATT-3′ and anti-sense, 5′-CACATTATTGCCAACATGAC-3′.18 IDH2 exon 4 was amplified using sense, 5′-CCACTATTATCTCTGTCCTC-3′ and anti-sense, 5′-GCTAGGCGAGGAGCTCCAGT-3′.19 Both reactions were performed in 25 μl volume containing 100 ng of DNA, 0.25 U Taq polymerase, 0.3 mM each of dATP, dCTP, dGTP and dTTP, 5 μl of a 10 × PCR buffer (Roche Diagnostics, Indianapolis, IN, USA) and 0.2 μM each of sense and anti-sense primers. The reaction was denatured at 94 °C for 3 min followed by 35 cycles of denaturing at 94 °C for 30 s, annealing at 57 °C for 30 s and extension at 72 °C for 40 s. After a final extension at 72 °C for 2 min, the products were confirmed by running on 1.3% agarose gel and purified using Qiagen's PCR Quick Purification Kit. The product was sequenced using the ABI PRISM 3730xl analyzer (Applied Biosystems Inc, Foster City, CA, USA) to screen for the presence of mutations.
High-resolution melting was performed using the LightCycler 480 real-time PCR system (Roche Diagnostics), using the above-mentioned primers for IDH1 mutations (R130) and the following primers for IDH2 mutations (R140 and R172): R140 sense, 5′-GCTGAAGAAGATGTGGAA-3′ and anti-sense, 5′-TGATGGGCTCCCGGAAGA-3′; R172 sense, 5′-CCAAGCCCATCACCATTG-3′ and anti-sense, 5′-CCCAGGTCAGTGGATCCC-3′ (Figure 1).

High-resolution melting (HRM) normalized and temperature-shifted difference plot for IDH1 (a) and IDH2 (b) and corresponding sequences (c and d).
Conventional statistical procedures were used (SAS Institute, Cary, NC, USA). All statistically analyzed data were obtained at time of IDH mutation analysis. All P-values were two-tailed and statistical significance was set at the level of P<0.05. Categorical variables were described as count and relative frequency and compared by χ2 statistics. Comparison of continuous variables between categories was performed by the Mann–Whitney U-test. Survival analysis was performed by the Kaplan–Meier method taking the interval from the date of diagnosis, for chronic-phase disease, or from the date of leukemic transformation, for blast-phase disease, to death or last contact. The log-rank test was used to compare survival data. Cox regression model was used for multivariable analysis.
Results
Disease- and stage-specific IDH mutational frequencies
A total of 1473 patients with BCR–ABL1-negative MPN were recruited from the Mayo Clinic, Rochester, MN, USA (n=629), University of Florence, Florence, Italy (n=522) and Harvard Medical Institute, Boston, Massachusetts, USA (n=322). Specific diagnoses included ET (n=594), PV (n=421), PMF (n=312), post-PV MF (n=54), post-ET MF (n=41), post-PV AML (n=12), post-ET AML (n=7) and post-PMF AML (n=32). Table 1 provides clinical and laboratory details of the study population including age and sex distribution, specific diagnoses and JAK2, MPL and TET2 mutational and JAK2 46/1 haplotype status, stratified by center of patient recruitment. A total of 38 IDH mutations were documented (Table 2): 18 involved IDH1 (10 R132S, 7 R132C and 1 R132G) and 20 IDH2 (18 R140Q, 1 R140W and 1 R172G). IDH mutations were infrequent in chronic- or fibrotic-phase disease and significantly more prevalent in blast-phase disease (P<0.01; Table 3): 5 (0.8%) in ET, 8 (1.9%) in PV, 13 (4.1%) in PMF, 1 (1%) in post-ET/PV MF, none in blast-phase ET, 3 (25%) in blast-phase PV and 8 (25%) in blast-phase PMF.
Correlation of IDH mutations with other MPN-associated mutations and JAK2 46/1 haplotype
Considering the preponderance of informative cases with centrally confirmed diagnosis and availability of a more complete laboratory data, the current analysis was limited to patients from the Mayo Clinic cohort (n=629). IDH mutational frequencies were similar among JAK2- (3.6%), MPL- (4.3%) and TET2 (3.2%)-mutated patients and their respective mutation-negative counterparts (4.2, 5.3 and 6.3%; Table 3). In other words, mutant IDH was shown to co-occur with a JAK2, MPL or TET2 mutation, and mutational frequency did not appear to be influenced by either the type of the coexisting mutation (P=0.96) or the presence or absence of each specific mutation (Table 3). However, IDH-mutated cases were more likely to be nullizygous for JAK2 46/1 haplotype, especially when analysis was restricted to informative (that is, with JAK2 46/1 haplotype information) patients with chronic- (n=158) or blast (n=23)-phase PMF, analyzed together (P=0.007) or separately (P=0.04; Table 4).
Clinical correlates and prognostic relevance
To avoid disease- or stage-specific confounding factors, as well as assure adequate sample size of informative cases, clinical correlative and prognostic studies were limited to PMF. In this patient cohort, detailed clinical information was available in 111 patients with chronic-phase PMF (including 7 IDH-mutated cases) and 27 patients with blast-phase PMF (including 8 IDH-mutated cases), both patient populations were accrued from the Mayo Clinic cohort. In both chronic- and blast-phase PMF, the presence of IDH mutations was not influenced by either age (P=0.51 and 0.70, respectively) or gender (P=0.09 and 0.3, respectively). In chronic-phase disease, comparison of prognostically relevant disease variables at diagnosis revealed that cytogenetic findings in IDH-mutated cases often belonged to a low- or intermediate-risk category,20 although the difference was not statistically significant (Table 4). Similarly, IDH-mutated blast-phase PMF was less likely to display complex karyotype (0 vs 64% in IDH-unmutated cases; P=0.001).
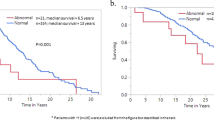
In addition to biological implications, the aforementioned associations of IDH mutations with favorable cytogenetic profile and JAK2 46/1 haplotype nullizygosity, both which have previously been shown to be prognostically relevant,19, 20 mandated their inclusion as covariates during multivariable survival analysis. In chronic-phase PMF, univariate analysis showed statistically significant adverse survival effect from JAK2 46/1 haplotype nullizygosity (P=0.0001; 34 nullizygous vs 74 not nullizygous), high-risk karyotype (P<0.0001; 13 high-risk vs 98 not high-risk) and higher International Prognostic Scoring System (IPSS; 27 high, 29 intermediate-2, 30 intermediate-1 and 25 low-risk patients)21 risk score (P<0.0001), but not from IDH mutational status (P=0.54; 7 mutated vs 104 unmutated; Figure 2). Multivariable analysis confirmed the independent prognostic value of JAK2 46/1 haplotype status (hazard ratio (HR) 2.2, 95% confidence interval (CI) 1.2–4.2), karyotype (HR 2.8, 95% CI 1.3–5.9) and IPSS risk score (HR 4.8, 95% CI 2.0–11.5).

Survival curves of 111 patients with chronic-phase primary myelofibrosis stratified by their isocitrate dehydrogenase (IDH) mutational status (a), cytogenetic risk (b), JAK2 46/1 haplotype status (c) or International Prognostic Scoring System risk category (d).
In blast-phase PMF, despite its association with noncomplex karyotype, the presence of mutant IDH predicted shortened survival, calculated from the time of disease transformation (P=0.04), and there was a similar trend for JAK2 non-46/1 haplotype (P=0.14; Figure 3). Significance was lost for both during multivariable analysis, probably because of small sample size. IDH mutation status also predicted worse survival when the analysis included all blast-phase MPN cases from the Mayo cohort (Figure 4; n=43; P=0.01). In this instance, significance was sustained during multivariable analysis that included JAK2 46/1 haplotype as a covariate.

Survival curves of patients with blast-phase primary myelofibrosis stratified by their isocitrate dehydrogenase (IDH) mutational (a; n=27 including 8 mutated cases) or JAK2 46/1 haplotype (b; n=23 including 8 nullizygous cases) status.

Survival curves of 43 patients with blast-phase myeloproliferative neoplasm stratified by their isocitrate dehydrogenase (IDH) mutational status.
Discussion
IDH1 point mutations involving exon 4 occur in the majority (60–90%) of patients with low-grade gliomas and secondary glioblastomas, and always affect the amino acid arginine at position 132 (∼93% R132H, 4% R132C, 2% R132S and <1% R132G, R132L or R132V).8, 22, 23 These mutations are relatively infrequent in primary glioblastoma (∼7%)22 and are usually not seen in other solid tumors.23, 24 A small fraction (∼4%) of glioma-associated IDH mutations involves IDH2, specifically the R132 analogous R172 residue on exon 4 (R172K, R172M, R172G, R172W).23, 25 IDH mutations in glioma are heterozygous, believed to constitute early genetic events and might be mutually exclusive of EGFR and PTEN, but not TP53 mutations. Clinical correlates of IDH mutations in glioma include younger age, longer survival and reduced risk of disease progression after conventional therapy.8, 22, 23, 26, 27
The first study on IDH mutations in AML included 188 patients with primary AML and reported IDH1, but not IDH2, mutations in 8.5% (n=16) of the cases and 16% of those with normal karyotype: R132C in 8 patients, R132H in 7 and R132S in 1.14 In a subsequent AML study of 493 patients,28 27 (5.5%) expressed IDH1 mutations (37% R132C, 26% R132H, 19% R132S, 15% R132G and 4% R132L). In both studies,14, 28 IDH1 mutations clustered with normal karyotype, NPM1 mutations and trisomy 8. IDH1 mutations are rare in pediatric AML.29 More recently, IDH2 mutations, affecting R172 (R172K)12, 13 or R140 (R140Q),13 were also shown to occur in primary AML.12, 13 In one of these studies, IDH1 or IDH2 mutations were seen in 18 (23%) of 78 AML cases and the majority of the mutations (12 of 18) involved IDH2, primarily R140Q.13 In general, survival in primary AML did not seem to be affected by the presence of IDH mutations.13, 14, 28, 29, 30 However, more recent studies suggest that specific IDH mutation variants might be prognostically relevant in certain molecular subsets of AML.31
The first reports of IDH mutations in MPN came from three independent groups.9, 32, 33 In one of these studies, IDH1 mutations were seen in ∼8% (5 of 63) of blast-phase MPN patients, mostly occurring in the absence of TET2 and ASXL1 mutations.32 The second study was focused on blast-phase MPN that arose from JAK2-mutated chronic-phase MPN.33 In this study, mutant IDH was seen in 5 (31%) of 16 blast-phase MPN (three cases with R132C and two with R140Q) and in none of the 180 PV or ET patients.33 The third study from the Mayo Clinic included 200 MPN patients and showed IDH mutational frequencies of ∼21% for blast-phase MPN, regardless of JAK2 mutational status, and ∼4% for PMF.9 The specific IDH1 mutations found in the particular study included R132C and R132S and the IDH2 mutations R140Q and R140W.
The current study is an extension of the above-mentioned Mayo Clinic study and involves a large number of patients (n=1473) recruited from three major MPN centers of excellence. The results of the study clarify a number of issues regarding IDH mutations in MPN. First, the study provides robust incidence figures for IDH1 and IDH2 mutations across different disease stages of specific MPN variants. Accordingly, we now show that both IDH1 and IDH2 mutations can occur in chronic-phase ET, PV or PMF, although infrequently. Mutational frequency was equally low in post-PV/ET MF and this fact combined with the significantly higher mutation incidence observed in blast-phase disease suggests a pathogenetic contribution to leukemic but not fibrotic disease transformation. Two additional observations support this contention (i) complex karyotype was infrequently encountered in IDH-mutated blast-phase MPN, which suggests an independent pathogenetic contribution that might be tied to distinct molecular alterations, such as, for example, overexpression of the APP (amyloid â (A4) precursor protein) gene, which has previously been shown in AML to be associated with either complex karyotype or IDHR172 mutation31 and (ii) the absence of mutual exclusivity between IDH and other MPN-associated mutations (for example, TET2, MPL), which is consistent with the suggestion that the former are later-arising cooperating mutations that are more involved in disease progression rather than disease initiation.
The types of IDH mutations seen in our patients with MPN (mostly IDH2R140Q and IDH1R132S/C) are distinctly different than those seen in gliomas (mostly IDH1R132H) and more similar to those seen in AML, although IDH1R132H was significantly more prevalent in AML. Within the context of MPN, IDH2R140Q was over represented in chronic-phase ET and PV, whereas IDH1 mutations were more prevalent in PMF and blast-phase MPN. More studies are needed to confirm this apparent trend. Regardless, there is currently no good explanation for the observed diversity in IDH mutation variants among gliomas and myeloid malignancies and current information suggests similar biological consequences.13 Whether or not different IDH mutations carry different prognostic relevance in MPN is currently not known and we did not attempt to address the particular issue because of our relatively small number of informative cases. Of note, in a recent study of primary AML with normal karyotype, different types of IDH mutations appeared to variably influence disease-free survival and complete remission rates.31
One particularly interesting observation from the current study was the significant association between mutant IDH and JAK2 non-46/1 haplotype. The latter phenomenon is further evidence for the JAK2 mutation specificity of the previously described association between the JAK2 46/1 haplotype and MPN.19, 34, 35 In other words, whereas JAK2 exon 1419, 35 or exon 1236 mutations have been shown to be associated with JAK2 46/1 haplotype, we did not see the same effect involving MPL mutations34 (although others have shown otherwise),37 and now show an association with JAK2 non-46/1 haplotype for IDH mutations. This latter observation is also consistent with our previous report on the prognostically detrimental effect of JAK2 non-46/1 haplotype in PMF;19 it is possible that patients with PMF who are nullizygous for JAK2 46/1 haplotype are susceptible to additional adverse molecular events, such as IDH mutations, which might lead to biologically more aggressive disease. Consistent with this possible scenario, in the current study, the negative prognostic impact of mutant IDH was accounted for by the JAK2 46/1 genotype in PMF but not in blast-phase MPN, in which risk factors other than JAK2 non-46/1 haplotype might have promoted the development of IDH mutations.
It is becoming increasingly evident that there are many more mutations than JAK2 and MPL mutations in BCR–ABL1-negative MPN including those that involve TET2,38, 39 ASXL1,40 IDH1,32, 33 IDH2,9, 33 CBL,41 IKZF142 and LNK.43 Some of these mutations might be later-arising and more prevalent in blast-phase disease. What is currently lacking is a composite evaluation (that is, concurrent analysis of all relevant mutations), which includes paired chronic- and blast-phase samples of a large number of patients with blast-phase MPN. Such an approach is essential for clarifying the individual pathogenetic or prognostic contribution of the aforementioned mutations and their chronological order of appearance. It is very likely that additional mutations in MPN will be described soon, but practical relevance in terms of either disease prognostication or value as drug targets has so far been limited.
References
James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007; 356: 459–468.
Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006; 3: e270.
Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006; 108: 3472–3476.
Kilpivaara O, Levine RL . JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia 2008; 22: 1813–1817.
Vannucchi AM, Antonioli E, Guglielmelli P, Pardanani A, Tefferi A . Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia 2008; 22: 1299–1307.
Tefferi A, Vardiman JW . Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2008; 22: 14–22.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807–1812.
Pardanani A, Lasho T, Finke C, Mai M, McClure R, Tefferi A . IDH1 and IDH2 mutation analysis in chronic and blast phase myeloproliferative neoplasms. Leukemia 2010 (in press).
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009; 324: 261–265.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462: 739–744.
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 2010; 207: 339–344.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17: 225–234.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361: 1058–1066.
Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008; 22: 437–438.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387–397.
Tefferi A, Lasho TL, Huang J, Finke C, Mesa RA, Li CY et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia 2008; 22: 756–761.
Tefferi A, Levine RL, Lim KH, Abdel-Wahab O, Lasho TL, Patel J et al. Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816V and FIP1L1-PDGFRA correlates. Leukemia 2009; 23: 900–904.
Tefferi A, Lasho TL, Patnaik MM, Finke CM, Hussein K, Hogan WJ et al. JAK2 germline genetic variation affects disease susceptibility in primary myelofibrosis regardless of V617F mutational status: nullizygosity for the JAK2 46/1 haplotype is associated with inferior survival. Leukemia 2010; 24: 105–109.
Hussein K, Pardanani AD, Van Dyke DL, Hanson CA, Tefferi A . International Prognostic Scoring System-independent cytogenetic risk categorization in primary myelofibrosis. Blood 2010; 115: 496–499.
Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895–2901.
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A . Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 2008; 116: 597–602.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 765–773.
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 2009; 30: 7–11.
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1010 diffuse gliomas. Acta Neuropathol 2009; 118: 469–474.
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H . IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 2009; 174: 1149–1153.
Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 2009; 27: 5874–5880.
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W et al. Distinct clinical and biological characteristics in adult acute myeloid leukemia bearing isocitrate dehydrogenase 1 (IDH1) mutation. Blood 2010; 115: 2749–2754.
Ho PA, Alonzo TA, Kopecky KJ, Miller KL, Kuhn J, Zeng R et al. Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 2010; 24: 909–913.
Wagner K, Damm F, Gohring G, Gorlich K, Heuser M, Schafer I et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 2010; 28: 2356–2364. JCO.2009.2027.6899.
Marcucci G, Maharry K, Wu Y-Z, Radmacher MD, Mrozek K, Margeson D et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group b study. J Clin Oncol 2010; 28: 2348–2355. JCO.2009.2027.3730.
Abdel-Wahab O, Manshouri T, Patel J, Harris K, Yao J, Hedvat C et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res 2010; 70: 447–452.
Green A, Beer P . Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 2010; 362: 369–370.
Patnaik MM, Lasho TL, Finke CM, Gangat N, Caramazza D, Siragusa S et al. MPL mutation effect on JAK2 46/1 haplotype frequency in JAK2V617F-negative myeloproliferative neoplasms. Leukemia 2010; 24: 859–860.
Pardanani A, Lasho TL, Finke CM, Gangat N, Wolanskyj AP, Hanson CA . et al. The JAK2 46/1 haplotype confers susceptibility to essential thrombocythemia regardless of JAK2V617F mutational status-clinical correlates in a study of 226 consecutive patients. Leukemia 2010; 24: 110–114.
Olcaydu D, Skoda RC, Looser R, Li S, Cazzola M, Pietra D et al. The ‘GGCC’ haplotype of JAK2 confers susceptibility to JAK2 exon 12 mutation-positive polycythemia vera. Leukemia 2009; 23: 1924–1926.
Jones AV, Campbell PJ, Beer PA, Schnittger S, Vannucchi AM, Zoi K et al. The JAK2 46/1 haplotype predisposes to MPL mutated myeloproliferative neoplasms. Blood 2010 (in press).
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360: 2289–2301.
Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009; 23: 905–911.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, Rey J et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009; 23: 2183–2186.
Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009; 113: 6182–6192.
Jager R, Gisslinger H, Berg T, Passamonti F, Cazzola M, Rumi E et al. Deletions of the transcription factor ikaros in myeloproliferative neoplasms at transformation to acute myeloid leukemia. ASH Annual Meeting Abstracts 2009; 114: 435.
Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, Gibbs Jr KD et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 2010 (in press).
Acknowledgements
This study is supported in part by grants from the ‘Myeloproliferative Disorders Foundation, Chicago, IL, USA’, ‘The Henry J. Predolin Foundation for Research in Leukemia, Mayo Clinic, Rochester, MN, USA’ and ‘Associazione Italiana per la Ricerca sul Cancro-AIRC Milan, Italy, to AMV’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tefferi, A., Lasho, T., Abdel-Wahab, O. et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 24, 1302–1309 (2010). https://doi.org/10.1038/leu.2010.113
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2010.113
Keywords
This article is cited by
-
Digital-droplet PCR assays for IDH, DNMT3A and driver mutations to monitor after allogeneic stem cell transplantation minimal residual disease of myelofibrosis
Bone Marrow Transplantation (2022)
-
Thrombocytosis in children and adolescents—classification, diagnostic approach, and clinical management
Annals of Hematology (2021)
-
IDH2 mutations in patients with normal karyotype AML predict favorable responses to daunorubicin, cytarabine and cladribine regimen
Scientific Reports (2021)
-
Philadelphia-Negative Myeloproliferative Neoplasms: Laboratory Workup in the Era of Next-Generation Sequencing
Current Hematologic Malignancy Reports (2019)
-
Blast phase myeloproliferative neoplasm: Mayo-AGIMM study of 410 patients from two separate cohorts
Leukemia (2018)
