
Abstract
Despite rapid advances in the identification of genes involved in disease, the predictive power of the genotype remains limited, in part owing to poorly understood effects of second-site modifiers. Here we demonstrate that a polymorphic coding variant of RPGRIP1L (retinitis pigmentosa GTPase regulator-interacting protein-1 like), a ciliary gene mutated in Meckel-Gruber (MKS) and Joubert (JBTS) syndromes, is associated with the development of retinal degeneration in individuals with ciliopathies caused by mutations in other genes. As part of our resequencing efforts of the ciliary proteome, we identified several putative loss-of-function RPGRIP1L mutations, including one common variant, A229T. Multiple genetic lines of evidence showed this allele to be associated with photoreceptor loss in ciliopathies. Moreover, we show that RPGRIP1L interacts biochemically with RPGR, loss of which causes retinal degeneration, and that the Thr229-encoded protein significantly compromises this interaction. Our data represent an example of modification of a discrete phenotype of syndromic disease and highlight the importance of a multifaceted approach for the discovery of modifier alleles of intermediate frequency and effect.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
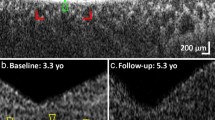

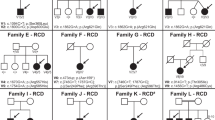
Accession codes
References
Badano, J.L., Mitsuma, N., Beales, P.L. & Katsanis, N. The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 7, 125–148 (2006).
Bergmann, C. et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am. J. Hum. Genet. 82, 959–970 (2008).
Olbrich, H. et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat. Genet. 34, 455–459 (2003).
Chang, B. et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 15, 1847–1857 (2006).
den Hollander, A.I. et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 79, 556–561 (2006).
Baala, L. et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 81, 170–179 (2007).
Leitch, C.C. et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443–448 (2008).
Sayer, J.A. et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 38, 674–681 (2006).
Valente, E.M. et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 38, 623–625 (2006).
Badano, J.L. et al. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 439, 326–330 (2006).
Tory, K. et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J. Am. Soc. Nephrol. 18, 1566–1575 (2007).
Gherman, A., Davis, E.E. & Katsanis, N. The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 38, 961–962 (2006).
Liu, Q. et al. The proteome of the mouse photoreceptor sensory cilium complex. Mol. Cell. Proteomics 6, 1299–1317 (2007).
Arts, H.H. et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 39, 882–888 (2007).
Delous, M. et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 39, 875–881 (2007).
Gerdes, J.M. et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350–1360 (2007).
Ross, A.J. et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 37, 1135–1140 (2005).
Corbit, K.C. et al. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 10, 70–76 (2008).
Beales, P.L. et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727–729 (2007).
Otto, E.A. et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 37, 282–288 (2005).
Shu, X. et al. RPGR mutation analysis and disease: an update. Hum. Mutat. 28, 322–328 (2007).
Breuer, D.K. et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 70, 1545–1554 (2002).
Hong, D.H. et al. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest. Ophthalmol. Vis. Sci. 44, 2413–2421 (2003).
Khanna, H. et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J. Biol. Chem. 280, 33580–33587 (2005).
Oprea, G.E. et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320, 524–527 (2008).
Giess, R. et al. Early onset of severe familial amyotrophic lateral sclerosis with a SOD-1 mutation: potential impact of CNTF as a candidate modifier gene. Am. J. Hum. Genet. 70, 1277–1286 (2002).
Badano, J.L. & Katsanis, N. Beyond Mendel: an evolving view of human genetic disease transmission. Nat. Rev. Genet. 3, 779–789 (2002).
Dryja, T.P. et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am. J. Hum. Genet. 68, 1295–1298 (2001).
Cheng, H. et al. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum. Mol. Genet. 13, 1563–1575 (2004).
He, S. et al. Retinitis pigmentosa GTPase regulator (RPGR) protein isoforms in mammalian retina: insights into X-linked retinitis pigmentosa and associated ciliopathies. Vision Res. 48, 366–376 (2008).
Acknowledgements
We thank the individuals affected with ciliopathies and their families for their continued support and encouragement. We also thank H. Arts for critical evaluation of the manuscript. This work was supported by grants R01EY007961 from the National Eye Institute (H.K. and A.S.), R01HD04260 from the National Institute of Child Health and Development (N.K.), R01DK072301, R01DK075972 (N.K.), R01DK068306, R01DK064614, R01DK069274 (F.H.), NRSA fellowship F32 DK079541 (E.E.D.) from the National Institute of Diabetes, Digestive and Kidney disorders, Intramural program of NEI (A.S.), the Macular Vision Research Foundation (N.K.), the Foundation for Fighting Blindness (H.K., S.S.B., A.S. and N.K.), the Foundation for Fighting Blindness Canada (R.K.K.), Le Fonds de la recherche en sante du Québec (FRSQ) (R.K.K.), Research to Prevent Blindness (A.S.), Harold Falls Collegiate Professorship (A.S.), the Midwest Eye Banks and Transplantation Center (H.K.), the Searle Scholars Program (M.A.B.), the Deutsche Forschungsgemeinschaft (DFG grant BE 3910/4-1; C.B.) the UK Medical Research Council (grant number G0700073; C.A.J.), NIHR Biomedical Research Centre for Ophthalmology (S.S.B.) and EU-GENORET Grant LSHG-CT-2005-512036 (S.S.B.). F.H. is an investigator of the Howard Hughes Medical Institute (HHMI) and a Doris Duke Distinguished Clinical Scientist (DDCF).
Author information
Authors and Affiliations
Contributions
H.K., E.E.D., A.S. and N.K. designed experiments and analyzed data; H.K., E.E.D., C.A.M.-Z., A.E.-C., I.L., A.I.d.H., M.N.Z., M.I.O., N.W., C.F.C., C.M., A.D.-F., C.V.L. and P.L.T. performed experiments; M.A.B. performed permutation analysis; C.F.I., R.A.L., S.G.J., C.B., P.L.B., T.A.-B., C.A.J., E.A.O., S.S.B., F.H. and R.K.K. provided subject DNA, genotypes, or clinical information; D.M.M., D.A.W., M.M., L.R.L. and R.A.G. generated and analyzed medical resequencing data; H.K., E.E.D., A.S. and N.K. wrote the manuscript.
Corresponding authors
Supplementary information
Supplementary Text and Figures
Supplementary Tables 1–3 and Supplementary Figures 1–5 (PDF 1817 kb)
Rights and permissions
About this article
Cite this article
Khanna, H., Davis, E., Murga-Zamalloa, C. et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet 41, 739–745 (2009). https://doi.org/10.1038/ng.366
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.366
This article is cited by
-
Proteomic profiling of retina and retinal pigment epithelium combined embryonic tissue to facilitate ocular disease gene discovery
Human Genetics (2023)
-
Genetic characteristics of 234 Italian patients with macular and cone/cone-rod dystrophy
Scientific Reports (2022)
-
Allelic overload and its clinical modifier effect in Bardet-Biedl syndrome
npj Genomic Medicine (2022)
-
A hypomorphic variant in EYS detected by genome-wide association study contributes toward retinitis pigmentosa
Communications Biology (2021)
-
The F220C and F45L rhodopsin mutations identified in retinitis pigmentosa patients do not cause pathology in mice
Scientific Reports (2020)
