
Abstract
Therapeutic cancer vaccines are being developed with the intention of treating existing tumors or preventing tumor recurrence. While the results of clinical trials, predominantly in the metastatic setting have been sobering, the central hypothesis of active immunotherapy i.e. that the human immune system can be activated to recognize and destroy tumor cells, remains a viable one. We believe that a fundamental shift in how clinical trials are performed, and what concepts they test is required to make meaningful strides towards future clinical use of cancer vaccines. First, we must reappraise whether the metastatic setting is the appropriate arena to test these agents. Second, we must arrive at a consensus on the most important biologic endpoints and rapidly test vaccines for their ability to achieve these endpoints. Third, we need to expend more effort on understanding how to manipulate the immune system beyond the initial stimulation provided by a vaccine. Fourth, in order to permit comparison of results across different studies, it would be helpful to narrow down the large number of vaccine platforms. We will discuss the current state of development of cancer vaccines and the relevance for future clinical use of these agents to treat and prevent cancers.
Similar content being viewed by others
Introduction: the gap between theory and reality in the clinical results for cancer vaccines
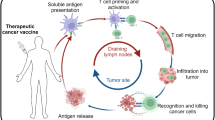
Therapeutic cancer vaccines, or so called active specific immunotherapies, are intended to activate the immune system to treat existing tumors or prevent tumor recurrence. While we (and others1) believe that the central hypothesis of active immunotherapy, i.e. that the human immune system can be activated to recognize and destroy tumor cells, remains viable, the field of active specific immunotherapy is clearly at a crossroads, with pessimism for current vaccines expressed by some leaders in the field,2 and more tempered views expressed by others.3,4 It is clear that there is a gap between the limited clinical activity of cancer vaccines (demonstrated thus far in clinical trials as defined by standard response criteria), and the promising preclinical findings that suggest much greater activity is achievable. Rosenberg2 surveyed 440 patients, mainly diagnosed with metastatic melanoma, who were treated with vaccines used by the National Cancer Institute Surgery Branch, and reported the overall objective response rate for all vaccine treatments was 2.6% (2.9% for peptide vaccines and 1.9% for viral-based vaccines). The majority of responders had disease limited to the skin or lymph nodes. Compiling the results from 35 representative vaccine studies published in the literature, they reported an objective response rate of 3.8%; 4.0% for peptide vaccines, 0% for pox viruses, 4.2% for modified tumor cells and 7.1% for dendritic cells (DC). Recent randomized studies have not demonstrated improved responses or overall survival benefits for patients with metastatic malignancies who were treated with specific vaccines compared with chemotherapy, non-specific vaccines, or best supportive care.5,6,7 Despite these results, periodic reports of more promising clinical data, particularly in selected situations such as low-grade lymphomas, continue to fuel the contention that these vaccines will have clinical practice applications in the future. In this review, we will describe why we remain optimistic that cancer vaccines will ultimately be clinically applicable. In particular, we will focus on how an evolving understanding of the necessary components of an immune response to cancer, and how testing of major hypotheses in clinical trials will continue to move the field forward. We will not present lists of published or ongoing clinical trials, as excellent reviews have recently been published;8,9,10,11 rather we will focus on the areas of development that we believe will translate into clinically relevant vaccines.
Are we testing cancer vaccines in the appropriate patients and clinical scenarios?
Most cancer vaccine studies are carried out in those with advanced disease, where the likelihood of response is low; and it has been suggested that more promising results would be seen in those with less advanced disease, such as in the adjuvant setting. Unfortunately, most studies of adjuvant therapy—extensively reviewed elsewhere11—are of non-randomized trials, and therefore it is difficult to determine their true efficacy. Nonetheless, one is more encouraged about the possibility of clinical efficacy of vaccines considering that some studies have shown benefit compared with historical controls, and at least two other studies12,13 have shown a disease-free survival benefit for a cancer vaccine. In the study by Jocham et al.12 patients who had undergone radical nephrectomies received an autologous renal-tumor-cell vaccine or no adjuvant treatment (control). Five-year progression-free survival was 77% in the vaccine group and 68% in the control. It is intriguing to consider that removal of the primary tumor permits greater activity of a vaccine against micrometastatic disease, in the same way that nephrectomy improves outcome with cytokine therapy for metastatic renal-cell carcinoma. We believe that the adjuvant setting will prove to be the most productive one for testing most cancer vaccines, except for those that require intratumoral injections in order to activate the immune response. This does not invalidate the metastatic setting, and initial testing of new vaccines may need to be performed in this group of patients for regulatory and safety reasons. Also, because true 'adjuvant' studies—such as those with resected locally-advanced cancers—may require substantial numbers of patients (i.e. many hundreds to thousands), we believe it is appropriate to consider the use of vaccines in patients with controlled metastatic disease as 'adjuvant therapy', regardless of how this was achieved.
It is also important to consider that there may be biologically plausible subgroups of patients who benefit from cancer vaccines. In some of the same studies mentioned earlier, which had overall negative results, subgroup analyses did detect groups with clinical benefit. For example, in Small's study7 that assessed the vaccine APC8015 (Provenge), an autologous DC product pulsed with a prostatic acid phosphatase-GM-CSF construct, prostate cancer patients with a Gleason Score of 7 or less had a longer median time to progression. In Mayordomo's report5 of the Theratope vaccine (tumor-associated antigen Sialyl Tn conjugated to the carrier protein keyhole limpet hemocyanin) for patients with metastatic breast cancer, there was a trend for a better time to progression for patients treated concomitantly with hormonal therapy, particularly in patients receiving aromatase inhibitors. In Sondak's study of the allogeneic melanoma vaccine Melacine in patients with resected melanoma,14 there was no overall relapse or survival benefit, but patients who expressed HLA-A2 and/or HLA-C3 had improved relapse-free survival and overall survival15,16 compared with controls with the same HLA types. There are biologically plausible explanations why these subgroups might have a better outcome. For example, the less aggressive prostate cancers might permit more time for the antitumor immune response to develop. It is also possible that less aggressive tumors express tumor antigens against which immune responses may be activated, whereas more aggressive, less differentiated tumors do not, as has been observed for melanomas.17 Estrogens may increase the frequency of regulatory T cells, which counteract immune responses. Perhaps, hormonal therapy inhibits the development of regulatory T cells and allows immune responses to proceed unimpeded. Certain HLA types may present more immunogenic peptide epitopes than others.
Finally, certain tumors, particularly hematologic malignancies, may have an inherently greater ability to respond to the immune system. Timmerman18 reported a response rate of 32% among patients with follicular lymphomas who received DC loaded with Id (IDIOTYPE) protein. Subsequently, Wen-Kai Weng19 reported that, among patients with follicular lymphoma who received Id vaccines, patients with the Fc RIIIa 158 valine/valine (V/V) genotype also had a longer progression-free survival than those with valine/phenylalanine (V/F) or phenylalanine/phenylalanine (F/F) genotypes. These results are not surprising given the fact that Id vaccines induce antibody responses, and response to the administered antibody rituximab also differs, depending on the Fc receptor polymorphism.
The challenge in the future is to use the information gleaned from these results and use laboratory testing to determine how vaccine strategies can be modified to take into account these possible influences on response. Also, it is prudent to perform the next clinical trial in the patient population with the apparent clinical benefit. For example, the Theratope vaccine will be tested in patients on hormonal therapy, Provenge is being tested in men with metastatic prostate cancer with a Gleason Score of 7 or less, and a follow-up study for melanoma patients with HLA-A2 and/or HLA-C3 using Melacine has been written.
Have we identified the right TUMOR ANTIGENS to target?
There is no lack of identified tumor antigens and epitopes.20 There has been extensive debate about whether patient-specific or universal tumor antigens are the best targets, and whether univalent or multivalent vaccines produce the greatest chance of clinical efficacy. A survey of clinical trial data does not settle the debate because there are examples of each type (e.g. univalent: Provenge, Theratope; multivalent: Melacine) that may have clinical activity. While some malignancies may lend themselves to vaccines that use patient-specific antigen targeting such, as the tumor-specific idiotype of non-Hodgkin's lymphoma (e.g. MyVax® idiotype vaccines), most tumors do not express such unique, foreign antigens. It is also not clear whether any peptides are preferentially expressed within the HLA class I molecules of tumors.21 Therefore, virtually any intracellular protein could be a potential target. For those vaccines in which a response to a particular antigen correlates with clinical outcome (e.g. the anti-TA90 IgM response to the allogeneic melanoma vaccine Canvaxin, which is an independent prognostic factor for overall survival and disease-free survival in immunized melanoma patients22), it may make sense to focus on the particular antigen. Otherwise, in our opinion there is no compelling reason to focus on any particular antigen or group of tumor antigens more than any other. In fact, if the theory of epitope or antigenic spreading is correct, an adequate immune response against one tumor-expressed antigen would lead to a series of T cells specific for other, possibly more relevant, target antigens.23 In addition, we must broaden our definition of what is likely to be a potential target antigen within a tumor. Vigneron et al. identified CD8 T lymphocytes that could recognize a nonameric peptide on melanoma cells that was derived from two noncontiguous segments of gp100, created by a proteosomal function.24 Use of standard computer algorithms for predicting epitopes within proteins would not have predicted the presence of such peptides.
The foregoing discussion also begs the question of whether we must target tumor antigens at all. Non-malignant tissues such as the tumor stroma and vasculature are critical for the survival of cancer cells. Also, the growth and survival factors transmitted by these tissues may also be targets for immune attack resulting in tumor death. For example, some gastrointestinal tumors express cholecystokinin-2 receptors that can bind amidated gastrins leading to transcriptional activation of epidermal growth factor receptor (EGFR) ligands, matrix metalloproteinases and anti-apoptotic factors, which increase tumorigenic potential. The vaccine, G17DT (Insegia™), activates neutralizing antibody responses against amidated and glycine-extended gastrin-17. In a recent randomized, double-blind, placebo-controlled trial of patients with advanced pancreatic cancer unsuitable or unwilling to take chemotherapy, median overall survival increased from 82 days to 151 days for patients receiving G17DT compared with placebo.25 Additionally, immune responses activated in response to receptors in the EGFR-regulated signaling pathways may have anticancer activity beyond cytolytic destruction. For example, HLA-A2-restricted epitopes of the EGFR have been identified, suggesting that it could be a target for immune responses.26 Tumor vasculature also represents a potential target for immune responses because it expresses some antigens not found on quiescent endothelium. Strategies have been developed to activate immune responses against these antigens.27
Do we need any more platforms for delivering tumor antigens to the immune system?
Just as we may be reaching the point at which there are diminishing returns for discovering more tumor antigens, so too we may be reaching a similar point for the development of new vaccine platforms. Modified and unmodified tumor cells and their lysates, DC, proteins and peptides, carbohydrates, anti-idiotype antibodies, viral, bacterial, and yeast vectors, and DC targeting agents are all in clinical development. Few have been compared to each other, and when they have been compared, little difference in clinical activity has been apparent. Slingluff et al.28 compared T-cell responses to melanoma peptides with GM-CSF in adjuvant or pulsed onto DC. T-cell responses to melanoma peptides were observed in 42% of peripheral blood lymphocytes and 80% of sentinel immunized nodes for patients receiving the peptides plus GM-CSF, but in patients vaccinated with DCs, they were observed in only 11% and 13%, respectively. Objective clinical responses were observed in two patients in the GM-CSF arm and one patient in the DC arm. We are performing a clinical trial comparing DC modified using a viral vector with the viral vector strategy alone to determine whether ex vivo generated DCs are necessary components of our immunization platform. While we cannot ignore the fact that differences in vaccine composition or administration may affect outcome, most vaccines in advanced development induce detectable tumor antigen-specific immune responses in a substantial fraction of patients. In a randomized clinical trial comparing different sequences of vaccinia virus and fowlpox virus expressing human prostate-specific antigen (PSA) as prime/boost vaccines, there was a trend showing a better clinical progression-free survival for those who received a priming dose of recombinant vaccinia virus expressing PSA.29 As we will discuss later, we believe that significant progress for cancer vaccines will come as we better understand how to manipulate the host regulatory mechanisms that mute the magnitude and durability of T-cell responses following immunization. Therefore, it is important to rapidly narrow down the vaccine platforms so that consistency across investigations may be achieved. Animal models and small exploratory studies could still be used to test new strategies that may have substantial promise. Then rapid assessment of the best dose and schedule are required. In one study, HER-2/neu intracellular domain protein vaccine activated HER-2/neu-specific T-cell and antibody immunity in breast and ovarian cancer patients.30 While the dose of vaccine did not affect the magnitude of T-cell or antibody immune response, higher doses were associated with more rapid activation of HER-2/neu-specific immunity.
How can we manipulate the hosts interaction with the vaccine?
It is well established that regulatory T cells, identified by their co-expression of CD4 and CD25 and the transcriptional regulator foxp3, are capable of modulating the immune response activated against specific antigens. Animal model studies have suggested that elimination of these T cells results in enhanced immune responses. Recently, it was demonstrated that potent tumor antigen-specific immune responses could be activated by a DC-based vaccine, when denileukin diftitox was administered prior to the vaccine to deplete the CD4+ and CD25+ T cells.31 Another regulatory effect is through cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), which transmits signals that inhibit proliferation of T cells when engaged by CD80 on antigen-presenting cells. Recent observations have suggested that blockade of CTLA-4 enhances antitumor T-cell responses, particularly during booster immunizations.32 Clinical trials combining anti-CTLA-4 antibody (MDX-010) and cancer vaccines have been initiated including a phase I clinical trial of the GVAX® prostate cancer vaccine, administered in combination with MDX-010, in patients with advanced prostate cancer. In addition to regulatory T cells, a number of alterations in the cancer patient or tumor milieu appear to alter immune responsiveness. For example, expression of cyclooxygenase-2 (COX-2) leads to increased synthesis of prostaglandin E2, an immune suppressor, and is associated with decreased T-cell stimulation.33 In vivo studies have demonstrated that COX-2 inhibition gave an additive effect to a cancer vaccine.34 Phase I studies at the National Cancer Institute are combining pox vector vaccines with COX-2 inhibition.
How will immunotherapy be integrated into multi-modality cancer treatment strategies?
Because chemotherapy remains a mainstay of cancer therapy, the ability to combine it with vaccines is of great interest. Doxorubicin and paclitaxel that were administered prior to HER-2 vaccines in mice, resulted in augmented anti-tumor activity.35 A number of groups are now performing phase I and II trials combining vaccines with systemic chemotherapy. Furthermore, combining more than one form of immunotherapy such as antibodies and vaccines might allow more complete tumor eradication. For example, in a neu-transgenic mouse model, a vaccine given in combination with neu-specific antibodies prevented tumor development in 70% of mice and eradicated established tumors in 30% of mice. The efficacy of the antibodies, alone or combined with the vaccine, was dependent on both CD4+ and CD8+ T cells.36
Are we measuring the correct endpoints?
The success of cancer vaccines is thought to lie in their ability to activate antigen-specific T cells; therefore, it has been argued that immunologic endpoints, particularly the frequency of T cells specific for the antigen of interest, would be critical to the further development of the vaccine strategy. Indeed, most published cancer vaccine studies make some attempt to measure an immunologic endpoint, whether it is an in vivo endpoint, such as delayed type hypersensitivity reaction to a tumor antigen or an in vitro endpoint, such as the ELISPOT ASSAY, which measures antigen-specific T cells by their cytokine secretion in response to antigen exposure. Frequently studies will report that immunologic response correlates with clinical outcome. For example, a recent study with the heat-shock protein preparation Oncophage™ (HSPPC-96), for patients who had undergone resection of hepatic metastases of colon cancer, reported that those with immune responses against colon-cancer cell lines experienced a better disease-free survival.37 Although such results are encouraging, in the absence of a controlled, randomized study, they do not prove that the peripheral blood immune response is the direct cause of the clinical outcome. Furthermore, there is not complete certainty that it is the antigen-specific T-cell response that is most critical. Some strategies, despite their antigen-specificity, may in fact activate natural killer cells. The best source of lymphocytes from which to measure the immune response is debated. Slingluff et al.38 recently observed that the T-cell responses against melanoma peptides were more frequently detected in the sentinel immunized lymph nodes, and with higher magnitude, than responses in the peripheral blood. Perhaps greater efforts should be expended on collecting tumor tissue from which T cells could be isolated. In our opinion, peripheral blood lymphocytes remain an important source of cells for immunologic analysis, and this analysis is important for future development of cancer vaccines. In the absence of clinical responses, there is no other way to determine if the vaccine had any biologic activity.
Conclusions
We have discussed the key issues that will need to be addressed in order to activate potent antigen-specific T cell and antibody responses. Once these potent effectors are reliably activated, the challenge of ensuring trafficking of these effectors to the site of the tumor and then recognition and destruction of the tumor, will remain. A major concern has been that tumors might be unresponsive despite activation of effective T-cell responses because of downregulation of tumor antigen or major histocompatibility complex (MHC) molecules, or by secreting or expressing inhibitory molecules. Interferon alpha can be used to upregulate MHC class I and other molecules. Tumor gene expression can be altered epigenetically.39 Finally, if we are successful in all these areas, will we ultimately see autoimmune disease? Animal models demonstrate that a potent immune response can occur in the absence of autoimmunity, although this lack of autoimmunity in preclinical studies does not guarantee that this effect will translate to humans.40
Review criteria
The data for this review were obtained using the MEDLINE database, which was searched for publications between 1 January 2001 and 15 November 2004. The search terms used were “cancer vaccine”, “immunotherapy”, “dendritic cell”, “GVAX”, “CANVAXIN”, “PANVAC” and “HSPCC”. In addition, the websites of manufacturers of cancer vaccines in late-stage clinical development and the NCI clinical trials website (http://clinicaltrials.gov/) were searched for the most recent information regarding the status of clinical trials with these vaccines.
References
Rosenberg SA et al. (2004) Reply to “Cancer vaccines: pessimism in check”. Nat Med 10: 1279–1280
Rosenberg SA et al. (2004) Cancer immunotherapy: moving beyond current vaccines. Nat Med 10: 909–915
Mocellin S et al. (2004) Correspondence 1: Cancer vaccines: pessimism in check. Nat Med 10: 1278–1279
Timmerman JM and Levy R (2004) Correspondence 2: Cancer vaccines: pessimism in check. Nat Med 10: 1279
Mayordomo J et al. (2004) Long-term follow-up of patients concomitantly treated with hormone therapy in a prospective controlled randomized multicenter clinical study comparing STn-KLH vaccine with KLH control in stage IV breast cancer following first-line chemotherapy. 2004 ASCO Annual Meeting Proceedings (Post-Meeting Edition). J Clin Oncol 22 (Suppl): [abstract 2603]
Schadendorf D et al. (2004) Dacarbacine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) as first-line treatment of patients with metastatic melanoma: Results of a prospective-randomized phase III study. ASCO Annual Meeting Proceedings (Post-Meeting Edition). J Clin Oncol 22 (Suppl): [abstract 7508]
Small EJ et al. (2003) A randomized, placebo-controlled phase III trial of APC8015 in patients with androgen-independent prostate cancer (AiPCa). Proc Am Soc Clin Oncol 22: 382 [abstract 1534]
Ridgway D (2003) The first 1000 dendritic cell vaccines. Cancer Invest 21: 876–886
Ribas A et al. (2003) Current developments in cancer vaccines and cellular immunotherapy. J Clin Oncol 21: 2415–2432
Mocellin S et al. (2004) Part II: Vaccines for haematological malignant disorders. Lancet Oncol 5: 727–737
Mocellin S et al. (2004) Part I: Vaccines for solid tumours. Lancet Oncol 5: 681–689
Jocham D et al. (2004) Adjuvant autologous renal tumour cell vaccine and risk of tumour progression in patients with renal-cell carcinoma after radical nephrectomy: phase III, randomised controlled trial. Lancet 363: 594–599
Bystryn JC et al. (2001) Double-blind trial of a polyvalent, shed-antigen, melanoma vaccine. Clin Cancer Res 7: 1882–1887
Sondak VK et al. (2002) Adjuvant immunotherapy of resected, intermediate-thickness node-negative melanoma with an allogeneic tumor vaccine: overall results of a randomized trial of the Southwest Oncology Group. J Clin Oncol 20: 2058–2066
Sosman JA et al. (2002) Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: impact of HLA class I antigen expression on outcome. J Clin Oncol 20: 2067–2075
Sosman JA et al. (2002) HLA-A2 and/or HLA-C3 expression defines a subset of T3N0 melanoma patients with improved overall survival from melacine vaccine: An updated analysis of SWOG 9035. Proc Am Soc Clin Oncol 20: 340 [abstract 1359]
Bittner M et al. (2000) Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 406: 536–540
Timmerman JM et al. (2002) Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: clinical and immune responses in 35 patients. Blood 99: 1517–1526
Weng W et al. (2004) Clinical outcome of lymphoma patients after idiotype vaccination is correlated with humoral immune response and immunoglobulin G Fc receptor genotype. J Clin Oncol 22: 4665–4672
Novellino L et al. (2004) A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother [10.1007/s00262-004-0560-6]
Hickman HD et al. (2004) Toward a definition of self: proteomic evaluation of the class I peptide repertoire. J Immunol 172: 2944–2952
DiFronzo LA et al. (2002) Enhanced humoral immune response correlates with improved disease-free and overall survival in American Joint Committee on Cancer stage II melanoma patients receiving adjuvant polyvalent vaccine. J Clin Oncol 20: 3242–3248
Disis ML et al. (2004) Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. J Clin Immunol 24: 571–578
Vigneron N et al. (2004) An antigenic peptide produced by peptide splicing in the proteasome. Science 304: 587–590
Gilliam AD et al. (2004) Randomised, double blind, placebo-controlled, multi-centre, group-sequential trial of G17DT for patients with advanced pancreatic cancer unsuitable or unwilling to take chemotherapy. ASCO Annual Meeting Proceedings (Post-Meeting Edition). J Clin Oncol 22 (Suppl): [abstract 2511]
Shomura H et al. (2004) Identification of epidermal growth factor receptor-derived peptides immunogenic for HLA-A2(+) cancer patients. Br J Cancer 90: 1563–1571
Niethammer AG et al. (2002) A DNA vaccine against VEGF receptor 2 prevents effective angiogenesis and inhibits tumor growth. Nat Med 8: 1369–1375
Slingluff CL Jr et al. (2003) Clinical and immunologic results of a randomized phase II trial of vaccination using four melanoma peptides either administered in granulocyte-macrophage colony-stimulating factor in adjuvant or pulsed on dendritic cells. J Clin Oncol 21: 4016–4026
Kaufman HL et al. (2004) Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): a trial of the Eastern Cooperative Oncology Group. J Clin Oncol 22: 2122–2132
Disis ML et al. (2004) Effect of dose on immune response in patients vaccinated with an her-2/neu intracellular domain protein-based vaccine. J Clin Oncol 22: 1916–1925
Vieweg J et al. (2004) Enhancement of antitumor immunity following depletion of CD4+CD25+ regulatory T cells. 2004 ASCO Annual Meeting Proceedings (Post-Meeting Edition). J Clin Oncol 22 (Suppl): [abstract 2506]
Gregor PD et al. (2004) CTLA-4 blockade in combination with xenogeneic DNA vaccines enhances T-cell responses, tumor immunity and autoimmunity to self antigens in animal and cellular model systems. Vaccine 22: 1700–1708
Pockaj BA et al. (2004) Reduced T-cell and dendritic cell function is related to cyclooxygenase-2 overexpression and prostaglandin E2 secretion in patients with breast cancer. Ann Surg Oncol 11: 328–339
Zeytin HE et al. (2004) Combination of a poxvirus-based vaccine with a cyclooxygenase-2 inhibitor (celecoxib) elicits antitumor immunity and long-term survival in CEA.Tg/MIN mice. Cancer Res 64: 3668–3678
Eralp Y et al. (2004) Doxorubicin and paclitaxel enhance the antitumor efficacy of vaccines directed against HER 2/neu in a murine mammary carcinoma model. Breast Cancer Res 6: R275–R283
Wolpoe ME et al. (2003) HER-2/neu-specific monoclonal antibodies collaborate with HER-2/neu-targeted granulocyte macrophage colony-stimulating factor secreting whole cell vaccination to augment CD8+ T cell effector function and tumor-free survival in Her-2/neu-transgenic mice. J Immunol 171: 2161–2169
Mazzaferro V et al. (2003) Vaccination with autologous tumor-derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clin Cancer Res 9: 3235–3245
Slingluff CL Jr et al. (2004) Immunologic and clinical outcomes of vaccination with a multiepitope melanoma peptide vaccine plus low-dose interleukin-2 administered either concurrently or on a delayed schedule. J Clin Oncol 22: 4474–4485
Khan AN et al. (2004) An epigenetically altered tumor cell vaccine. Cancer Immunol Immunother 53: 748–754
Hodge JW et al. (2003) Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res 9: 1837–1849
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- GM-CSF
-
Granulocyte-macrophage colony-stimulating factor
- Id (IDIOTYPE)
-
A specific protein antigen made by B lyphocyte cells, which distinguishes a clone of immunoglobulin-producing cells from other clones
- ELISPOT ASSAY
-
Enzyme-linked immunospot assay is a highly sensitive tool for analyzing immunological secretions of peripheral blood cell populations
Rights and permissions
About this article
Cite this article
Morse, M., Chui, S., Hobeika, A. et al. Recent developments in therapeutic cancer vaccines. Nat Rev Clin Oncol 2, 108–113 (2005). https://doi.org/10.1038/ncponc0098
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/ncponc0098
This article is cited by
-
CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients
Scientific Reports (2016)
-
Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer
Breast Cancer Research and Treatment (2014)
-
In vivo imaging of therapy-induced anti-cancer immune responses in humans
Cellular and Molecular Life Sciences (2013)
-
Invasion and destruction of a murine fibrosarcoma by Salmonella-induced effector CD8 T cells as a therapeutic intervention against cancer
Cancer Immunology, Immunotherapy (2011)
-
Synergistic antitumor effects of CpG oligodeoxynucleotide and STAT3 inhibitory agent JSI‐124 in a mouse melanoma tumor model
Immunology & Cell Biology (2008)
