
Abstract
Phagocytosis has essential functions in immunity. Here we highlight the presence of a subcellular level of self–non-self discrimination in dendritic cells that operates at the level of individual phagosomes. We discuss how engagement of Toll-like receptor signaling controls distinct programs of phagosome maturation. An inducible mode of phagosome maturation triggered by these receptors ensures the selection of microbial antigens for presentation by major histocompatibility class II molecules during the simultaneous phagocytosis of self and non-self.
Similar content being viewed by others


Main
Uptake of external materials is an essential process for all eukaryotic cells. Cells internalize nutrients and growth factors for their intrinsic needs, and specialized cell types also remove unwanted or excessive materials from the circulation or tissue fluids, thus contributing to tissue homeostasis. Four principal endocytic pathways mediate the internalization of macromolecules and particles: clathrin-mediated endocytosis (CME), macropinocytosis, phagocytosis and caveolae-mediated endocytosis1,2. Which endocytic pathway is used depends both on the receptors involved and on particle size. CME proceeds through clathrin-coated pits and is involved in a variety of essential cellular functions. Phagocytosis, in contrast, is generally restricted to several motile cells of hematopoietic origin known as 'professional phagocytes', including macrophages, neutrophils and dendritic cells (DCs). Unlike CME, both phagocytosis and macropinocytosis are actin-dependent processes controlled by the Rho-family GTPases Rho, Rac and Cdc42 (ref. 3). Given the importance of endocytic activities in cellular and organismal physiology, it is not surprising that multiple regulatory mechanisms have evolved to control various stages of endocytic pathways4,5. Here, we focus our discussion on the regulation of phagocytosis and phagosome maturation and its implications for antigen presentation.
Phagocytosis in 'housekeeping' and host defense
In complex metazoans, macrophages and to some extent other phagocytes have two essential functions that rely on phagocytosis: a 'housekeeping' or scavenging function and a function in host defense against infection6. Although there are dozens of different types of tissue-resident macrophages7, as far as is known at present the scavenging and host defense functions are not performed by specialized cell lineages: the same macrophage, in other words, can carry out both functions.
The scavenging function is essential for tissue homeostasis of complex metazoans and involves removal of apoptotic cells, components of the extracellular matrix and other by-products of metazoan physiology. Apoptotic cell recognition is mediated through the detection of a number of molecules that are normally absent on live and healthy cells8. These molecules include surface expression of calreticulin and exposure of phosphatidylserine on the outer leaflet of the plasma membrane, as well as the expression of amino sugars and lysophospholipids, which are recognized by various types of collectins, scavenger receptors, integrins and bridging molecules that link these structures to macrophage receptors. In contrast, viable cells express specialized signals such as CD47 that engage inhibitory receptors, such as SIRPα, that inhibit phagocytosis8,9.
Although phagocytosis of microbial cells and of apoptotic cells seem to rely on the same phagocytic machinery, the cellular responses that accompany the two forms of phagocytosis are markedly different. Phagocytosis of apoptotic cells is accompanied by triggering of anti-inflammatory responses, such as production of transforming growth factor (TGF)-β10. Apoptotic cells can also specifically inhibit the expression of several cytokines, including interleukin (IL)-12 (ref. 11). The receptors and signaling pathways involved in the anti-inflammatory effects of apoptotic cells are poorly characterized, but they seem to involve the Tyro-3-family receptor tyrosine kinases12. Phagocytosis of microbial cells, in contrast, is designed to eliminate pathogens and trigger the inflammatory response, including the production of tumor necrosis factor (TNF), IL-1 and IL-6, to alert the immune system to the presence of infection13. Pathogen killing is mediated by a variety of inducible antimicrobial defenses, including phagocyte oxidase14 and nitric oxide synthase15. The difference in outcome for apoptotic cell versus microbial cell phagocytosis is determined primarily through the engagement of Toll-like receptors (TLRs) during inflammatory phagocytosis of microbial pathogens and not during noninflammatory phagocytosis of apoptotic cells. TLRs recognize conserved macromolecules of microbial origin and can distinguish apoptotic cells from bacterial cargo on the basis of their specificities16. However, the uptake of microbial cells is not mediated by TLRs but rather by a number of phagocytic receptors, including receptors of the C-type lectin family and scavenger receptors6. Notably, these receptors generally do not induce inflammatory responses on their own, but they have been reported to cooperate with TLRs in the induction of some of these responses17,18.
Particle recognition and uptake is one of the regulated steps in phagocytosis, as evidenced by the evolution of a number of opsonins—secreted proteins that tag microbial and apoptotic cells for internalization mediated by receptors specific for the opsonins. The opsonization strategy is put to particularly good use in the case of antibody- and Fc receptor (FcR)-mediated phagocytosis19. Another regulatory mechanism at the level of particle uptake is through the transcriptional induction of the receptors involved in phagocytosis. TLRs, for example, can trigger expression of a number of phagocytic receptors in macrophages20.
Phagosome maturation: basic cell biology and signaling
Phagocytosis relies on a network of endocytic vesicles to deliver cargo from nascent phagosomes to lysosomes for degradation. Phagosomes form de novo at the plasma membrane and maintain an asymmetric lipid bilayer with associated proteins and lipids21. Once inside cells, the composition of these membranes gains a unique character determined primarily through the types of receptors engaged during phagocytosis. The nature of the ligands present on internalized cargo dictates the types of receptors engaged. For example, a particle opsonized with immunoglobulin will engage FcR, whereas a particle opsonized with complement will engage complement receptors. Similarly, mannose residues and phosphatidylserine will engage distinct receptors. Particle internalization involves the focal delivery of endomembranes containing vesicle-associated membrane protein (VAMP3, or cellubrevin) to newly forming phagosomes through ADP-ribosylation factor-6 (ARF6), 'zippering' of membranes along the internalized particle, actin polymerization after the activation of Rho GTPases and particle engulfment associated with phosphatidylinositol-3-kinase activity13,22. With time, phagosomes acquire new components through sequential fusions with endosomes, a process described as 'phagosome maturation'21. Maturing phagosomes ultimately fuse with lysosomes for terminal degradation of the cargo and killing of internalized microorganisms. Notably, phagosome maturation is accompanied by a progressive decrease in phagosomal pH, which drops from around pH 5.5 in nascent phagosomes to around pH 4.5 in lysosomes23. This process occurs within most cell types, including non–'professional antigen-presenting cells' such as fibroblasts, but not including immature DCs, which seem to actively maintain a more alkaline pH within their phagosomes (as discussed below)24. Hydrolytic enzymes resident in late endosomes and lysosomes have low pH optima, which ensures that their activities are confined to a particular stage in the endocytic pathway25. Low pH in late endosomes and lysosomes is generated by the vacuolar ATPase (V-ATPase), which consists of a peripheral V1 domain and an integral V0 domain26. Phagosome acidification seems to be tailored to the functions of the particular cell type in which the phagosome forms24. Whereas the outcome of phagosome maturation in macrophages is the killing of internalized microorganisms and complete degradation and clearance of phagocytic cargo, maturation of phagosomes in DCs serves to prevent complete degradation of cargo antigens such that major histocompatibility complex (MHC) molecules can present them. Accordingly, a progressive decrease in phagosomal pH occurs over time in macrophages, whereas no significant acidification seems to occur in phagosomes maturing in DCs24.
Although phagosome maturation presumably always proceeds through similar steps, aside from the reported cell type–specific differences in phagosomal pH, phagosomes are not created equal, and there is a significant degree of phagosome heterogeneity and individuality that is dictated primarily by the cargo contained in phagosomes, as well as by external signals and the activation and/or differentiation state of phagocytes27. Phagosome complexity is further underscored by recent proteomic analyses, which have revealed hundreds of distinct proteins associated with phagosomes28. Only a fraction of these have known functions related to phagosome biology, indicating that many of the basic principles and the majority of the details of phagosome maturation are presently unknown.
Do TLRs control phagosome maturation?
At the subcellular level, receptor-ligand interactions during phagocytosis have distinct consequences to the phagosome itself. Upon phagocytosis of microbial pathogens, surface TLRs, including TLR4 and TLR2, are recruited to the phagosome and become activated by microbial cell wall components29,30. Phagosome maturation is then regulated by signals from TLRs through the adaptor protein MyD88 and the mitogen-associated protein kinase (MAPK) p38 (ref. 31; Fig. 1). When TLR signaling is engaged by ligands present in the cargo, an inducible mode of phagosome maturation occurs that has distinct kinetics and functional consequences. Microbial cells, including Escherichia coli, Staphylococcus aureus and Salmonella typhimurium, engage TLRs during internalization and are delivered to lysosomes at an inducible rate manifested by increased clearance and phagolysosomal fusion. This inducible rate was evident in two situations: first, when phagocytosis of bacteria was compared in the presence or absence of TLR signaling (the concomitant absence of both TLR2 and TLR4 or MyD88) and, second, when phagocytosis of bacteria was compared to that of apoptotic cells that do not engage TLRs. In both cases, it was evident that there were two modes of phagocytosis, constitutive and inducible, the latter being triggered by the TLR-MyD88-p38 signaling pathway. In addition, stimulation of DCs with LPS triggers activation of actin cytoskeletal rearrangements and increases the rate of uptake at early time points32. Stimulation of DCs with TLR ligands also 'shuts off' actin-dependent endocytic activity at later time points33,34. The mechanism of the late shutoff is mediated by the inactivation of Cdc42 (ref. 35).

The molecular targets of regulation are most likely the Rab family of small GTPases. Rabs serve as master regulators of endocytic traffic, and constitute the rate-limiting step in vesicular fusion. The pool of p38 MAPK species specifically phosphorylated by signals from TLRs can increase the rate of endocytic traffic by phosphorylating the guanyl-nucleotide dissociation inhibitor GDI, which in phosphorylated form acquires an increased affinity for exhausted GDP-bound Rabs, and recycles them through the cytosolic phase of their activation back to donor vesicles for reloading with GTP. This serves as a mechanism for increasing the rate of endocytic traffic.
As TLRs are not the only regulators of DC and macrophage function, one would anticipate that other receptors for microbial cargo would engage a similar pathway of inducible phagosome maturation. Indeed, macrophages derived from mice deficient in Syk tyrosine kinase or in all three members of the Src-family kinases, Hck, Fgr and Lyn, which are activated 'downstream' of FcR signaling, show slower phagocytosis of IgG-coated sheep erythrocytes36,37. However, one recent study examining the phagolysosomal fusion of beads coated with either mannose or IgG found no differences in the rate of phagolysosomal fusion of phagosomes containing mannose- or IgG-coated beads in the presence or absence of ligands that stimulate TLRs38. One interpretation of such results is that TLRs do not regulate phagosome maturation38.
A different interpretation, however, is that particle opsonization often leads to a maximal rate of phagocytic processing that cannot be increased by addition of TLR ligands. To illustrate this point, imagine that the constitutive mode of phagosome maturation proceeds at a rate 10 (in some arbitrary units), whereas an inducible mode proceeds at the rate 100. Let us also assume that 100 is a maximal possible rate of maturation given that it can be triggered by receptors such as FcR and macrophage mannose receptor, which presumably evolved to optimize the phagocytic process. Now imagine two distinct receptors that control the inducible mode of phagosome maturation, receptors A and B, and imagine that either receptor can trigger the maximal rate of maturation when its cognate ligands are present on the cargo. If, however, cognate ligands for both A and B are present on the same cargo, the rate of phagosome maturation would still be the maximal rate, as each individual receptor triggers the maximal possible rate. Given such a scenario, one could not conclude that neither receptor A nor receptor B was involved in phagosome maturation. Definitive conclusions about the potential for inducing phagocytosis of a given receptor can only be made, in other words, by comparing phagocytosis of particles (for example, latex beads) that either engage no receptors or engage only a particular cognate receptor of interest.
Returning now to the situation under discussion, in which no difference in the kinetics of phagolysosomal fusion was seen for mannose- or IgG-coated beads when TLR ligands were also present on these beads: because both mannose and IgG engage phagocytic receptors—macrophage mannose receptor (MMR) and FcR, respectively—the kinetics of phagolysosomal fusion observed for mannose- or IgG-coated beads would be maximal and therefore the addition of a TLR ligand (ligand of receptor B) to the mannose- or IgG-opsonized particles (ligands of receptor A) would not have an additive effect. A more informative way to evaluate the inducible properties of TLR ligands, therefore, would be to compare particles that are not opsonized in the presence or absence of TLR ligands.
Whether 'defects' in phagosome maturation in Myd88−/− macrophages truly exist has been cast into question on the basis of possible developmental defects in Myd88−/− macrophages38. In one study, 578 genes were indeed differentially expressed between wild-type and Myd88−/− macrophages in the absence of deliberate stimulation39. Notably, the downregulated genes in Myd88−/− macrophges all encoded TLR-MyD88–dependent inflammatory cytokines and chemokines, and no intrinsic differences in expression were seen in genes encoding Rab, SNARE, ARF or other trafficking proteins, for example. Thus, Myd88−/− cells express lower basal amounts of several TLR-regulated genes, and it is likely that the differences observed reflect the basal level of TLR activation in tissue culture (by traces of endotoxin present in the medium, for example). In our opinion, this is not an indication of a developmental defect.
In short, we believe that there are no discrepancies in the reports indicating TLR involvement, or lack thereof, in phagocytosis. The differences lie instead in experimental design and interpretation of results. Indeed, recent follow-up studies have shown that TLR-mediated control of phagosome maturation strongly influences the efficiency of antigen presentation (as discussed below)40.
Phagosome-autonomous maturation
Recent data have shown that TLR control of the kinetics and function of phagosome maturation is phagosome autonomous31,40 and most likely occurs because of the compartmentalized assembly of TLR signaling complexes specifically on phagosome membranes containing cargo that carry TLR ligands. Thus, the maturation fate of individual phagosomes is dictated by the cargo they contain, and not by cargo in other phagosomes within the same cell.
When a single cell internalizes distinct particles—for example, bacteria and apoptotic cells—at the same time, each cargo is dealt with separately within the confines of distinct phagosomes. Disparate ligands present on these different cargos naturally engage unique corresponding receptors during internalization. The types of receptor-ligand interactions and signaling pathways that ensue provide the first opportunity for generating functionally distinct phagosomes within the same DC27. As the nascent phagosome matures, this heterogeneity can be further amplified by differential and signal-dependent recruitment of various protein and lipid mediators that directly affect the molecular profile, fusion properties and functions of that phagosome. We speculate that this heterogeneity during maturation in DCs may serve to isolate the phagosome even further, precluding its membrane components from nonspecifically interacting with other phagosomal membranes. Although phagosomal membrane components have been found to recycle efficiently among phagosomes carrying similar contents, whether this occurs among phagosomes bearing different cargos has not been examined41,42. In addition, inter-phagosomal exchange as well as exchange with the plasma membrane in DCs may be restricted so that it occurs only after the particular phagosome has already relayed its information and exhausted its purpose. Obviously, all these possibilities need to be tested experimentally.
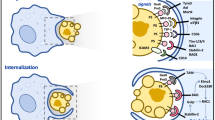
The detailed molecular mechanisms responsible for phagosome-autonomous maturation pathways are currently unknown. In the case of cargo that engages TLRs, the MyD88-mediated activation of p38 MAP kinase seems to have a crucial role in marking phagosomes for a distinct mode of maturation (Fig. 2); the p38 MAP kinase activity, moreover, has to be confined to the phagosomal membrane, which can be achieved by one or more phagosome-associated scaffolding proteins43. Phosphorylation of a (currently unknown) phagosome-associated target by p38 then marks the individual phagosome for a distinct maturation pathway in a phagosome-autonomous manner (Fig. 2). This type of 'marking' could in turn dictate the assembly or activity of endosomal fusion machinery. It is tempting to speculate that the assembly of the V-ATPase may be regulated by the p38 MAP kinase in a phagosome-autonomous manner. Rab GTPases are another possible target for control of phagosomal regulation. Indeed, stress-induced activation of the MAPK p38 results in p38-dependent phosphorylation of the guanyl-nucleotide dissociation inhibitor (GDI)44 (Fig. 1). Phosphorylated GDI acquires increased affinity for the GDP-bound form of Rab5 present on target endocytic membranes and then shuttles GDP-Rab5 through the cytosol 'back' to donor membranes where nucleotide exchange takes place. This type of mechanism allows prompt recycling of Rab5, which overcomes what is considered to be a rate-limiting component in endocytic transport. A distinct signaling complex may thus assemble around the TLR-activated p38 scaffold, the components of which may differ when TLRs are engaged as compared to when they are not engaged. Such TLR-initiated complex formation would be likely to endow a phagosome with a TLR-based 'molecular signature' that could dictate the immediate fate of the cargo and immune responses tailored to that cargo.

Upon simultaneous phagocytosis of microbial and apoptotic cells, each cargo is internalized into individual phagosomes having distinct biochemical profiles. Engagement of TLRs selectively along phagosomal membranes carrying pathogens results in MyD88-dependent phosphorylation of the MAPK p38, which marks those phagosomes for an inducible mode of maturation. p38 kinase activity can specifically be confined to TLR signaling phagosomes by one or more phagosome-associated scaffolding proteins. Phosphorylation of phagosome-associated targets by p38 may then regulate the assembly or activity of endosomal fusion machinery. These events dictate the fate of cargo-derived peptides and their presentation by MHC class II, which subsequently results in activation of microbial antigen-specific CD4+ T cells.
Another potential mechanism whereby TLRs could trigger inducible phagosome maturation is by increasing the rate at which phagosomes move along microtubules. The mobility of endocytic organelles, including phagosomes, along microtubules occurs in both directions guided by motor protein complexes, kinesin-kinectin and dynein-dynactin45,46. The predominant direction of movement of phagosomes is toward the minus-end of microtubules and depends on the dynein-dynactin complex. The presence of dynactin on phagosome membranes recruits the cytosolic dynein, and this seems to require a post-translational event such as phosphorylation by a dynactin-associated kinase47,48. Similarly, microtubule-associated proteins (MAPs) such as CLIP-115 (ref. 49) and CLIP-170 (ref. 50) may increase the rate of phagosome motility by dislodging from microtubules either upon their phosphorylation or by 'intrinsic timing'51. Regulation of motor protein activity by signal-dependent phosphorylation, therefore, may be another mechanism for regulating the kinetics of phagosome maturation.
Signaling proteins have indeed recently been shown to be associated with motor complexes. Adenoviruses efficiently traverse the cytoplasm along microtubules en route to the nucleus by stimulating both the cAMP-dependent protein kinase A (PKA) and the MAPK p38, which thereby increases minus-end-directed motility mediated by the dynein-dynactin motor complex52. In another example, salmonella alter phagosome mobility to create an ideal intracellular niche for bacterial replication. This effect is accomplished through modulating the activity of motor proteins by the salmonella pathogenicity island 2 (SPI-2)-encoded type III secretion system effector protein SifA, which forms a complex with Rab 7 and uncouples it from Rab7-interacting lysosomal protein (RILP)–dynein. This process additionally targets the host protein SKIP, which functions to downregulate kinesin recruitment53.
Phagosome-autonomous control of antigen presentation
An important immunological consequence of phagosome maturation is the processing and presentation of peptides derived from internalized cargo. In DCs, the antigen presentation process is highly regulated at several levels and can be triggered by TLR ligands54. When DCs encounter microbial cells in peripheral tissues, engagement of TLRs induces DC activation, pathogen uptake, killing, migration to draining lymph nodes, processing and presentation of pathogen-derived antigens, and expression of a variety of cytokines and co-stimulatory molecules55,56. Together, these effects result in a highly immunogenic state for the DC, which facilitates the activation of naive T cells. DCs can also phagocytose apoptotic cells, which does not activate them in the absence of TLR engagement, but rather results in a tolerogenic state that often leads to deletion of autoreactive T cells57,58.
One potential problem arises, however, when DCs simultaneously encounter and phagocytose both microbial and apoptotic cells. On the one hand, microbial cells normally induce DC activation; on the other hand, self-antigens derived from simultaneously engulfed apoptotic cells would, in the case of activated DCs, be processed and presented59, which could potentially lead to stimulation of naive T cells by self-antigens. Our recent findings suggest, however, that DCs have an additional level of self–non-self discrimination that operates at the level of phagosome maturation40. Even though all cargos are delivered into the same late endosomal and lysosomal compartments where antigen processing and presentation take place, only cargo containing TLR ligands significantly 'remodels' these compartments for efficient antigen processing and MHC class II presentation. The hydrolytic environment in maturing phagosomes is important not only for processing of the cargo, but also for processing of the MHC class II–associated invariant chain (Ii) through a series of proteolytic steps such that an N-terminal fragment called the 'class II–associated invariant chain derived peptide' (CLIP) occupies the peptide-binding groove of particular MHC II molecules60,61. CLIP is then exchanged for antigenic peptides derived from cargo proteins through a reaction catalyzed by the nonclassical MHC molecules H2-DM and DO. TLR signals trigger the processing of Ii within individual phagosomes only when phagocytic cargo stimulates TLR signaling. Phagosomes that do not engage TLR signaling fail to process Ii40.
On the basis of these findings, we propose a model where upon simultaneous phagocytosis of microbial and apoptotic cells, phagosomes that contain microbial cargo and engage TLRs are 'favored' for MHC class II presentation (Fig. 3). Such phagosomes mature with enhanced kinetics of fusion with the endocytic pathway and activate hydrolytic enzymes that process Ii, thus making MHC class II molecules amenable to binding of antigenic peptides. Apoptotic cells, in contrast, do not stimulate TLR signaling and thus phagosomes with apoptotic cell cargo mature into terminal lysosomes, where they become degraded. In those phagosomes, specific proteases that cleave Ii are not activated and MHC class II molecules bound to unprocessed Ii are targeted to lysosomes for degradation.

Microbial-derived antigens are selected for presentation by MHC class II. TLR signals allow these phagosomes to mature with enhanced kinetics, and activate hydrolytic enzymes that process Ii, making MHC class II molecules amenable to binding of antigenic peptides. Apoptotic cells, in contrast, do not engage TLR signaling, and their phagosomes mature into terminal lysosomes where apoptotic cell proteins are completely degraded. See text for discussion.
Phagosome autonomous control by TLRs thus ensures that processing of Ii occurs only within phagosomes containing microbial cells where ligands on the cargo engage compartmentalized TLR signaling pathways. The resulting peptide-MHC complexes that form specifically within such phagosomes are then transported to the plasma membrane and presented to T cells in the context of TLR-induced co-stimulation. Peptides derived from apoptotic cell antigens are excluded from these MHC class II complexes and are instead targeted for degradation.
How can signaling from TLRs regulate antigen processing and MHC class II presentation? One possibility is that this occurs through selective recruitment of Ii-processing cathepsins to endocytic compartments marked by TLR signals. However, we found no evidence for selective recruitment of cathepsins to TLR-regulated phagosomes40. Another possibility is that cathepsin (and perhaps other protease) activity is somehow regulated in a phagosome-autonomous manner. Cathepsins are regulated both by acidic pH, which is required for their catalytic activity, and by negative regulators such as cystatins25. Phagosome autonomous control of phagosome acidification is a particularly attractive possibility, because, as mentioned above, the assembly of V-ATPase is regulated in DCs by maturation signals, including TLR signals23. It is thus conceivable that engagement of TLRs in individual phagosomes may trigger V-ATPase assembly specifically on that phagosome, resulting in acidification and compartment-specific proteolytic cleavage of Ii. It also seems that phagosomes in immature DCs actively maintain an alkaline pH through NADPH oxidase NOX2 recruitment and only succumb to the default pathway of acidification upon DC maturation62. We speculate that, at minimum, TLR signals would have to counteract the actions of NOX2 by fortifying V-ATPase assembly on individual phagosomes23. An equally likely possibility is that in DCs, TLR signals temporarily increase the recruitment of the cytosolic subunits of NOX2, such as p47phox, to TLR-marked DC phagosomes to induce a respiratory burst that kills microbial pathogens within those phagosomes. One study has indeed reported that NOX2 activity is increased in DCs upon TLR stimulation63. Ultimately, TLRs may fine-tune phagosomal pH in DCs by coordinately and temporally regulating the recruitment of NOX2 and V-ATPase subunits through a process more complex than originally expected.
Such considerations do not imply that TLR signaling is required for the MHC class II presentation of all exogenous antigens internalized through various endocytic pathways. Our discussion above with regard to self-antigens applies only to exogenous self-antigens internalized by phagocytosis—that is, antigens that are derived from phagocytosed apoptotic cells. We suggest that TLR-mediated recognition controls selection of phagocytosed antigens for presentation such that microbial antigens are favored over self-antigens—somewhat in the way that, in tennis tournaments, the mediocre tennis players rarely reach the finals not because it is impossible in principle, but because they are far more likely to get knocked out of the competition by the top players. It should also be mentioned that endogenous self-antigens, that is, antigens synthesized in DCs, are presented constitutively and do not seem to require extra regulation by TLRs (except for the induction of surface MHC class II expression). Notably, the immune system is tolerant of DC-expressed endogenous self-antigens resulting from thymic deletion of T cells specific to those antigens. Moreover, presentation of endogenous self-antigens is required for activation of T cells specific for foreign antigens64,65. In addition, the requirements for regulating antigen presentation may be different for different DC subsets and under different conditions in vivo. It has been reported that a subset of lymphoid DCs characterized as B220−PDCA1−CD8+ and their B220−CD8− precursors, but not granulocyte-macrophage colony-stimulating factor (GM-CSF)-derived DCs, produce type I interferon through a MyD88- and TRIF-independent pathway and effectively prime CD4+ and CD8+ T cells specific to apoptotic cell derived antigen24. Perhaps myeloid DCs (represented by the GM-CSF DCs) as well as macrophages are specialized in the execution of immunologically silent clearance of apoptotic cells.
Concluding remarks
Regulation of phagosome maturation by TLRs and other immune receptors has several interesting implications and raises a number of questions for future studies. One obvious implication related to antigen immunogenicity and vaccine development is that the antigen and TLR ligands have to be associated within the same particle or macromolecule to be 'interpreted' by the immune system (and by DCs in particular) as 'microbial'. This may explain why immunodominant antigens often either are TLR ligands themselves or are physically associated with TLR ligands. The best demonstration of this is the immunodominant nature of the TLR11 ligand profilin from Toxoplasma gondii66. The immunodominance of profilin was shown to be TLR11 dependent, thus proving the link between immunogenicity and TLR stimulation66. In addition, the TLR5 ligand flagellin, and a variety of lipidated outer membrane proteins that activate TLR2, are all known to be potent immunodominant antigens, presumably for the reasons discussed above. An implication for vaccine design is that targeting an antigen of interest and a TLR ligand to the same phagosome would enhance immunogenicity of the antigen. The simplest way to achieve this is by physically associating them through conjugation, fusion or absorption on the same particle. Indeed, recent studies have shown that conjugation of TLR ligands and antigens resulted in a superior activation of T cell responses67.
One interesting problem not accounted for by the phagosome autonomous maturation model is the handling of infected apoptotic cells. Infected cells commonly undergo apoptosis, and their phagocytosis could result in DC activation by microbial stimuli and presentation of self-antigens by immunogenic DCs. It is unknown whether this occurs in vivo and, if so, to what extent. It is possible that infected apoptotic cells are primarily removed by macrophages that do not induce naive T cell activation. Whether this is true is currently unknown, indicating that there are additional levels of self–non-self discrimination that operate at the level of phagocytosis and antigen presentation.
References
Conner, S.D. & Schmid, S.L. Regulated portals of entry into the cell. Nature 422, 37–44 (2003).
Mellman, I. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 12, 575–625 (1996).
Hall, A. & Nobes, C.D. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Phil. Trans. R. Soc. Lond. B 355, 965–970 (2000).
Gruenberg, J. The endocytic pathway: a mosaic of domains. Nat. Rev. Mol. Cell Biol. 2, 721–730 (2001).
Pelkmans, L. et al. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436, 78–86 (2005).
Taylor, P.R. et al. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 23, 901–944 (2005).
Gordon, S. & Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964 (2005).
Gardai, S.J., Bratton, D.L., Ogden, C.A. & Henson, P.M. Recognition ligands on apoptotic cells: a perspective. J. Leukoc. Biol. 79, 896–903 (2006).
Oldenborg, P.A. et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000).
Henson, P.M. & Hume, D.A. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 27, 244–250 (2006).
Kim, S., Elkon, K.B. & Ma, X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity 21, 643–653 (2004).
Scott, R.S. et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 (2001).
Aderem, A. & Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17, 593–623 (1999).
Cross, A.R. & Segal, A.W. The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochim. Biophys. Acta 1657, 1–22 (2004).
MacMicking, J., Xie, Q.W. & Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350 (1997).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Brown, G.D. et al. Dectin-1 mediates the biological effects of beta-glucans. J. Exp. Med. 197, 1119–1124 (2003).
Gantner, B.N., Simmons, R.M., Canavera, S.J., Akira, S. & Underhill, D.M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 197, 1107–1117 (2003).
Swanson, J.A. & Hoppe, A.D. The coordination of signaling during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 76, 1093–1103 (2004).
Doyle, S.E. et al. Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199, 81–90 (2004).
Vieira, O.V., Botelho, R.J. & Grinstein, S. Phagosome maturation: aging gracefully. Biochem. J. 366, 689–704 (2002).
Greenberg, S. & Grinstein, S. Phagocytosis and innate immunity. Curr. Opin. Immunol. 14, 136–145 (2002).
Trombetta, E.S., Ebersold, M., Garrett, W., Pypaert, M. & Mellman, I. Activation of lysosomal function during dendritic cell maturation. Science 299, 1400–1403 (2003).
Janssen, E. et al. Efficient T cell activation via a Toll-Interleukin 1 Receptor-independent pathway. Immunity 24, 787–799 (2006).
Trombetta, E.S. & Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 23, 975–1028 (2005).
Nishi, T. & Forgac, M. The vacuolar (H+)-ATPases—nature's most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 3, 94–103 (2002).
Griffiths, G. On phagosome individuality and membrane signalling networks. Trends Cell Biol. 14, 343–351 (2004).
Garin, J. et al. The phagosome proteome: insight into phagosome functions. J. Cell Biol. 152, 165–180 (2001).
Ozinsky, A. et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. USA 97, 13766–13771 (2000).
Underhill, D.M. et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401, 811–815 (1999).
Blander, J.M. & Medzhitov, R. Regulation of phagosome maturation by signals from toll-like receptors. Science 304, 1014–1018 (2004).
West, M.A. et al. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science 305, 1153–1157 (2004).
Sallusto, F., Cella, M., Danieli, C. & Lanzavecchia, A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J. Exp. Med. 182, 389–400 (1995).
Inaba, K., Inaba, M., Naito, M. & Steinman, R.M. Dendritic cell progenitors phagocytose particulates, including bacillus Calmette-Guerin organisms, and sensitize mice to mycobacterial antigens in vivo. J. Exp. Med. 178, 479–488 (1993).
Garrett, W.S. et al. Developmental control of endocytosis in dendritic cells by Cdc42. Cell 102, 325–334 (2000).
Crowley, M.T. et al. A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 186, 1027–1039 (1997).
Fitzer-Attas, C.J. et al. Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J. Exp. Med. 191, 669–682 (2000).
Yates, R.M. & Russell, D.G. Phagosome maturation proceeds independently of stimulation of toll-like receptors 2 and 4. Immunity 23, 409–417 (2005).
Shi, S. et al. MyD88 primes macrophages for full-scale activation by interferon-gamma yet mediates few responses to Mycobacterium tuberculosis. J. Exp. Med. 198, 987–997 (2003).
Blander, J.M. & Medzhitov, R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440, 808–812 (2006).
Steinman, R.M., Mellman, I.S., Muller, W.A. & Cohn, Z.A. Endocytosis and the recycling of plasma membrane. J. Cell Biol. 96, 1–27 (1983).
Berthiaume, E.P., Medina, C. & Swanson, J.A. Molecular size-fractionation during endocytosis in macrophages. J. Cell Biol. 129, 989–998 (1995).
Morrison, D.K. & Davis, R.J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19, 91–118 (2003).
Cavalli, V. et al. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol. Cell 7, 421–432 (2001).
Blocker, A., Griffiths, G., Olivo, J.C., Hyman, A.A. & Severin, F.F. A role for microtubule dynamics in phagosome movement. J. Cell Sci. 111, 303–312 (1998).
Karcher, R.L., Deacon, S.W. & Gelfand, V.I. Motor-cargo interactions: the key to transport specificity. Trends Cell Biol. 12, 21–27 (2002).
Blocker, A. et al. Molecular requirements for bi-directional movement of phagosomes along microtubules. J. Cell Biol. 137, 113–129 (1997).
Hamm-Alvarez, S.F., Kim, P.Y. & Sheetz, M.P. Regulation of vesicle transport in CV-1 cells and extracts. J. Cell Sci. 106, 955–966 (1993).
De Zeeuw, C.I. et al. CLIP-115, a novel brain-specific cytoplasmic linker protein, mediates the localization of dendritic lamellar bodies. Neuron 19, 1187–1199 (1997).
Pierre, P., Scheel, J., Rickard, J.E. & Kreis, T.E. CLIP-170 links endocytic vesicles to microtubules. Cell 70, 887–900 (1992).
Rickard, J.E. & Kreis, T.E. CLIPs for organelle-microtubule interactions. Trends Cell Biol. 6, 178–183 (1996).
Suomalainen, M., Nakano, M.Y., Boucke, K., Keller, S. & Greber, U.F. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 20, 1310–1319 (2001).
Henry, T., Gorvel, J.P. & Meresse, S. Molecular motors hijacking by intracellular pathogens. Cell. Microbiol. 8, 23–32 (2006).
Chow, A.Y. & Mellman, I. Old lysosomes, new tricks: MHC II dynamics in DCs. Trends Immunol. 26, 72–78 (2005).
Banchereau, J. & Steinman, R.M. Dendritic cells and the control of immunity. Nature 392, 245–252 (1998).
Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 (2004).
Heath, W.R. & Carbone, F.R. Cross-presentation, dendritic cells, tolerance and immunity. Annu. Rev. Immunol. 19, 47–64 (2001).
Steinman, R.M., Hawiger, D. & Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 21, 685–711 (2003).
Steinman, R.M. & Nussenzweig, M.C. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA 99, 351–358 (2002).
Cresswell, P. Invariant chain structure and MHC class II function. Cell 84, 505–507 (1996).
Watts, C. Antigen processing in the endocytic compartment. Curr. Opin. Immunol. 13, 26–31 (2001).
Savina, A. et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126, 205–218 (2006).
Vulcano, M. et al. Toll receptor-mediated regulation of NADPH oxidase in human dendritic cells. J. Immunol. 173, 5749–5756 (2004).
Krogsgaard, M. et al. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature 434, 238–243 (2005).
Stefanova, I., Dorfman, J.R. & Germain, R.N. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature 420, 429–434 (2002).
Yarovinsky, F., Kanzler, H., Hieny, S., Coffman, R.L. & Sher, A. Toll-like receptor recognition regulates immunodominance in an anti-microbial CD4+ T cell response. Immunity (in the press) (2006).
Wille-Reece, U. et al. Toll-like receptor agonists influence the magnitude and quality of memory T cell responses after prime-boost immunization in nonhuman primates. J. Exp. Med. 203, 1249–1258 (2006).
Acknowledgements
We thank J. Kagan for critical reading of the manuscript and A. Sher for sharing unpublished results. Supported by Arthritis Foundation Investigator Award (to J.M.B.), US National Institutes of Health grant AI-46688 and the Howard Hughes Medical Institute (to R.M.). We regret that many relevant papers could not be cited owing to space limitations.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Blander, J., Medzhitov, R. On regulation of phagosome maturation and antigen presentation. Nat Immunol 7, 1029–1035 (2006). https://doi.org/10.1038/ni1006-1029
Issue Date:
DOI: https://doi.org/10.1038/ni1006-1029
This article is cited by
-
Systematic review of type 1 diabetes biomarkers reveals regulation in circulating proteins related to complement, lipid metabolism, and immune response
Clinical Proteomics (2023)
-
Targeting SLC7A11 improves efferocytosis by dendritic cells and wound healing in diabetes
Nature (2022)
-
Expanding cross-presenting dendritic cells enhances oncolytic virotherapy and is critical for long-term anti-tumor immunity
Nature Communications (2022)
-
Macrophage mediated recognition and clearance of Borrelia burgdorferi elicits MyD88-dependent and -independent phagosomal signals that contribute to phagocytosis and inflammation
BMC Immunology (2021)
-
Rab20 is critical for bacterial lipoprotein tolerization-enhanced bactericidal activity in macrophages during bacterial infection
Science China Life Sciences (2020)
