
Abstract
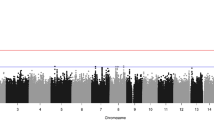
Sagittal craniosynostosis is the most common form of craniosynostosis, affecting approximately one in 5,000 newborns. We conducted, to our knowledge, the first genome-wide association study for nonsyndromic sagittal craniosynostosis (sNSC) using 130 non-Hispanic case-parent trios of European ancestry (NHW). We found robust associations in a 120-kb region downstream of BMP2 flanked by rs1884302 (P = 1.13 × 10−14, odds ratio (OR) = 4.58) and rs6140226 (P = 3.40 × 10−11, OR = 0.24) and within a 167-kb region of BBS9 between rs10262453 (P = 1.61 × 10−10, OR = 0.19) and rs17724206 (P = 1.50 × 10−8, OR = 0.22). We replicated the associations to both loci (rs1884302, P = 4.39 × 10−31 and rs10262453, P = 3.50 × 10−14) in an independent NHW population of 172 unrelated probands with sNSC and 548 controls. Both BMP2 and BBS9 are genes with roles in skeletal development that warrant functional studies to further understand the etiology of sNSC.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


Accession codes
Accessions
GenBank/EMBL/DDBJ
References
Cohen, M.M. Craniosynostosis: Diagnosis, Evaluation, and Management (Oxford University Press, New York, 2000).
Johnson, D. et al. A novel mutation, Ala315Ser, in FGFR2: a gene-environment interaction leading to craniosynostosis? Eur. J. Hum. Genet. 8, 571–577 (2000).
Seto, M.L. et al. Isolated sagittal and coronal craniosynostosis associated with TWIST box mutations. Am. J. Med. Genet. A 143, 678–686 (2007).
Weber, I. et al. Molecular analysis of 74 patients with craniosynostosis. Eur. J. Hum. Genet. 9 (suppl. 1), P0409, 179 (2001).
Merrill, A.E. et al. Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum. Mol. Genet. 15, 1319–1328 (2006).
Wilkie, A.O. et al. Clinical dividends from the molecular genetic diagnosis of craniosynostosis. Am. J. Med. Genet. A 143A, 1941–1949 (2007).
Mefford, H.C. et al. Copy number variation analysis in single-suture craniosynostosis: multiple rare variants including RUNX2 duplication in two cousins with metopic craniosynostosis. Am. J. Med. Genet. A 152A, 2203–2210 (2010).
Vissers, L.E. et al. Heterozygous mutations of FREM1 are associated with an increased risk of isolated metopic craniosynostosis in humans and mice. PLoS Genet. 7, e1002278 (2011).
Kim, S.-D. et al. Leucine-rich repeat, immunoglobulin-like and transmembrane domain 3 (LRIT3) is a modulator of FGFR1. FEBS Lett. 586, 1516–1521 (2012).
Melville, H. et al. Genetic basis of potential therapeutic strategies for craniosynostosis. Am. J. Med. Genet. A 152A, 3007–3015 (2010).
Wilkie, A.O. et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics 126, e391–e400 (2010).
Passos-Bueno, M.R. et al. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front. Oral Biol. 12, 107–143 (2008).
Kimonis, V. et al. Genetics of craniosynostosis. Semin. Pediatr. Neurol. 14, 150–161 (2007).
Kolar, J.C. An epidemiological study of nonsyndromal craniosynostoses. J. Craniofac. Surg. 22, 47–49 (2011).
Lajeunie, E. et al. Genetic considerations in nonsyndromic midline craniosynostoses: a study of twins and their families. J. Neurosurg. 103 (suppl.), 353–356 (2005).
Lajeunie, E. et al. Syndromal and nonsyndromal primary trigonocephaly: analysis of a series of 237 patients. Am. J. Med. Genet. 75, 211–215 (1998).
Lajeunie, E. et al. Genetic study of scaphocephaly. Am. J. Med. Genet. 62, 282–285 (1996).
Sanchez-Lara, P.A. et al. Fetal constraint as a potential risk factor for craniosynostosis. Am. J. Med. Genet. A 152A, 394–400 (2010).
Zeiger, J.S. et al. Genetic and environmental risk factors for sagittal craniosynostosis. J. Craniofac. Surg. 13, 602–606 (2002).
Gardner, J.S. et al. Maternal exposure to prescription and non-prescription pharmaceuticals or drugs of abuse and risk of craniosynostosis. Int. J. Epidemiol. 27, 64–67 (1998).
Källén, K. Maternal smoking and craniosynostosis. Teratology 60, 146–150 (1999).
Patterson, N., Price, A.L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Dudbridge, F. Pedigree disequilibrium tests for multilocus haplotypes. Genet. Epidemiol. 25, 115–121 (2003).
Cordell, H.J. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 11, 2463–2468 (2002).
Browning, B.L. & Browning, S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 84, 210–223 (2009).
Rosen, V. BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev. 20, 475–480 (2009).
Jabs, E.W. et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell 75, 443–450 (1993).
Spruijt, L. et al. A novel mutation in the MSX2 gene in a family with foramina parietalia permagna (FPP). Am. J. Med. Genet. A 139, 45–47 (2005).
Wilkie, A.O. et al. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat. Genet. 24, 387–390 (2000).
Howard, T.D. et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 15, 36–41 (1997).
Stankiewicz, P. et al. Phenotypic findings due to trisomy 7p15.3-pter including the TWIST locus. Am. J. Med. Genet. 103, 56–62 (2001).
Mundlos, S. et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89, 773–779 (1997).
Desai, J. et al. Nell1-deficient mice have reduced expression of extracellular matrix proteins causing cranial and vertebral defects. Hum. Mol. Genet. 15, 1329–1341 (2006).
Zhang, X. et al. The role of NELL-1, a growth factor associated with craniosynostosis, in promoting bone regeneration. J. Dent. Res. 89, 865–878 (2010).
Yagnik, G. et al. ALX4 gain-of-function mutations in nonsyndromic craniosynostosis. Hum. Mutat. published online, doi:10.1002/humu.22166 (24 July 2012).
Styrkarsdottir, U. et al. Linkage of osteoporosis to chromosome 20p12 and association to BMP2. PLoS Biol. 1, E69 (2003).
Dathe, K. et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 84, 483–492 (2009).
Tobin, J.L. & Beales, P.L. Bardet-Biedl syndrome: beyond the cilium. Pediatr. Nephrol. 22, 926–936 (2007).
Marshall, W.F. & Nonaka, S. Cilia: tuning in to the cell's antenna. Curr. Biol. 16, R604–R614 (2006).
Schneider, L. et al. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 15, 1861–1866 (2005).
Haycraft, C.J. et al. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53 (2005).
Ma, R. et al. PKD2 functions as an epidermal growth factor–activated plasma membrane channel. Mol. Cell. Biol. 25, 8285–8298 (2005).
Brailov, I. et al. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res. 872, 271–275 (2000).
Jenkins, D. et al. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 80, 1162–1170 (2007).
Ehlen, H.W., Buelens, L.A. & Vortkamp, A. Hedgehog signaling in skeletal development. Birth Defects Res. C Embryo Today 78, 267–279 (2006).
Miettinen, P.J. et al. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat. Genet. 22, 69–73 (1999).
Ding, H. et al. A specific requirement for PDGF-C in palate formation and PDGFR-α signaling. Nat. Genet. 36, 1111–1116 (2004).
Choi, D.S. et al. 5–HT2B receptor-mediated serotonin morphogenetic functions in mouse cranial neural crest and myocardiac cells. Development 124, 1745–1755 (1997).
Neugebauer, J.M. et al. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature 458, 651–654 (2009).
Keen, T.J. et al. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur. J. Hum. Genet. 10, 245–249 (2002).
Marinaki, A.M. et al. Genetic basis of hemolytic anemia caused by pyrimidine 5′ nucleotidase deficiency. Blood 97, 3327–3332 (2001).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Mills, J.L. et al. Folate and vitamin B12–related genes and risk for omphalocele. Hum. Genet. 131, 739–746 (2012).
Barrett, J.C. et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Schwender, H. et al. Rapid testing of SNPs and gene-environment interactions in case-parent trio data based on exact analytic parameter estimation. Biometrics 68, 766–773 (2012).
Koeleman, B.P. et al. Adaptation of the extended transmission/disequilibrium test to distinguish disease associations of multiple loci: the conditional extended transmission/disequilibrium test. Ann. Hum. Genet. 64, 207–213 (2000).
Kazeem, G.R. & Farrall, M. Integrating case-control and TDT studies. Ann. Hum. Genet. 69, 329–335 (2005).
Acknowledgements
The authors thank all families who contributed to this study. S.A.B. is partially funded through a Children's Miracle Network Endowed Chair and through grants K23 DE00462, R03 DE016342 and R01 DE016886 from the National Institute of Dental and Craniofacial Research (NIDCR)/NIH and M01-RR00052 from the National Center for Research Resources/NIH and was fully supported by Zlatka, Anton and Alec Boyadjiev. Partial funding was also obtained from grants to E.W.J. (US Centers for Disease Control and Prevention (CDC) 5 R01 DD000350), M.L.C. (R01 DE018227), A.O.M.W. (Wellcome Trust 093329), P.A.R. (CDC 5U01DD000492), J.K. (NIDCR/NIH R21DE022419), J.L.M. (Intramural Research Program (IRP) of the NIH, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD); IRP HHSN267200703431C; NICHD N01-DK-7-3431), P.A.S.-L. (Robert Wood Johnson Foundation 3R37DE012711-13S1 and Children's Hospital Los Angeles–University of Southern California Child Health Research Career Development Program; NIH K12-HD05954), J.T.R. (NIDCR/NIH and the American Recovery and Reinvestment Act R01 DE018500 and 3R01 DE018500-02S1) and I.P. (National Center for Advancing Translational Sciences, NIH UL1TR000067). This project was also supported in part by the Division of Intramural Research Program of the National Human Genome Research Institute, NIH (C.M.J., Y.K. and A.F.W.). Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the NIH to Johns Hopkins University, contract number HHSN268200782096C. We thank B. Wilson, N. Issac, C. Nauta, E. Goude, E. Cherkez, L. Peters and J. Harrison for patient recruitment, C. Boehm and A. Scott for coordination of discovery-phase genotyping, C. Stevens, A. Stoner, J.L. Liu, A. Gearhart, A. Atkins and E. McGrath for bench work, D. Mortlock for bioinformatic analysis and informative discussion and J. Graham (Cedars-Sinai Hospital, Los Angeles, California, USA), J. Bernstein (Stanford University, Palo Alto, California, USA), J. Marsh (Washington University, St. Louis, Missouri, USA), J. Panchal (University of Oklahoma Health Science Center, Oklahoma, USA), T. Tollefson (University of California Davis, Sacramento, California, USA) and M. Passos-Bueno (University of São Paolo, Brazil) for contributing clinical information and biospecimens for this project.
Author information
Authors and Affiliations
Contributions
C.M.J. and G.Y. are the first coauthors of the manuscript. C.M.J., Y.K. and I.P. performed statistical analyses. G.Y., M.E., X.Y., E.A. and L.S. performed experiments. C.M.J., G.Y., Y.K., I.P., M.L.C., V.K., T.R., A.O.M.W., J.S., J.T.R., Y.H., P.A.S.-L., M.F.B., J.K., A.F.W. and S.A.B. analyzed data. C.M.J. and G.Y. wrote the manuscript, with contributions from Y.K., I.P., J.T.R., Y.H., P.A.R., A.F.W. and S.A.B. E.W.J., M.L.C., V.K., S.A.W., J.S., P.A.S.-L., M.F.B., C.M.D., J.L.M., M.C., P.A.R., D.M.K., C.S., P.J.T., O.D.K., J.B., M.Z.-L. and C.N. contributed materials and reagents. E.W.J., A.O.M.W., M.F.B., D.M.K. and P.J.T. contributed to editing of the manuscript. E.W.J., C.M.D., J.L.M., M.C., P.A.R., D.M.K., C.S., P.J.T., O.D.K., J.B., M.Z.-L. and C.N. helped with experimental design. J.K. and A.F.W. supervised the research. S.A.B. was the principal investigator, designed and supervised research and recruited and evaluated participants.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–4 and Supplementary Tables 1–6 (PDF 2165 kb)
Rights and permissions
About this article
Cite this article
Justice, C., Yagnik, G., Kim, Y. et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat Genet 44, 1360–1364 (2012). https://doi.org/10.1038/ng.2463
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2463
This article is cited by
-
SMAD6 variants in nonsyndromic craniosynostosis
European Journal of Human Genetics (2023)
-
The current understanding of germline predisposition in non-syndromic sagittal craniosynostosis: a systematic review
Child's Nervous System (2023)
-
Retroperitoneal liposarcoma and craniosynostosis: possible genomic relationship, case report, and literature review
Functional & Integrative Genomics (2023)
-
SMAD6-deficiency in human genetic disorders
npj Genomic Medicine (2022)
-
An Axin2 mutation and perinatal risk factors contribute to sagittal craniosynostosis: evidence from a Chinese female monochorionic diamniotic twin family
Hereditas (2021)
