
Abstract
The rapid onset of massive, systemic viral replication during primary HIV or simian immunodeficiency virus (SIV) infection and the immune evasion capabilities of these viruses pose fundamental problems for vaccines that depend upon initial viral replication to stimulate effector T cell expansion and differentiation1,2,3,4,5. We hypothesized that vaccines designed to maintain differentiated effector memory T cell (TEM cell) responses5,6 at viral entry sites might improve efficacy by impairing viral replication at its earliest stage2, and we have therefore developed SIV protein-encoding vectors based on rhesus cytomegalovirus (RhCMV), the prototypical inducer of life-long TEM cell responses7,8,9. RhCMV vectors expressing SIV Gag, Rev-Tat-Nef and Env persistently infected rhesus macaques, regardless of preexisting RhCMV immunity, and primed and maintained robust, SIV-specific CD4+ and CD8+ TEM cell responses (characterized by coordinate tumor necrosis factor, interferon-γ and macrophage inflammatory protein-1β expression, cytotoxic degranulation and accumulation at extralymphoid sites) in the absence of neutralizing antibodies. Compared to control rhesus macaques, these vaccinated rhesus macaques showed increased resistance to acquisition of progressive SIVmac239 infection upon repeated limiting-dose intrarectal challenge, including four macaques who controlled rectal mucosal infection without progressive systemic dissemination. These data suggest a new paradigm for AIDS vaccine development—vaccines capable of generating and maintaining HIV-specific TEM cells might decrease the incidence of HIV acquisition after sexual exposure.
Similar content being viewed by others


Main
According to current concepts of T cell–directed HIV vaccines3,4,10, a broadly targeted, high-frequency, HIV-specific CD8+ memory T cell response would restrict acute-phase HIV replication and decrease the subsequent chronic-phase HIV replication set point. CD8+ T cell vaccines are not expected to completely prevent or eliminate infection. Rather, by lowering viral load set points, these vaccines would ameliorate the AIDS epidemic by substantially reducing both the rate of disease progression in infected individuals and the likelihood that such individuals would transmit the infection to others. Although experiments in monkey models have supported these principles11,12,13,14, the recent STEP phase 2b clinical trial of adenovirus-5 (Ad5) vectors, widely viewed as a test of the CD8+ T cell vaccine concept, was a clear failure, causing many to question the ability of T cell responses to contribute to an effective HIV/AIDS vaccine3,10.
In assessing the lack of efficacy seen in the STEP trial, it is instructive to consider whether the characteristics of the T cell responses elicited by current vaccine strategies are well matched to the goal of lentiviral containment. One key consideration is the influence of differentiation state on the efficacy of vaccine-generated memory T cells. Most current T cell vaccine strategies use nonpersistent vectors that produce antigen for a limited time, and, as the antigen provided by these vectors wanes, the memory T cell responses they elicited become increasingly secondary lymphoid tissue–based, or central memory T cell (TCM cell) in character5. TCM cells have limited immediate effector function and require antigen-induced expansion, differentiation and migration to produce peak effector responses at viral replication sites5,6. TCM cell–derived effector responses are accelerated over primary responses but still seem to develop too slowly to prevent the initial systemic dissemination and extensive early replication of SIV in nonhuman primate models11,12. In contrast, agents or vectors that provide a controlled, persistent level of antigen maintain functionally differentiated TEM cells in extralymphoid sites5,15,16. Given that TEM cells are the predominant type of T cell in mucosal effector sites8,17, and that mucosal HIV transmission is associated with small seed populations of limited genetic diversity18, we hypothesized that a vaccine capable of generating and maintaining a potent HIV-specific TEM-type response might improve efficacy by initiating adaptive antiviral immunity at the outset of mucosal infection (the initial 24–48 h), when the virus is probably most susceptible to immune control.
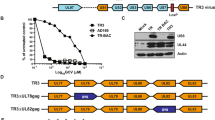
Primate CMVs comprise a family of closely related, but species-specific, β-herpes viruses that are ubiquitous, persistent and, for competent immune systems, largely benign19,20,21. CMV infections are associated with lifelong, high-frequency and highly TEM-biased CD4+ and CD8+ T cell responses that protect against disease development but do not eliminate the CMV infection or prevent CMV superinfection7,8,9,20,22,23,24,25. Therefore, we explored the possibility that RhCMV could be exploited as a persistent vaccine vector for generation of durable SIV-specific, TEM-biased CD4+ and CD8+ T cell responses in rhesus macaques, thereby allowing assessment of the ability of these responses to prevent or completely contain SIV infection during repeated limiting-dose intrarectal challenge. RhCMV-SIV vectors encoding SIV Gag, a Rev-Tat-Nef fusion protein, and Env (Fig. 1a) were constructed and shown to express high amounts of these SIV antigens during lytic infection in vitro and to possess wild-type RhCMV growth kinetics (Supplementary Figs. 1, 2, 3 online). Administration of RhCMV-SIV vectors to either RhCMV-naive or RhCMV+ rhesus macaques resulted in clinically benign primary infection and superinfection, respectively. Virological analysis showed the appearance of RhCMV-SIV vectors in urine and saliva by day 56 after inoculation (Fig. 1b), but little to no measurable viremia (data not shown). The persistence of these infections and the genetic stability of the three RhCMV-SIV vectors in vivo were indicated by our ability to culture SIV antigen–expressing RhCMV from urine and saliva co-cultures for up to 664 d after inoculation (Fig. 1b). Notably, despite the presence of potentially competing RhCMV-derived T cell epitopes and the preexisting, high-frequency T cell responses to these epitopes in RhCMV+ rhesus macaques, all rhesus macaques inoculated with these RhCMV-SIV vectors developed both CD4+ and CD8+ T cell responses to the recombinant SIV protein(s) (Fig. 1c). These responses remained detectable for more than 3 years after vector administration (details below and data not shown), suggesting that the RhCMV-vectored, SIV-specific responses persist indefinitely, similar to native RhCMV-specific T cell responses. In contrast to these robust SIV-specific T cell responses, RhCMV-SIV vectors elicited only low-titer antibodies to SIV (detectable at ≤1:10 dilution of plasma) that were unable to neutralize either SIVmac239 or a highly neutralization-sensitive, tissue culture–adapted variant of SIVmac251 (data not shown).

(a) Schematic of the SIV protein expression cassettes inserted into the RhCMV genome in the intergenic region between Rh213 and Rh214 to create the RhCMV-Gag, RhCMV-Retanef (Rev-Tat-Nef fusion protein) and RhCMV-Env vectors. EF1-α, elongation factor-1α; Myc, V5 and Flag, distinct epitope tags; pA, SV40 polyadenylation site (see Supplementary Fig. 1). (b) Western blot analysis of telomerized rhesus fibroblasts co-cultured for 4 weeks with virus pelleted from urine collected at the designated intervals from initially RhCMV+ rhesus macaques after their inoculation with RhCMV-Gag, RhCMV-Retanef and RhCMV-Env. 'Anti' indicates the antibody specific for the designated protein. IE2, RhCMV immediate early protein-2. The molecular weight in kDa is shown at left. (c) FCICA of peripheral blood T cells responding to wild-type RhCMV lysate, SIV Gag or Rev-Tat-Nef–overlapping 15-mer peptides in a representative initially RhCMV-positive rhesus macaque after inoculation with the RhCMV-Gag and RhCMV-Retanef vectors. The values in the upper and lower right quadrants of the flow cytometric profiles indicate the net percentage (minus background) of the total CD4+ or CD8+ T cell population responding to the designated antigen with production of both TNF and IFN-γ or TNF alone, respectively.
We next studied the differentiation and functional characteristics of established SIV-specific memory T cells induced by RhCMV-SIV vector vaccination. The markers CD28 and CCR7 define the TCM to TEM cell differentiation axis among rhesus macaque T cells, with TCM, transitional TEM1 and fully differentiated TEM2 cells expressing CD28+CCR7+, CD28+CCR7− and CD28−CCR7− phenotypes, respectively8,17. Both RhCMV-specific and RhCMV-vectored, SIV-specific T cells in peripheral blood showed a nearly exclusive (>90%) TEM phenotype, with CD8+ T cells having an almost entirely TEM2 phenotype and CD4+ T cells being about equally split between TEM1 and TEM2 phenotypes (Fig. 2a,b and Supplementary Fig. 4 online). In sharp contrast, a protein prime–Ad5 boost vaccine induced a predominantly TCM phenotype among SIV-specific CD4+ and CD8+ T cells (P < 0.0001; Fig. 2a and Supplementary Fig. 4). In keeping with TEM localization in extralymphoid sites8,17, both RhCMV-specific and RhCMV-vectored, SIV-specific T cells were highly enriched among bronchoalveolar lavage (BAL) lymphocytes (Fig. 2c,d). Finally, RhCMV-specific and RhCMV-vectored, SIV-specific T cells showed essentially identical functional potential (Fig. 3 and Supplementary Fig. 5 online). Specifically, CD4+ T cells were predominantly polyfunctional, capable of simultaneous production of tumor necrosis factor (TNF), interferon-γ (IFN-γ), interleukin-2 (IL-2) and macrophage inflammatory protein-1β (MIP-1β), the latter being a CCR5-binding chemokine capable of blocking HIV and SIV infection26. CD8+ T cells were also capable of producing TNF, IFN-γ and MIP-1β, but, consistent with a highly polarized TEM2 phenotype, lacked substantial IL-2 production. The polarized TEM2 phenotype of the RhCMV-vectored, SIV-specific CD8+ T cells would predict cytotoxic potential6, and, accordingly, these cells efficiently extruded cytotoxic granules (externalized CD107) upon antigen recognition (Fig. 3c).

(a,b) Combined FCICA and surface phenotype analysis of CD4+ (a) and CD8+ (b) peripheral blood T cells responding to wild-type (WT) RhCMV lysate, SIV Gag or Rev-Tat-Nef–overlapping 15-mer peptides. The graphs compare the CD28 versus CCR7 phenotype of RhCMV and SIV antigen-responsive CD4+ or CD8+ T cells (CD69+TNF+) in a representative initially RhCMV-positive rhesus macaque that was inoculated 595 d and 330 d earlier with RhCMV-Retanef and RhCMV-Gag, respectively (left and middle). They also compare the SIV Gag response of this rhesus macaque to another rhesus macaque that received a Gag protein prime and Ad5(Gag) boost (105 d after the boost; right). (c) FCICA of the response of peripheral blood versus BAL T cells (CD4+ or CD8+) to SIV Rev-Tat-Nef–overlapping 15-mer peptides in a rhesus macaque that received the RhCMV-Retanef vector 192 and 94 d earlier. The values in the profiles indicate the net percentage (minus background) of the total CD4+ or CD8+ T cell population responding to the Rev-Nef-Tat peptides with the production of the indicated cytokines. (d) Comparison of the mean frequencies (± s.e.m.) of RhCMV-vectored SIV Gag-specific, Rev-Tat-Nef (RTN)–specific and Env-specific T cell responses in the peripheral blood memory compartment versus the BAL (memory) T cell compartment in a group of six rhesus macaques 192 d after initial inoculation and 94 d after a second inoculation with the RhCMV-Gag, RhCMV-Retanef and RhCMV-Env vectors.

(a–c) Representative FCICA of peripheral blood CD4+ or CD8+ T cells responding to SIV Gag or Rev-Tat-Nef–overlapping peptides from rhesus macaques that were inoculated with the corresponding RhCMV vector more than 500 d earlier. The graphs show coordinate analysis of TNF versus IFN-γ versus IL-2 (a), TNF versus IFN-γ versus MIP-1β (b) or TNF versus CD107 externalization (indicating degranulation of cytoplasmic cytotoxic granules) (c). The left and middle profiles are gated on the overall CD4+ or CD8+ T cell populations with the percentage of the indicated responding populations (CD69+, cytokine+ or CD107+) shown in each profile. The right profiles are Boolean-gated on total responding T cells (those making any of the indicated cytokines or expressing CD107, alone or in combination), with either the percentage of triple producers (a and b, colored blue), or the percentage of responding cells showing CD107 and TNF reactivity alone or in combination (c).
These results indicate that the RhCMV-SIV vectors elicit and maintain SIV-specific T cells that are highly skewed toward TEM differentiation. To test whether this TEM-skewed anti-SIV T cell response would protect against SIV infection, we assembled a cohort of 12 long-term RhCMV-SIV vector–vaccinated rhesus macaques (which we used >486 d from their last RhCMV-SIV vaccination) and 16 unvaccinated wild-type RhCMV+ control rhesus macaques for repeated limiting-dose intrarectal challenge with the highly pathogenic SIVmac239 (Fig. 4a). At the time of challenge, the CMV-SIV vector–vaccinated rhesus macaques had approximately equivalent CD4+ and CD8+ responses to Gag, Rev-Tat-Nef and Env, totaling, on average, 1.5% and 2.0% of blood memory T cells for CD4+ and CD8+ responses, respectively (Fig. 4b). All 16 control rhesus macaques showed progressive infection over the course of challenge, with 50% of these macaques becoming infected with the first or second dose of virus and the last control macaque infected with the twelfth dose (Fig. 4c). In contrast, the median number of doses to achieve progressive, systemic infection was eight for the vaccinated group (Fig. 4c). Most notably, four of the twelve vaccinees failed to show progressive infection (P = 0.024). Two of these four rhesus macaques showed transient plasma virus levels of 60–80 SIV RNA copy equivalents per ml after the first challenge, whereas the other two underwent 13 SIVmac239 challenges without measurable plasma virus (Fig. 4c). All four rhesus macaques, however, seemed to be infected with SIVmac239 on the basis of their development of reproducible de novo CD8+ T cell responses to two SIV proteins, Pol and Vif, that were not included in the vaccine (Fig. 4d). Such new SIV-specific T cell responses did not develop in naive rhesus macaques repeatedly challenged with equivalent doses of aldrithiol-2–inactivated SIVmac239 or in three unvaccinated control macaques that were not overtly infected in the six initial challenges with competent SIVmac239 (Supplementary Fig. 6 online). These controls argue against the possibility that the de novo CD8+ T cell responses to Pol and Vif in the four protected rhesus macaques were related to cross-presentation of SIV proteins in the challenge virus preparation in the absence of infection and, in agreement with previous work27, indicate that the typical outcome of repeated limiting-dose intrarectal SIV challenge of unvaccinated rhesus macaques is either no infection or overt progressive infection, with no immune sensitization in the absence of overt infection.

(a) Vaccination and challenge protocol for efficacy assessment of RhCMV vectors. RhCMV-Gag, RhCMV-Retanef and RhCMV-Env vectors were given individually at 133-d intervals in the following orders for four rhesus macaques each: Gag, Retanef and Env; Retanef, Env and Gag; or Env, Gag and Retanef. Half of each group was subsequently provided with a combined boost of all three vectors. The long-term anti-SIV T cell responses did not differ between these subgroups, and all 12 of these rhesus macaques were therefore combined into one vaccinated group for challenge (in comparison to 16 unvaccinated, but RhCMV-positive, control rhesus macaques). RM, rhesus macaque. (b) Mean prechallenge frequencies (± s.e.m.) of RhCMV-vectored, SIV Gag-specific, Rev-Tat-Nef–specific and Env-specific responses among blood CD4+ and CD8+ memory T cells of the 12 vaccinated rhesus macaques. (c) Plasma viral loads of the control (left) and vaccinated (right) rhesus macaque cohorts over the course of, and subsequent to, limiting-dose intrarectal SIVmac239 challenge. Four of twelve vaccinated rhesus macaques resisted progressive infection; these protected rhesus macaques were treated with the humanized CD8-specifc monoclonal antibody cM-T807 at days 133, 136, 140 and 143 after initial challenge (10, 5, 5 and 5 mg per kg body weight doses, respectively) and were profoundly depleted of CD8+ T cells from blood (<2.5% of baseline absolute counts) for 14–21 d. (d) FCICA of peripheral blood CD8+ T cells from the four protected vaccinees, examining the response of these cells to SIV proteins that were (Rev-Tat-Nef) or were not (Pol and Vif) expressed by the administered RhCMV vectors before (left) and 133 d after (right) initiation of the SIVmac239 intrarectal challenge protocol. Percentages indicate the net fraction (minus background) of the responding CD8+ T cells in the indicated quadrants.
At 19 weeks after the first challenge, the four protected rhesus macaques were depleted of CD8+ cells by administration of monoclonal antibody cM-T807, a treatment capable of transiently reversing CD8+ T cell-mediated control of SIV replication28. This treatment caused profound depletion of circulating CD8+ T cells but did not result in measurable viremia (Fig. 4c). Moreover, cell-associated SIV DNA or RNA was not detected by sensitive PCR assays in isolated peripheral blood and lymph node CD4+ T cells from these four rhesus macaques, both before and during maximal CD8+ cell depletion (data not shown). Thus, four of twelve RhCMV-SIV vector–vaccinated rhesus macaques were infected with SIVmac239 via a mucosal route but showed no systemic evidence of infection months later, strongly suggesting that infection was effectively contained locally, before dissemination and establishment of typical systemic infection. This study did not directly compare the efficacy of RhCMV-SIV vectors to other vaccine regimens that induce TCM-biased responses, but it should be noted that the degree of protection observed in this study was not achieved in a test of a potent DNA prime–Ad5 boost vaccine regimen that used the same challenge virus and limiting-dose intrarectal challenge procedure12. Our data also do not directly demonstrate the mechanism of this protection, but among the possibilities—T cell, antibody or innate immunity—the most likely candidate is certainly the robust, SIV-specific TEM responses observed in the vaccinees. SIV-specific antibody responses were weak with no neutralization activity, and both vaccinated and control monkeys were chronically infected with RhCMV and >486 d removed from any overt RhCMV exposure, making it highly unlikely that innate immune activation contributed to protection (although it should be noted that the control rhesus macaques were not experimentally re-infected with RhCMV via vector administration as were the vaccinated macaques).
The presence of class I major histocompatibility variants such as Mamu-B*08 and Mamu-B*17, previously associated with spontaneous, CD8+ T cell–mediated control of viral replication in the chronic phase of SIV infection4, did not correlate with the acute phase protective effects we observed (only one of four protected versus five of eight unprotected rhesus macaques were B*17+ or B*08+), suggesting potential differences in the mechanism(s) mediating early and local versus late and systemic protection. We speculate that in the four early-protected rhesus macaques reported here, RhCMV vector–generated, SIV-specific TEM cells in rectal lamina propria responded to challenge by creating adverse local conditions for viral replication and spread, effectively lowering the basic reproductive ratio of the infection below that required for sustained infection (<1)2,29. In the eight nonprotected rhesus macaques, the local TEM cell responses may have been insufficient to suppress the reproductive ratio to <1, perhaps owing to intrinsic genetic differences among these rhesus macaques or to stochastic variation in local TEM cell distribution and function. In future studies, it will clearly be necessary to quantify the distribution and function of SIV-specific TEM cells in the rectal mucosa so as to define precise correlates of local protection. Not surprisingly, given the extralymphoid localization and relatively low expansion potential of TEM cells, the RhCMV-vectored, SIV-specific TEM cell responses were unable to provide substantial protection once progressive, systemic infection was initiated (Supplementary Fig. 7 online). However, it should also be noted that the vaccine-generated, SIV-specific CD4+CCR5+ TEM cells did not accelerate or enhance viral replication in these 'break-through' infections.
Consistent with our original hypothesis, these results suggest that TEM-biased T cell immunity, in the absence of neutralizing antibody, can prevent establishment of progressive systemic infection after mucosal challenge with a highly pathogenic SIV, presumably by interfering with viral replication at its earliest stages. CMV-based vectors offer one strategy to generate such a TEM cell component, but other strategies that provide persistent, low-level antigen expression could also be explored. It may also be possible to engineer HIV/AIDS vaccines that generate and maintain both TEM and TCM cell components, the latter serving as a second line of defense if the initial TEM cell barrier is breached. Such TEM cell or combination TEM-TCM cell vaccines will probably not provide absolute protection against all HIV exposure (against parenteral routes, for example). However, our data suggest that vaccine-generated T cell responses are able to do more than simply lower viral replication set points. Specifically, HIV/AIDS vaccines with a TEM component may have the ability to protect against the sexual transmission of HIV.
Methods
Rhesus macaques. We used 49 purpose-bred juvenile and adult male rhesus macaques (Macaca mulatta) of Indian genetic background in this study, including 12 RhCMV+ and six RhCMV− rhesus macaques inoculated with RhCMV-SIV vectors, nine rhesus macaques that received a Gag protein prime followed by an Ad5(Gag) boost vaccine protocol and 22 RhCMV+ rhesus macaques used as unvaccinated controls. All rhesus macaques were free of cercopithicine herpesvirus 1, D-type simian retrovirus, simian T-lymphotrophic virus type 1 and SIV infection at the start of the study, and they were used with approval of the Oregon National Primate Research Center Animal Care and Use Committee, under the standards of the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. We administered RhCMV-SIV vectors subcutaneously at a dose of 1 × 107 plaque-forming units. The Gag protein prime–Ad5(gag) boost protocol consisted of 450 μg SIV Gag protein (Protein Sciences) emulsified in montanide ISA-51 carrier (SEPPIC), with or without 1 mg of polyI:C (Invitrogen), 3M-12 (3M Pharmaceuticals) or class C CpG (Pfizer) adjuvant at weeks 0 and 10, followed by Ad5(gag) (1 × 1010 particles, from National Institutes of Health Vaccine Research Center) at week 20. We performed SIVmac239 challenge by administering 300 focus-forming units of virus weekly via the intrarectal route for the first eight challenges and then, if necessary, five additional weekly challenges at 1,000 focus-forming units. We challenged all of the vaccinated macaques and 12 of the control macaques concurrently, and we challenged the remaining four control rhesus macaques as part of a titration performed immediately before the primary challenge, using the same viral dose and challenge procedure. We followed plasma viral loads weekly, with challenge being discontinued the week after detection of >30 copy equivalents per ml of SIV Gag RNA. For CD8+ cell depletion, we treated rhesus macaques with 10, 5, 5 and 5 mg per kg body weight of the humanized monoclonal antibody cM-T807 (ref. 30) on days 1, 3, 5 and 10, respectively. We collected BAL cells as previously described7.
Viruses and vectors. We prepared RhCMV constructs expressing SIV proteins (designated RhCMV-SIV viruses; Fig. 1a) with a bacterial artificial chromosome (BAC) containing the entire RhCMV strain 68-1 genome (pRhCMV–BAC-Cre), as previously described31,32 (see Supplementary Fig. 1). We confirmed the genomic integrity of the recombinant RhCMV-SIV BACs by restriction enzyme digestion, Southern analysis and direct DNA sequence analysis of the SIV open reading frames (data not shown). We reconstituted RhCMV-SIV viruses by transfection of BAC DNA into RhCMV-permissive rhesus fibroblasts and confirmed expression of SIV antigens by western blot analysis of RhCMV-SIV–infected cell lysates (Supplementary Fig. 2). Multistep growth analysis of RhCMV-SIV viruses31 showed growth kinetics comparable to wild-type RhCMV (Supplementary Fig. 3). The pathogenic SIV challenge stock (kindly provided by C.J. Miller) was generated by expanding the SIVmac239 clone33 in rhesus macaque peripheral blood mononuclear cells and quantified by sMAGI cell assay34 and quantitative RT-PCR for SIV genomic RNA35. We prepared AT-2–inactivated SIVmac239 (lot P4146) as previously described36 and normalized this stock to the infectious SIVmac239 stock by real-time RT-PCR quantification of SIV genome copy number.
Viral detection assays. We centrifuged filter-sterilized (0.4 μm) urine at 16,000g for 1 h at 4 °C to concentrate virus for co-culture on rhesus fibroblasts. After we observed extensive cytopathic effects, or after 42 d of co-culture if cytopathic effects were minimal or not observed, we prepared cell lysates and assessed RhCMV-SIV vector replication on the basis of expression of SIV antigen-specific epitope tags by western immunoblotting. We measured whole-blood RhCMV DNA, plasma SIV RNA, and cell-associated SIV DNA and RNA by real-time PCR and RT-PCR assays24,35,37, with the cell-associated SIV quantification with immunobead-isolated CD4+ T cells before and after a 48-h stimulation with an immobilized antibody to CD3 and recombinant IL-2 and including RT-PCR analysis of supernatants of the stimulated cells.
Immunologic assays. We measured RhCMV- and SIV-specific CD4+ and CD8+ T cell responses by flow cytometric intracellular cytokine analysis (FCICA) of peripheral blood mononuclear cells and BAL cells, as previously described7,24,25,38 (see Supplementary Methods online). We measured antibody binding to SIV by ELISA of purified SIVmac239 viral lysates39. We measured neutralizing antibodies against SIVmac239 and tissue culture–adapted SIVmac251 in luciferase reporter gene assays with the TZM-bl and M7 Luc cell lines, respectively40.
Statistical analyses. We performed statistical analyses with SAS version 9.1 (Statistical Analysis System). We assessed the significance of differences in the phenotype or functional properties of RhCMV-specific T cells versus RhCMV-vectored, SIV-specific T cells versus prime-boost–elicited SIV-specific T cells by mixed-effects analysis; the proportion of rhesus macaques within the vaccinated versus control groups that completely controlled SIV infection after repeated, limiting-dose mucosal challenge by a two-sided Fischer's exact test; and the peak and mean plateau phase (day 42 to day 91) plasma viral loads of the control versus vaccinated rhesus macaques with progressive infection by general linear models with log10-transformed data. In all analyses, we used a two-sided significance level of 0.05, with correction made for multiple comparisons using the Bonferroni method.
Note: Supplementary information is available on the Nature Medicine website.

Comparison of amino acid sequences of Retanef(Hel) and Retanef(int)

Immunogenicity profiles of RhCMV/retanef(int)-vaccinated controllers before and after SIV challenge. Flow cytometric intracellular cytokine analysis (FCICA) (Nat. Med. 15, 293–299, 2009) of peripheral-blood CD8+ T cells responding to 15-mer overlapping peptides comprising amino acid sequences common or unique to Retanef(Hel) and Retanef(int) in the four RhCMV/retanef(int)-vaccinated, SIV controllers we initially reported before and after SIV challenge. The percentage of responding SIV antigen–specific CD8+ T cells (producing tumor necrosis factor-α, interferon-γ or both) within the overall memory subset is shown. Responses before challenge were restricted to sequences included in the RhCMV/retanef(int) vector, whereas after challenge responses to sequences that are only expressed by the SIV challenge virus are identified. The only exception to this pattern is the int(Lys159–Ala293) sequence, which, though included in the RhCMV/retanef(int) vector and in SIV itself, is nonimmunogenic for T cells both before and after challenge.
Change history
06 April 2009
In the version of this article initially published, a “left” and “right” designation was switched in the legend for Figure 4d. The legend should read “FCICA of peripheral blood CD8+ T cells from the four protected vaccinees, examining the response of these cells to SIV proteins that were (Rev-Tat-Nef) or were not (Pol and Vif) expressed by the administered RhCMV vectors before (left) and 133 d after (right) initiation of the SIVmac239 intrarectal challenge protocol.” The error has been corrected in the HTML and PDF versions of the article.
07 November 2011
In Supplementary Figure 1 of our paper, the reference cited (Hel et al. in Vaccine 20, 3171–3186, 2002) and the description for the Retanef fusion gene expressed by RhCMV-Retanef were incorrect. The correct description for the fusion gene (here designated Retanef(int)) and citation are now contained in the Supplementary Information. Retanef(int) is a fusion comprised of simian immunodeficiency virus (SIV) rev (Met1–Leu19), int (Lys159–Ala293), full-length nef (starting at Ala4) and tat (Glu2–Arg82), mutagenized to decrease toxicity, essentially as previously described by Kulkarni et al. (Vaccine 29, 6742–6754, 2011). Table 1 shows a comparison between Retanef(Hel) (Vaccine 20, 3171–3186, 2002) and our Retanef(int) construct. In Figure 4d of our paper, the appearance of de novo CD8+ T cell responses to overlapping peptides comprising the full-length SIV pol protein in the four stringently protected RhCMV-SIV–vaccinated rhesus macaques after SIVmac239 challenge was used as evidence of occult SIV infection. Although the presence of a pol component—int(Lys159–Ala293)—within the RhCMV-Retanef(int) vector would bring this conclusion into question, several lines of evidence strongly support our original interpretation. First, as we initially indicated, two of the four protected rhesus macaques showed transient SIV viremia, and all four of these rhesus macaques showed the appearance of de novo CD8+ responses to peptides comprising full-length SIV vif (a protein not included in the vaccine). Second, peripheral blood mononuclear cells from a total of 23 RhCMV-Retanef(int)–vaccinated and three SIV-infected rhesus macaques were found to be negative for both CD4+ and CD8+ T cell responses to peptide mixes comprising int (Lys159–Ala293), indicating this region is poorly immunogenic or nonimmunogenic for T cells in rhesus macaques. Finally, further analysis of cryopreserved T cells from the four protected monkeys in our study specifically shows the absence of CD8+ T cell responses to full-length pol, int(Lys159–Ala293) and the SIV rev/tat regions unique to the retanef(Hel) construct (and not in the RhCMV-retanef(int) vector administered to these rhesus macaques) prior to SIVmac239 challenge, as well as the appearance of such responses to full-length pol and the unique retanef(Hel) regions, but not the int(Lys159–Ala293), in all monkeys after SIV challenge (Fig. 1). Thus, challenge was associated with the induction of de novo CD8+ T cell responses to multiple distinct SIV sequences that were not included in the vaccine (that is, full-length vif, pol other than int(Lys159–Ala293), rev(Leu20–Asp100) and tat(Arg83–Arg131)), consistent, as we originally suggested, with controlled SIV infection. Thus, none of the conclusions of our original report are affected by the use of RhCMV-Retanef(int) reported in this addendum. (See PDF)
References
Johnson, W.E. & Desrosiers, R.C. Viral persistence: HIV's strategies of immune system evasion. Annu. Rev. Med. 53, 499–518 (2002).
Haase, A.T. Perils at mucosal front lines for HIV and SIV and their hosts. Nat. Rev. Immunol. 5, 783–792 (2005).
Walker, B.D. & Burton, D.R. Toward an AIDS vaccine. Science 320, 760–764 (2008).
Goulder, P.J. & Watkins, D.I. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat. Rev. Immunol. 8, 619–630 (2008).
Robinson, H.L. & Amara, R.R. T cell vaccines for microbial infections. Nat. Med. 11, S25–S32 (2005).
Sallusto, F., Geginat, J. & Lanzavecchia, A. Central memory and effector memory T cell subsets: function, generation and maintenance. Annu. Rev. Immunol. 22, 745–763 (2004).
Pitcher, C.J. et al. Development and homeostasis of T cell memory in rhesus macaque. J. Immunol. 168, 29–43 (2002).
Picker, L.J. et al. IL-15 induces CD4 effector memory T cell production and tissue emigration in nonhuman primates. J. Clin. Invest. 116, 1514–1524 (2006).
Chan, K.S. & Kaur, A. Flow cytometric detection of degranulation reveals phenotypic heterogeneity of degranulating CMV-specific CD8+ T lymphocytes in rhesus macaques. J. Immunol. Methods 325, 20–34 (2007).
Watkins, D.I., Burton, D.R., Kallas, E.G., Moore, J.P. & Koff, W.C. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans. Nat. Med. 14, 617–621 (2008).
Horton, H. et al. Immunization of rhesus macaques with a DNA prime/modified vaccinia virus Ankara boost regimen induces broad simian immunodeficiency virus (SIV)-specific T-cell responses and reduces initial viral replication but does not prevent disease progression following challenge with pathogenic SIVmac239. J. Virol. 76, 7187–7202 (2002).
Wilson, N.A. et al. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J. Virol. 80, 5875–5885 (2006).
Letvin, N.L. et al. Preserved CD4+ central memory T cells and survival in vaccinated SIV-challenged monkeys. Science 312, 1530–1533 (2006).
Liu, J. et al. Immune control of an SIV challenge by a T-cell–based vaccine in rhesus monkeys. Nature 457, 87–91 (2008).
Gauduin, M.C. et al. Induction of a virus-specific effector memory CD4+ T cell response by attenuated SIV infection. J. Exp. Med. 203, 2661–2672 (2006).
Pipeling, M.R. et al. Differential CMV-specific CD8+ effector T cell responses in the lung allograft predominate over the blood during human primary infection. J. Immunol. 181, 546–556 (2008).
Grossman, Z. & Picker, L.J. Pathogenic mechanisms in simian immunodeficiency virus infection. Curr. Opin. HIV AIDS 3, 380–386 (2008).
Keele, B.F. et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 105, 7552–7557 (2008).
Pass, R.F. Cytomegalovirus. in Fields Virology 4th edn (eds. Knipe, D.M. & Howley, P.M.) 2675–2706 (Lippincott Williams & Wilkins, Philadelphia, 2001).
Britt, W. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. in Human Cytomegalovirus (eds. Shenk, T. & Stinski, M.F.) 417–470 (Springer-Verlag, Heidelberg, Germany, 2008).
Powers, C. & Fruh, K. Rhesus CMV: an emerging animal model for human CMV. Med. Microbiol. Immunol. 197, 109–115 (2008).
Sylwester, A.W. et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 202, 673–685 (2005).
Kern, F. et al. Distribution of human CMV-specific memory T cells among the CD8pos. subsets defined by CD57, CD27, and CD45 isoforms. Eur. J. Immunol. 29, 2908–2915 (1999).
Price, D.A. et al. Induction and evolution of cytomegalovirus-specific CD4+ T cell clonotypes in rhesus macaques. J. Immunol. 180, 269–280 (2008).
Casazza, J.P. et al. Acquisition of direct antiviral effector functions by CMV-specific CD4+ T lymphocytes with cellular maturation. J. Exp. Med. 203, 2865–2877 (2006).
DeVico, A.L. & Gallo, R.C. Control of HIV-1 infection by soluble factors of the immune response. Nat. Rev. Microbiol. 2, 401–413 (2004).
Letvin, N.L. et al. No evidence for consistent virus-specific immunity in simian immunodeficiency virus–exposed, uninfected rhesus monkeys. J. Virol. 81, 12368–12374 (2007).
Friedrich, T.C. et al. Subdominant CD8+ T cell responses are involved in durable control of AIDS virus replication. J. Virol. 81, 3465–3476 (2007).
Petravic, J. et al. Estimating the impact of vaccination in acute SHIV/SIV infection. J. Virol. 82, 11589–11598 (2008).
Schmitz, J.E. et al. A nonhuman primate model for the selective elimination of CD8+ lymphocytes using a mouse-human chimeric monoclonal antibody. Am. J. Pathol. 154, 1923–1932 (1999).
Rue, C.A. et al. A cyclooxygenase-2 homologue encoded by rhesus cytomegalovirus is a determinant for endothelial cell tropism. J. Virol. 78, 12529–12536 (2004).
Chang, W.L. & Barry, P.A. Cloning of the full-length rhesus cytomegalovirus genome as an infectious and self-excisable bacterial artificial chromosome for analysis of viral pathogenesis. J. Virol. 77, 5073–5083 (2003).
Kestler, H. et al. Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science 248, 1109–1112 (1990).
Chackerian, B., Haigwood, N.L. & Overbaugh, J. Characterization of a CD4-expressing macaque cell line that can detect virus after a single replication cycle and can be infected by diverse simian immunodeficiency virus isolates. Virology 213, 386–394 (1995).
Cline, A.N., Bess, J.W., Piatak, M. Jr . & Lifson, J.D. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol. 34, 303–312 (2005).
Rossio, J.L. et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J. Virol. 72, 7992–8001 (1998).
Venneti, S. et al. Longitudinal in vivo positron emission tomography imaging of infected and activated brain macrophages in a macaque model of human immunodeficiency virus encephalitis correlates with central and peripheral markers of encephalitis and areas of synaptic degeneration. Am. J. Pathol. 172, 1603–1616 (2008).
Walker, J.M., Maecker, H.T., Maino, V.C. & Picker, L.J. Multi-color flow cytometric analysis in SIV-infected rhesus macaques. in Cytometry 4th edn (eds. Darzynkiewicz, Z., Roederer, M. & Tanke, H.) 535–557 (Academic Press, San Diego, 2004).
Lu, X. et al. Targeted lymph-node immunization with whole inactivated simian immunodeficiency virus (SIV) or envelope and core subunit antigen vaccines does not reliably protect rhesus macaques from vaginal challenge with SIVmac251. AIDS 12, 1–10 (1998).
Montefiori, D.C. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. Ch.12, 12.11 (2005).
Acknowledgements
This work was supported by the US National Institute of Allergy and Infectious Diseases, the International AIDS Vaccine Initiative, the Bill & Melinda Gates Foundation–supported Collaboration for AIDS Vaccine Discovery, the US National Center for Research Resources and the US National Cancer Institute. We thank J. Edgar, A. Keech, J. Ford, J. Cook, M. Rohankhedkar, T. Ha, A. Sylwester and J. Dewane for technical assistance; P. Barry (University of California–Davis) for the RhCMV bacterial artificial chromosome, G. Pavlakis (National Cancer Institute) for the SIV Gag and Env constructs, G. Franchini (National Cancer Institute) for the SIV Retanef construct, R. Seder (Vaccine Research Center, National Institutes of Health) for the Gag protein immunogens, C. Miller (University of California–Davis) for the pathogenic SIVmac239 challenge stock, K. Reimann and Centocor for the cM-T807 antibody, M. Mori and J. O'Malley for statistical assistance and K. Frueh and S. Wong for helpful discussion and advice.
Author information
Authors and Affiliations
Contributions
S.G.H., assisted by C.V., N.W., D.C.S. and L.C.-J., planned and performed experiments and analyzed data. A.W.L. and M.K.A. managed the animal protocols. M.A.J. designed, constructed and characterized the RhCMV vectors, assisted by D.D.D. M.P. and J.D.L., assisted by K.O. and C.M.T., planned and performed SIV quantification studies. J.A.N. was involved in conception of the RhCMV vector strategy. L.J.P. conceived the RhCMV vector strategy, supervised experiments, analyzed data and wrote the paper (assisted by M.A.J.).
Corresponding author
Supplementary information
Supplementary Text and Figures
Supplementary Figs. 1–7 and Supplementary Methods (PDF 982 kb)
Rights and permissions
About this article
Cite this article
Hansen, S., Vieville, C., Whizin, N. et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 15, 293–299 (2009). https://doi.org/10.1038/nm.1935
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm.1935
This article is cited by
-
‘Stem-like’ precursors are the fount to sustain persistent CD8+ T cell responses
Nature Immunology (2022)
-
MCMV-based vaccine vectors expressing full-length viral proteins provide long-term humoral immune protection upon a single-shot vaccination
Cellular & Molecular Immunology (2022)
-
Long-term protective immunity induced by an adjuvant-containing live-attenuated AIDS virus
npj Vaccines (2021)
-
Influenza- and MCMV-induced memory CD8 T cells control respiratory vaccinia virus infection despite residence in distinct anatomical niches
Mucosal Immunology (2021)
-
Immunologic Control of HIV-1: What Have We Learned and Can We Induce It?
Current HIV/AIDS Reports (2021)
