
Abstract
Protein–protein interactions are a crucial element in cellular function. The wealth of information currently available on intracellular-signaling pathways has led many to appreciate the untapped pool of potential drug targets that reside downstream of the commonly targeted receptors. Over the last two decades, there has been significant interest in developing therapeutics and chemical probes that inhibit specific protein–protein interactions. Although it has been a challenge to develop small molecules that are capable of occluding the large, often relatively featureless protein–protein interaction interface, there are increasing numbers of examples of small molecules that function in this manner with reasonable potency. This article will highlight the current progress in the development of small molecule protein–protein interaction inhibitors that have applications in the treatment or study of central nervous system function and disease. In particular, we will focus upon recent work towards developing small molecule inhibitors of amyloid-β and α-synuclein aggregation, inhibitors of critical components of G-protein-signaling pathways, and PDZ domain inhibitors.
Similar content being viewed by others

INTRODUCTION
Protein–protein interactions (PPIs) are essential components of virtually all cellular processes. The binding of two or more proteins in a cell can have a wide array of effects, including modulating or initiating signal transduction, regulating patterns of gene transcription, providing cytoskeletal stability, and promoting cellular replication or death. Because the cellular network of PPIs is vast and essential, in theory it should contain many potential sites at which a drug may be targeted. In the past several years, there has been much effort placed upon identifying specific inhibitors of PPIs. Currently, there are a number of clinically relevant therapies that target PPI interfaces. Most currently used PPI inhibitors (PPIIs) in the clinic are based upon humanized monoclonal antibodies. Although this class of therapeutics possesses some very desirable drug properties (eg high specificity, low toxicity) it also has several drawbacks that make the approach less applicable to the widespread development of PPIIs (eg lack of cell/blood–brain barrier permeability, poor oral bioavailability, high cost of manufacture). This review will highlight recent work that has identified small molecule inhibitors of PPIs, with a focus on those compounds that have the potential to affect neural functioning. The central nervous system (CNS), in particular, is ripe for targeting of PPIs as the highly organized nature of CNS signal transduction relies heavily on localization and compartmentalization of signaling functions. Blocking the PPIs underlying this compartmentalization (eg PDZ domain targets) could provide more subtle tissue-specific therapeutic actions than does blocking the signal pathway itself. Furthermore, highly specific neural transcriptional patterns of regulatory molecules (eg RGS proteins) provide great opportunities for cell-type selective modulation of signaling. This burgeoning field is only starting to be developed and entails a large number of unexplored potential drug targets of which we discuss some of the best-developed examples.
INHIBITING PROTEIN–PROTEIN INTERACTIONS OUTSIDE OF THE CNS
Directly targeting PPIs with small molecules has only recently become a feasible approach to drug development. Over the past several years significant progress has been made in the development of small, drug-like molecules that are capable of inhibiting the interaction between two proteins. However, this progress has not come easily—PPI interfaces have proven to be particularly difficult drug targets and have been deemed intractable in many instances (Hajduk and Burns, 2002; Whitty and Kumaravel, 2006). The difficulties encountered in targeting a PPI are substantial and it takes a great deal of work to develop useful lead compounds. The most obvious obstacle is the sheer size and geometry of the standard protein interaction interface. These regions are often relatively featureless expanses of protein surface that cover 750–1500 Å2 (Arkin and Wells, 2004) and are devoid of traditional ‘pockets’ into which a small molecule can dock in an energetically favorable manner. While developing a cell-permeant, bioavailable small molecule that is capable of occluding such a large interaction surface was deemed intractable by many, recent advances in the field have shown that this conclusion was premature. Numerous families of small molecule protein–protein inhibitors have been developed for a number of targets, the majority of which are directed towards potential application for cancer therapy. For example, much progress has been made in the development of inhibitors of the p53/MDM2 interaction, the Bak/Bcl interaction, or the Myc/Max interaction (Arkin and Well, 2004; Pagliaro et al, 2004). Although the development of these inhibitors is of great academic and clinical interest, they are beyond the scope of this chapter and as such will not be discussed further. Several good reviews have been published on small molecule PPIIs that function as cancer therapeutics (Arkin and Wells, 2004; Fotouhi and Graves, 2005; Laurie et al, 2007; Pagliaro et al, 2004; Sarek and Ojala, 2007), so we will focus here on CNS-related targets.
A major breakthrough in the development of small molecule PPIIs was the discovery of ‘hot spots’ on protein interaction surfaces (Figure 1). These small regions of the interaction interface, often identified by alanine scanning (Bogan and Thorn, 1998; Wells, 1991), are responsible for a disproportionate contribution to the binding energy of the two proteins. An extensive database of single alanine mutations has shown that these hot spots are often enriched in aromatic and positively charged residues (Bogan and Thorn, 1998). The discovery that many PPIs are primarily governed by a relatively small section of the dimer interface has given renewed life to the idea that large, relatively flat protein interaction interfaces could bind small molecules in such a way that occludes protein dimerization. By identifying and targeting these sites, a small molecule has a much greater chance of binding to and directly occluding a PPI. To this end, at least two independent web servers have been developed that analyze PPIs or predict interaction ‘hot spots’ (Darnell et al, 2008; Shulman-Peleg et al, 2008). One of these servers, the knowledge-based fade and contacts (KFC) server, has been developed to predict protein interaction ‘hot spots’ based upon the three-dimensional structure of the PPI (Figure 1). This prediction software functions primarily upon a structure of the PPI complex, but is also capable of incorporating information from Robetta's alanine scanning (Kortemme et al, 2004), ConSurf sequence conservation (Glaser et al, 2003; Landau et al, 2005), the alanine scanning energetics database (Thorn and Bogan, 2001), or the binding interface database (Fischer et al, 2003) to aid in the prediction. Although clearly still just a prediction requiring experimental confirmation, algorithms such as these may provide a rapid mechanism to determine if a particular PPI contains a well-defined ‘hot spot’ that may be amenable to small molecule targeting.

Predicted ‘hot spots’ on the regulator of G-protein signaling 4 (RGS4)/Gαi1 protein–protein interaction interface. The highlighted residues on both surfaces are predicted by the KFC Server to be energetically important for the protein–protein interaction (Darnell et al, 2008). Structure from PDB ID 1AGR (Tesmer et al, 1997).
RATIONALE FOR TARGETING PROTEIN–PROTEIN INTERACTIONS IN THE CNS
The importance of PPIs in proper cellular function is particularly striking in the nervous system. In the CNS, a host of PPIs is required for virtually all cellular processes, including neurite outgrowth, synaptic formation and modulation, neurotransmission, signal transduction, and the induction of apoptosis (Collins and Grant, 2007; Morrison et al, 2002; Planas et al, 2006; Redies, 1997). Indeed, the highly specialized structures and discrete localization of signaling molecules in the synapse are dependent on a large network of PPIs. Targeting specific PPIs in the CNS may provide novel mechanisms to modulate neural function downstream of receptor activation or to disrupt localization signals that contribute to the efficiency or specificity of signaling. Furthermore, by targeting these processes, it may be possible to more subtly and specifically tune neural functioning than can be achieved by administering a receptor agonist/antagonist. Most receptor-targeted drugs do not have the ability to selectively act upon receptors in a particular region of the body. For example, μ-opioid receptor (MOP) agonists (eg morphine, codeine) commonly cause constipation due to their effects on MOP receptors in the intestine. The benefit of targeting localization signals or downstream members of the pathway is that, in many instances, those factors are tissue specific. By using this approach, it may be possible to provide a measure of tissue specificity in the intrinsic mechanism of a drug. This benefit could be particularly important in the development of centrally acting drugs, as many broadly acting drugs in the CNS tend to have serious side effects limiting their use (Brunton et al, 2006). Theoretically, this selectivity could be achieved at various points in the signaling cascade, as there are often several steps in a signal transduction pathway that are dependent on PPIs. Another mechanism that targeting PPIs affords is the potential ability to localize two important signaling molecules with a bifunctional molecule that facilitates the interaction (Gestwicki and Marinec, 2007). Such a molecule is comprised of two protein-binding moieties joined by a short linker region and functions to localize the two potential binding partners by noncovalently tethering them together. Although these bifunctional molecules are more of a PPI facilitator (or agonist) than an inhibitor, they may also provide a mechanism to specifically modulate neural signaling. Overall, targeting a downstream-signaling modulator is likely to provide an increase in tissue specificity of the therapeutic effect and may also provide a mechanism to subtly modulate neural firing downstream of natural neurotransmission.
INHIBITING PROTEIN AGGREGATION IN THE CNS
Amyloid-β Aggregation
Alzheimer's disease (AD), Parkinson's disease (PD), and other ‘plaqueopathies’ are becoming increasingly prevalent in our society and there is growing interest in the mechanism, prevention, and treatment of these protein aggregation diseases. Therapies for these diseases, typified by accumulation of aggregated protein plaques, have largely dealt solely with the symptoms of the disease (ie dyskinesias, decline of cognitive abilities). Although these treatments can offer some benefit, they offer no real chance of disease reversal nor can they halt its progression. There has been great interest, however, in understanding the biochemistry and pathophysiology of the plaque development and in discovering methods to inhibit or reverse plaque formation. Emphasis recently has shifted to finding compounds that inhibit the development of the small oligomeric species that both lead to the macroscopic plaques and are believed to be the pathogenic factor in these diseases (Aisen, 2005). Several of these methods rely upon directly inhibiting the aggregation of the protein, whereas a subset are focused upon modulating the expression levels of the plaque-forming protein or the chaperones that assist it into its native conformation. We will focus on the former.
Identifying compounds that selectively disrupt protein aggregates or that prevent plaque formation by inhibiting protein aggregation could be a viable approach to the treatment of protein aggregation diseases. As such, there has been a push for the discovery and development of compounds that selectively inhibit protein aggregation. Compounds have been identified that inhibit the aggregation of a variety of proteins including, huntingtin (Heiser et al, 2002, 2000), amyloid-β (Aisen, 2005; Aisen et al, 2007; Cavalli et al, 2007, 2008; Gestwicki et al, 2004), and amyloid-τ (Necula et al, 2005). Particular attention has been paid to the proteins that form the basis of plaque formation in AD, namely amyloid-β and τ. It has long been known that a variety of dyes bind to and can destabilize or inhibit plaque formation (for an extensive list, see Necula et al, 2007). Histopathological evaluation of brains from AD patients has shown at least two distinct types of plaques form during this disease. In the brain of an AD patient, aggregates of amyloid-β form in the extracellular matrix and neurofilbrillary tangles of aggregated τ-protein form intracellularly. Both of these aggregates are correlated with AD, but it has yet to be conclusively shown that these plaques cause the observed neurodegeneration and are not merely coincident with it or even a result of it. In fact, significant plaque development has been observed in a population of cognitively normal 70-year olds (Dickson and Rogers, 1992). A current hypothesis states that it is not the mature plaques that are the triggering factor for neurodegeration, but rather the protofibrils—small oligermeric complexes of the protein—that are the basis of (or are at least correlated with) disease progression (Lansbury and Lashuel, 2006). Owing to the lack of in vivo imaging methods for visualizing protofibril formations, this hypothesis has yet to be tested in living human patients. This suggests that by inhibiting the development of protofibrils it might be possible to slow the disease progression. Indeed, several drugs that inhibit amyloid-β fibril formation by distinct mechanisms are currently in or have been tested in clinical trials (Aisen, 2005; Aisen et al, 2007; Wright, 2006). One of these drugs, Alzhemed (Figure 2a, tramiprosate) is a PPII that functions by sequestering monomeric amyloid-β protein (Aisen, 2005; Aisen et al, 2007; Wright, 2006). This drug passed through phase II clinical trials, but failed in phase III clinical trials (Hébert, 2007). Although tramiprosate ultimately failed in the clinical trials, it provides a proof of concept that small molecule inhibitors of amyloid-β protofibril formation are capable of reaching late stage development and may still be a viable approach to AD treatment.

Examples of amyloid-β aggregation inhibitors. (a) Alzhemed (tramiprosate, homotaurine) (Aisen et al, 2007). (b) Memoquin, a multifactorial compound for the treatment of Alzheimer's disease (Bolognesi et al, 2005; Melchiorre et al, 1998; Piazzi et al, 2003; Rosini et al, 2005). (c) SLF-CR, a bifunctional molecule that recruits the FKBP family of chaperones to aggregating amyloid-β (Gestwicki et al, 2004).
Another recent development in amyloid-β aggregation inhibitors was the development of the multifunctional compound memoquin (Figure 2b) (Bolognesi et al, 2005; Melchiorre et al, 1998; Piazzi et al, 2003; Rosini et al, 2005). This compound was rationally designed by incorporating a radical scavenging moiety (the benzoquinone fragment of coenzyme Q10) into a series of cholinesterase inhibitor derivatives (Cavalli et al, 2007). Along with possessing antioxidant activity, coenzyme Q10 and other benzoquinones have been shown to directly inhibit amyloid-β aggregation (Bragin et al, 2005; Ono et al, 2005). It was therefore expected that this compound would be a multifactorial therapy for the treatment of AD, acting as an AchE inhibitor, a free-radical scavenger, and an inhibitor of amyloid-β aggregation. Indeed, it was found that memoquin is a potent AchE inhibitor (2.6 nM Ki) and is capable of inhibiting both the AchE-induced and intrinsic aggregation of amyloid-β (Cavalli et al, 2007, 2008). This compound is orally bioavailable, crosses the blood–brain barrier, and rescues the amyloid plaque accumulation in TG2576 APP mice. It was also shown to prevent AD-like neurodegeneration in the anti-NGF transgenic mice (AD11), another model of AD (Capsoni and Cattaneo, 2006; Capsoni et al, 2000; Cavalli et al, 2007). As of June 2008, there was no clinical data readily available on the efficacy of the compound in human subjects.
Although the previous small molecule inhibitors discussed have directly inhibited amyloid-β aggregation, a different approach was taken by Gestwicki et al (2004). They developed a bifunctional molecule that recruits the FK506 binding protein (FKBP) family of chaperones to a developing amyloid-β aggregate (Figure 2c). This series of molecules was created by using various linkers to join Congo red—a dye known to bind to amyloid-β—and SLF, a synthetic ligand for FKBPs. By recruiting the chaperone, the molecule dramatically increases its steric bulk and is capable of inhibiting the aggregation of amyloid-β. The recruitment of FKBP by this molecule is essential for its activity, suggesting that this particular scaffold does not disrupt the amyloid-β interaction energy as much as some of the previously mentioned compounds. Tethering large molecules together with selective bifunctional small molecules may be an important and powerful mechanism to modulate PPIs in the CNS, as many of the current small molecule inhibitors are bulky and may not have good permeability across the blood–brain barrier. Furthermore, this approach allows for the development of not just PPIIs, but also for the development of PPI facilitators. There are instances where it would clearly be desirable to promote PPIs in a cell rather than inhibit them and through this general schema it may be possible to selectively colocalize different molecules in a single cell by varying one component of the bifunctional molecule.
Although the pathophysiological mechanism behind the development of AD has yet to be fully understood, it seems reasonable to hypothesize that amyloid-β protofibril formation is important in the progression of the disease. Several small molecules have been developed that inhibit the oligomerization of amyloid-β either in vitro or in vivo. Although inhibiting amyloid-β aggregation may provide therapeutic benefit on its own, the development of multifactorial agents such as memoquin has the potential to be much more efficacious in terms of treating the underlying disease.
α-Synuclein Aggregation
PD is the second most common neurodegenerative disorder in most Western countries (Elbaz and Tranchant, 2007; Weintraub et al, 2008). This disease is characterized by the loss of dopaminergic neurons in several brain regions, including the substantia nigra pars compacta and other regions important for higher order functioning (Lee and Trojanowski, 2006). Histopathological evaluation of the post-mortem brains of Parkinson's patients has revealed the presence of large intraneuronal aggregates termed ‘Lewy bodies.’ These aggregates are primarily composed of a 140 amino-acid protein, α-synuclein, although they are generally not as homogenous as amyloid-β plaques (Lee and Trojanowski, 2006). It has been shown that overexpression of α-synuclein in several model organisms causes the development of Parkinsonian-like symptoms (Bilen and Bonini, 2005; Feany, 2004; Giasson et al, 2004). Further study of α-synuclein has shown that the protein contains a highly amyloidogenic domain that, when misfolded, oligomerizes and forms a series of self-associating β-pleated sheets that spontaneously form Lewy bodies (Duda et al, 2000; Norris et al, 2004). Like amyloid-β oligomers in AD, it is believed that it is the α-synuclein oligomers and not the fully formed Lewy bodies that are the pathological factor in PD. The current hypothesis states that α-synuclein oligomers are capable of forming membrane pores that disrupt organelle function, leading to cell dysfunction and death (Lee and Trojanowski, 2006).
Several inhibitors of α-synuclein aggregation have been identified (Conway et al, 2001; Norris et al, 2005; Rochet et al, 2004; Savitt et al, 2006; Skovronsky et al, 2006) (Figure 3). An intriguing finding is that catecholamines are capable of inhibiting α-synuclein aggregation (Conway et al, 2001; Norris et al, 2005). This has also been shown in a mouse model of α-synuclein aggregation, where Lewy bodies were dissolved in brain slices by the addition of l-dopa (Li et al, 2004a). It is known that the oxidation state of the catecholamine was important for the activity, whereby the several oxidation products of dopamine are more potent at inhibiting α-synuclein aggregation than the parent neurotransmitter (Li et al, 2004a) (Figure 3). The link, if any, between dopaminergic neuron loss and the ability of catecholamines to inhibit α-synuclein aggregation has yet to be fully understood, but remains an intriguing concept in the pathophysiology of PD.

Examples of small molecule α-synuclein aggregation inhibitors. (a) One member of the series of catechols known to inhibit α-synuclein aggregation (Conway et al, 2001). (b) A noncatechol inhibitor from the same series (Conway et al, 2001). (c) Baicalein, a natural product from the Chinese skullcap (Scutellaraia baicalensis) (Johnston and Brotchie, 2006). (d) The presumed active oxidation product of baicalein that inhibits α-synuclein aggregation (Johnston and Brotchie, 2006). (e) Dopamine, a known inhibitor of α-synuclein aggregation (Conway et al, 2001; Li et al, 2004), and (f) dopaminochrome, one of the oxidation products of dopamine that has anti-aggregation properties (Li et al, 2004).
A series of peptide inhibitors of α-synuclein aggregation were identified by developing a library of overlapping heptapeptides that span the α-synuclein sequence. The active peptides were centered around residues 69–72 of α-synuclein, suggesting that this region of the molecule was important for self-association (Bodles et al, 2004; El-Agnaf et al, 2004). It appears that short peptide fragments of α-syncuclein also occur naturally, as the serine protease neurosin degrades α-synuclein into fragments that are capable of inhibiting α-synuclein polymerization (Iwata et al, 2003). Current work is focused upon developing peptidomimetics and identifying small molecule inhibitors of α-synuclein using both high-throughput screening (HTS) and rational design from the information obtained in the peptide library study (Amer et al, 2006).
Other small molecule inhibitors of α-synuclein have been identified. Rifampicin and several of its derivatives can inhibit both α-synuclein (Li et al, 2004b) and amyloid-β (Tomiyama et al, 1994, 1996) aggregation in a concentration-dependent manner with reasonable potency (<10 μM IC50). A conclusive mechanism of rifampicin action has not been fully elucidated, but it has been suggested that it could act by binding directly to the developing plaque (Tomiyama et al, 1994) and/or by acting as a free radical scavenger (Tomiyama et al, 1996). Panacea Pharmaceuticals had also developed a pair of α-synuclein inhibitors, PAN-408 and PAN-527, that had progressed to preclinical trials, however, there have been no recent reports of compounds with these names (Johnston and Brotchie, 2004, 2006).
Polyphenolic compounds, like flavenoids or Congo red, have been proposed to be α-synuclein aggregation inhibitors (Porat et al, 2006). Many of these compounds are derived from natural sources and have low micromolar IC50 values for protein aggregation inhibition. Baicalein (Figure 3), a flavinoid isolated from the Chinese Skullcap plant (Scutellaraia baicalensis), has been shown to directly bind to a single site on α-synuclein with submicromolar affinity and inhibit oligomerization (Zhu et al, 2004). It is likely that a quinone oxidation product of this compound is responsible for the observed inhibitory activity (Figure 3) (Zhu et al, 2004). Interestingly, this compound has been shown to inhibit α-synuclein aggregate nucleation but not affect fibril elongation, suggesting that the molecule may be acting by stabilizing the monomeric α-synuclein, but not by dissolving aggregates themselves (Zhu et al, 2004). This mechanism could be beneficial, as plaque disruption could generate free protofibrils and lead to increased cellular damage. Circular dichroism confirmed that binding of baicalein stabilized the semifolded state of α-synuclein (Zhu et al, 2004). Unfortunately, it was also found that baicalein is capable of stabilizing an oligomeric species of α-synuclein as well as monomer (Zhu et al, 2004). It is not known whether the oligomeric species stabilized by baicalein has neurodegenerative properties, however this finding does not bode well for the family of polyphenoic compounds as inhibitors of α-synuclein function. It is unfortunately possible that these molecules could stabilize the formation of the protofibrils that, as the current hypothesis states, are the pathogenic factor in protein aggregation diseases.
Protein aggregation diseases are a major cause of morbidity in the elderly population of First World countries. Although there are a number of therapeutics currently in use to treat the symptoms of these diseases (Brunton et al, 2006; Johnston and Brotchie, 2004, 2006; Zhu et al, 2004), there are very few if any actual treatments that stop or reverse disease progression. If the hypothesis that protein oligomers are the primary pathogenic factor in these diseases is correct, then small molecules that prevent or reverse protein oligomerization may provide a mechanism to target the actual cause of the disease. There has been substantial work put forth to develop inhibitors of protein oligomerization and significant progress has been made. There is, however, much more work that needs to be performed in this field before a clinically useful agent will be available for general use.
MODULATING SIGNAL TRANSDUCTION THROUGH INHIBITING PROTEIN PROTEIN INTERACTIONS
Signal transduction cascades are required for nearly all biological functions. The importance of these systems is further illustrated by the fact that a large proportion of all clinically used therapeutics modulate signaling (Brunton et al, 2006). The most common method to modulate information processing through a signal transduction pathway is to alter activity of the most upstream molecule in the system: the receptor. These receptors come in many forms including G-protein-coupled receptors, intracellular steroid/glucocorticoid receptors, and tyrosine kinase-linked receptors. Currently ∼30 to 50% of all clinically used drugs target GPCRs and a substantial portion of the remaining drugs target other receptor systems (Brunton et al, 2006). Although many of these drugs are effective therapeutics, targeting regulation systems or molecules further downstream in the signaling pathway may provide advantages not readily available when solely modulating receptor activity.
Targeting downstream-signaling molecules in a signal transduction pathway requires overcoming several significant hurdles in drug development, including cell permeability of the compound, achieving pathway specificity, and avoiding unwanted or unexpected side effects. There are currently several examples of clinically used drugs or drug candidates that target downstream-signaling molecules in a pathway. The majority of these are kinase inhibitors, exemplified by Gleevec, that inhibit an enzymatic step in a signal transduction cascade (Traxler et al, 2001). As compared to a standard PPI, these enzymes are much more amenable to small molecule targeting due to their possession of a well-defined active site binding pocket. Furthermore, they represent a critical step in the signal transduction pathway that can be selectively inhibited. With all of these qualities, it is easy to understand why a kinase inhibitor could be a useful and safe therapeutic.
Many signal transduction steps do not rely upon an enzymatic process but rather use PPIs to relay information. Targeting these steps requires the development of small molecules that inhibit the PPIs required for signal transduction. Common examples of this are the heterotrimeric G-protein subunits, of which the α and βγ subunits each interact through PPIs with a large number of downstream effectors, including adenylate cyclase, PI3K, PLCβ, voltage-gated Ca2+ channels, GIRK channels, and others. G-proteins, especially Gα subunits, also bind to regulatory proteins that can alter the temporal and spatial-signaling pattern of the subunit. Developing specific inhibitors of various G-protein-effector or G-protein-regulator interactions could provide a mechanism to selectively modulate GPCR-signaling pathways. It is not difficult to imagine several scenarios whereby modulating GPCR signaling could provide significant therapeutic benefit, either by potentiating positive actions of a drug or by inhibiting undesirable side effects.
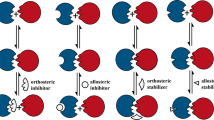
Selective Gβγ Inhibitors
Owing to the prevalence of clinically important drugs that target GPCRs, there has been great interest in the therapeutic modulation of signaling downstream of these receptors. Canonical signaling through GPCRs (Figure 4) progresses through the activation of a receptor by ligand binding, which stimulates the exchange of GDP for GTP by the Gα subunit of a heterotrimeric G protein. The GTP bound Gα and Gβγ subunits of the G protein then dissociate or at least undergo a conformational change to act upon downstream effectors in the signaling pathway. As the first signaling molecule downstream of a GPCR is the G-protein heterotrimer, it has become an interesting target for small molecule inhibition. Although there have been no published reports of a small molecule inhibitor of Gα/effector PPIs, there have been a family of compounds that bind to Gβγ and selectively inhibit it's interactions with downstream effectors (Bonacci et al, 2006; Lehmann et al, 2008; Smrcka et al, 2008). The strategy used to identify these inhibitors provides a clear example of a protocol being used to identify small molecule PPIIs. The first step that the investigators took was to screen a random-peptide phage display library to identify binding sites on Gβγ (Scott et al, 2001). A series of peptides were identified, one of which was capable of inhibiting the Gβγ regulation of PI3K and PLCβ, but not of type I adenylate cyclase or N-type Ca2+ channels, suggesting that effector selectivity may be possible with small molecule modulators of Gβγ activity. Analysis of the crystal structure of Gβ1γ2 bound to this selective peptide inhibitor, it was possible to define the binding pocket into which this peptide docks. By using this site as a binding pocket in virtual screening, the investigators were able to identify 85 small molecules (top 1% in the screen) that were predicted to bind to the Gβγ ‘hotspot’ (Bonacci et al, 2006). Analysis of these compounds using an ELISA assay identified nine actives that inhibited Gβγ binding to their lead peptide with reasonable IC50 values (100 nM–60 μM). One of these compounds, M119 (Figure 5a), was able to inhibit the Gβγ stimulation of PLCβ and PI3Kγ activity in vitro and inhibited calcium release from activation of the Gαi-linked N-formyl peptide receptor in differentiated HL-60 cells. The compound had no inhibitory activity upon the calcium mobilization initiated by carbachol in HEK cells stably expressing the Gαq-linked M3 muscarinic receptor, suggesting that M119 is selective for Gβγ-dependent calcium mobilization. M119 also showed in vivo activity when tested in a morphine antinociception assay in mice. PLCβ3−/− mice have been shown to be 10 times more sensitive to the antinociceptive effects of morphine and an intracerebroventricular injection of M119 recapitulated this augmentation of morphine activity in wild type animals (Bonacci et al, 2006; Xie et al, 1999). As opioid receptors have many Gβγ-dependent functions, the fact that M119 potentiates morphine-induced antinociception instead of inhibiting it provides evidence that this compound is not globally inhibiting Gβγ activity. Another structurally distinct compound identified by this approach, M201, also showed an interesting selectivity profile in its ability to inhibit Gβγ–effector interactions. This compound showed no ability to inhibit PLCβ2 activity, potentiated PLCβ3 and PI3K activity, and inhibited GRK2 binding. The discovery of effector-selective inhibitors of Gβγ signaling M119 and M201, has thus provided a clear example of how targeting downstream-signaling molecules can be a viable approach to modulating the pharmacological properties of a common drug (eg morphine).

Canonical G-protein-signaling pathway. Note that the hydrolysis of GTP by Gα is accelerated by GTPase-activating proteins (GAPs) such as members of the RGS family.

(a) Structure of M119, a pathway-selective inhibitor of Gβγ signaling (Bonacci et al, 2006). (b) Crystal structure of Gβγ bound to peptide SIGK. This peptide, identified by phage display, binds to the identified ‘hot spot’ on Gβγ (Davis et al, 2005). Structure from PDB ID 1XHM.
Potentiating GPCR Signaling by Inhibiting RGS Proteins
Another approach to modulating GPCR signaling downstream of the receptor is to modulate the activity of the key proteins that regulate signal transduction. The G-protein pathways are regulated by a number of proteins, including a family of GTPase accelerating proteins (GAP), the regulators of G-protein signaling (RGS). These proteins bind directly to the Gα subunit and accelerate the intrinsic hydrolysis rate of GTP (Figure 4) (Hollinger and Hepler, 2002; Tesmer et al, 1997; Zhong and Neubig, 2001). The discovery of these proteins provided a solution to the paradox of how rapid regulation of GPCR signaling could occur given the slow intrinsic rate of GTP hydrolysis by purified Gα subunits. They explain the subsecond regulation of G-protein signals observed in excitable cells (Garzon et al, 2004; Neubig and Siderovski, 2002; Tesmer et al, 1997; Zhong and Neubig, 2001) and they can strongly inhibit cellular responses (Bodenstein et al, 2007; Clark et al, 2003; Fu et al, 2006). There are over 20 identified RGS proteins that interact with limited selectivity for all Gα subtypes (Neubig and Siderovski, 2002; Siderovski and Willard, 2005; Tosetti, 2004; Woulfe and Stadel, 1999; Zhong and Neubig, 2001). The only exception to this is Gαs, for which no RGS interaction has been confirmed. There are increasing reports of RGS selectivity for specific GPCR signaling, suggesting that targeting an RGS may provide a mechanism to selectively augment signaling through a particular GPCR (Bernstein et al, 2004; Hague et al, 2005). It is also being increasingly appreciated that RGS proteins are very heterogeneously expressed throughout the body, including in individual neuron tracks in given brain regions (Doupnik et al, 2001; Grafstein-Dunn et al, 2001; Nunn et al, 2006). The distinct expression patterns, presumed GPCR selectivity, and the dependence on an active-signaling pathway for function all suggest that small molecules that modulate RGS activity could be potentially useful therapeutics. Indeed, it has been found that mice expressing a mutated (G184S) form of Gαi2 or Gαo that render these G proteins insensitive to RGS effects exhibit markedly enhanced potency of agonists and substantial physiological phenotypes (Fu et al, 2004; Huang et al, 2006, 2008; Lan et al, 1998). Specifically, mice with the Gαi2 G184S mutation knocked-in show reduced fat mass and resistance to high-fat diet, possibly due to CNS actions (Huang et al, 2008). They also show behaviors consistent with enhanced 5HT1a signaling and a spontaneously antidepressant-like state as well as 10-fold increased potency of 5HT-based antidepressant drugs (Talbot JN et al, submitted for publication). Mice with the RGS-insensitive mutant Gαo show increased antiepileptiform activity in hippocampal slices by α2a agonists (Doze VA et al, in preparation). Strikingly, the effects are quite specific where the 5HT1a potentiation is only seen for antidepressant-like and not for hypothermia effects (Talbot JN et al, submitted for publication).
These data suggest that RGS inhibitors could be of significant interest therapeutically. Given alone, an RGS inhibitor would be expected to accentuate signaling initiated by endogenous ligands, a treatment that could be useful in a variety of neurological conditions such as depression (by enhancement of serotonin signaling), early stage Alzheimer's or PDs (by enhancement of cholinergic or dopaminergic signaling, respectively). They could also be used as an adjunct with a GPCR agonist. An RGS inhibitor could increase the potency or selectivity of the drug by accentuating signal transduction through the receptor. One could imagine that an RGS9 inhibitor that accentuated dopaminergic signaling selectively in the striatum where RGS9 is expressed could be a useful adjunctive therapy with l-dopa or synthetic dopamine agonists in PD (Figure 6). Furthermore, an RGS inhibitor that selectively accentuated opioid signaling in neurons in the pain pathway may provide a mechanism to selectively increase the analgesic properties of opioids but might leave alone the undesirable actions of these drugs (ie constipation, abuse liability) which might be regulated by different RGS proteins. Thus, RGS inhibitors could serve as GPCR agonist potentiators but would also enhance agonist specificity in a cell-type or pathway-specific manner.

RGS inhibitors increase the tissue specificity of an agonist. (Left) Graphical representation of the RGS4 and RGS9 protein expression patterns in the brain (Gold et al, 1997). The red/green color denotes regions of high expression, specifically cortex and thalamic regions for RGS4, and basal ganglia for RGS9. (Center) Upon addition of a low or high concentration of agonist, a response would be seen across all brain regions that express the receptor. (Right) In the presence of an RGS inhibitor, there would be tissue-specific enhancement of agonist effect in the tissues where the RGS is expressed.
RGS4 is a prototypical RGS protein that is widely expressed throughout the CNS but has limited expression in peripheral tissues. It controls a variety of signaling systems, and has been implicated as a risk factor for schizophrenia (Bakker et al, 2007; Bowden et al, 2007; Campbell et al, 2008; Chowdari et al, 2002, 2008; Gu et al, 2007; Guo et al, 2006; Ishiguro et al, 2007; Lane et al, 2008; Lang et al, 2007; Levitt et al, 2006; Li and He, 2006; Mirnics et al, 2001; Morris et al, 2004; Nicodemus et al, 2007, 2008; O’Tuathaigh et al, 2007; Sanders et al, 2008; So et al, 2008; Sutrala et al, 2007; Talkowski et al, 2006; Vilella et al, 2008; Williams et al, 2004; Wood et al, 2007). RGS4, like other RGS proteins interacts strongly with several members of the Gαi/o and Gαq families and shows limited selectivity between these proteins in in vitro binding and functional studies (Roman et al, 2007; Roof et al, 2006; Tesmer et al, 1997; Zhong et al, 2003). The interaction interface between RGS4 and Gαi1 has been clearly mapped in the crystal structure by Tesmer et al (1997). This relatively flat interface is comprised of the three switch regions in Gαi1 (residues 179–185 in switch 1, 204–213 in switch 2, and 235–237 in switch 3) and several of the core helices in the RGS box domain of RGS4 (Tesmer et al, 1997). The interaction interface covers approximately 1100 Å2 and is composed primarily of van der Waals and hydrogen bond interactions (Figure 7). The binding affinity of the Gα/RGS complex is dependent upon the nucleotide bound to the Gα. There is very little to no interaction when Gα is in the GDP-bound state, some affinity when bound with GTP, and a strong interaction (∼1 to 5 nM) when the Gα is bound to GDP-aluminum fluoride, a transition state mimic (Berman et al, 1996; Roman et al, 2007).

(a) Crystal structure of RGS4 (blue ribbon) in complex with Gαi1 (red surface). This stereotypical protein–protein interaction buries approximately 1100 Å2 and is relatively featureless in terms of readily identifiable small molecule binding sites. (b) Structure of RGS4 that has been rotated to display the regions of the surface (red) that lies within 5 Å of Gαi1. Note the large diffuse contact interface. Structures from PDB ID 1AGR (Tesmer et al, 1997).
There has been significant interest in the literature regarding the development of RGS inhibitors (Burchett, 2005; Neubig, 2002; Neubig and Siderovski, 2002; Riddle et al, 2005; Traynor and Neubig, 2005; Zhong and Neubig, 2001). Two groups have published independent series of peptide inhibitors of RGS function. One series was rationally designed to mimic the switch 1 region of Gαi and expanded by screening of a constrained peptide library (Jin et al, 2004; Roof et al, 2006, 2008). The other peptide inhibitor series was developed by a random yeast two-hybrid screening campaign (Wang et al, 2008). The lead peptide from this latter campaign bears no resemblance to the sequence of any known RGS4 interacting protein and it's mechanism of action is unclear. Both of these series produced lead peptides with modest (mid–low micromolar) activity in both binding and functional assays, suggesting that small molecule inhibition of RGS function may be more tractable than previously thought (Roof et al, 2006; Wang et al, 2008). The first small molecule inhibitor of RGS4 function was published in 2007 (Roman et al, 2007). This compound, CCG-4986 (4-chloro-N-(N-(4-nitrophenyl)methoxysulfanyl)benzene-1-sulfonamide), was identified through a flow-cytometry protein interaction assay (FCPIA) based high-throughput screening campaign from a random compound library. This compound was shown to have a single digit micromolar IC50 value for the inhibition of RGS4 binding to Gαo and showed significant selectivity for RGS4 over its closest relative based upon sequence homology, RGS8. The activity of the compound was confirmed by single turnover GTP hydrolysis assays, which showed that CCG-4986 was capable of blocking the RGS-stimulated increase in GTP hydrolysis rate by Gαo. The compound bound directly to RGS4 as determined by changes in intrinsic fluorescence of the RGS upon compound binding. Further study of the mechanism of CCG-4986 action showed that it did not function in a cellular environment and it was determined that the compound irreversibly formed a covalent adduct with the RGS in both orthosteric (ie at the site of Gα binding) (Kimple et al, 2007) (Roman DL, in preparation) and allosteric interaction sites (Roman DL, in preparation). Although this limits the utility of the compound as a pharmacological tool, the development of CCG-4986 is nonetheless exciting, as it clearly shows that RGS proteins are susceptible to small molecule inhibition and also to allosteric modulation which may provide greater specificity among RGS proteins. Current efforts toward developing small molecule RGS inhibitors using high-throughput screening and rational design approaches are ongoing. Hopefully, specific, reversible small molecule inhibitors of RGS proteins will be available in the not too distant future.
Although RGS inhibitors have the potential to accentuate signaling through GPCRs, it is also possible to imagine a scenario where augmenting RGS activity with ‘RGS agonists’ could be therapeutically beneficial. RGS4 has been shown to be negatively regulated by acidic phospholipids (Ishii et al, 2005a, 2005b; Popov et al, 2000). This inhibition can be reversed by the binding of calmodulin to what is presumed to be the same site (Ishii et al, 2005a, 2005b; Popov et al, 2000). The binding site for these molecules is distinct from the Gα binding site and functions to allosterically inhibit the GAP activity of RGS4 (Ishii et al, 2005b; Zhong and Neubig, 2001). A small molecule that inhibits the interaction between RGS4 and acidic phospholipids and/or calmodulin could provide a mechanism to ‘activate’ RGS4. By activating RGS4, it may be possible to attenuate aberrant or overactive GPCR neurotransmitter receptors. There is another approach by which the development of agonists at some RGS proteins may be possible. Phosphodiesterase-γ (PDEγ) has been shown to selectively potentiate the GTPase accelerating activity of RGS9 but not RGS4, 16, or 19. It does this in part by forming a ternary complex with Gα and the RGS9/Gβ5 complex (Natochin et al, 1997; Nekrasova et al, 1997; Slep et al, 2001; Wieland et al, 1997). It may be possible to develop compounds that mimic PDEγ or alter its ability to bind to the Gα–RGS complex providing a novel mechanism to enhance RGS activity.
Whether the goal is to produce RGS inhibitors or RGS activators, there are clear challenges as most actions do require alterations in PPIs. However, small molecules that regulate RGS function would provide novel approach to the treatment of diseases stemming from or benefited by changes in GPCR signaling.
Inhibition of PDZ Interactions
PDZ domains are important scaffolding components in many signaling systems, with an extensive role in the development and maintenance of both pre- and postsynaptic structures (Garner et al, 2000; Jelen et al, 2003). Development of reversible small molecule inhibitors that target neuron-specific PDZ domains would provide useful tools to probe the many functions of these important scaffolds (Lamprecht and Seidler, 2006). Of all canonical PPIs, PDZ domains are possibly the most similar to a traditional ligand–receptor interaction, as the interaction interface is comprised of a groove on the PDZ domain binding to the last few (3–5) amino-acid residues in its partner (Figure 8a) (Jelen et al, 2003). The small interaction interface requires that the few amino acids compromising the PDZ ligand contribute a great deal to the energetics of binding. Having such a small PPI interface might suggest that these interactions would be amenable to small molecule disruption. To this end, there have been a few PDZ inhibitors described based either upon rational design or from random high-throughput screening (Figure 8b) (Chen et al, 2007; Fujii et al, 2003, 2007a, 2007b, 2007c; Hammond et al, 2006; Mayasundari et al, 2008; Shan et al, 2005). Rational design of PDZ inhibitors would appear to be relatively straightforward, as the PDZ ligand is comprised of so few residues and the binding pockets of many PDZ domains have been characterized structurally by NMR or crystallography. Indeed, several peptidomimetic scaffolds have been developed that inhibit PDZ interactions (Figure 8b) (Fujii et al, 2007a, 2003; Hammond et al, 2006; Mayasundari et al, 2008).

(a) Crystal structure of the first PDZ domain from MAGI (surface) bound to the PDZ ligand of HPV18 E6 (ball and stick). Note how in this protein–protein interaction, only a few (generally 3–5) residues are important in the binding energetics. Structure from PDB ID 2I04 (Zhang et al, 2007). (b) Examples of small molecule PDZ inhibitors. (i) General scaffold for a wide array of PDZ domains (Fujii et al, 2003, 2007a, 2007b). Analogs of this structure have been shown to inhibit the second PDZ domain of NHERF1 (Fujii et al, 2007b). (ii) β-Hairpin peptidomimetic developed to inhibit the α1-syntrophin PDZ domain (Hammond et al, 2006). (iii) Peptidomimetic developed to inhibit the NHERF1 PDZ domains (Mayasundari et al, 2008).
Cell permeant small molecule inhibitors of PDZ domains will provide a mechanism with which to probe the complex functions of these scaffolding proteins. For example, the Na+/H+ exchanger regulatory factor 1 (NHERF1) contains two PDZ domains and has been shown to have altered expression in many cancers (Cardone et al, 2007; Kreimann et al, 2007; Pan et al, 2006; Shibata et al, 2003; Voltz et al, 2001). The role of NHERF1 in cancer is complicated and appears to be dependent upon cellular context. Outside the realm of oncology, NHERF1 has been shown to be a multifunctional scaffolding protein that is capable of regulating the trafficking and localization of many membrane associated proteins (Shenolikar and Weinman, 2001). Clearly, a tool which would allow for the acute and reversible inhibition of NHERF1 could provide a powerful mechanism with which to determine the physiological role of this protein in different cellular contexts.
Currently, the best-defined PDZ inhibitors are directed against the disheveled and the NHERF1 PDZ domains. These compounds were originally designed as a treatment for β-catenin-dependent tumor growth or to study the controversial role of NHERF1 in cancer progression, respectively. Although these compounds have limited utility as centrally acting agents, they provide a clear example of how a PDZ inhibitor could be developed for a more centrally important PDZ domain containing proteins, of which there are many (for reviews, see Collins and Grant, 2007; Dev et al, 2003; Garner et al, 2000; Jelen et al, 2003; Kennedy, 1998; Nagano et al, 1998; O’Brien et al, 1998; Sarrouilhe et al, 2006).
The first cell permeable PDZ inhibitor was developed by Fujii et al (2003). This irreversible inhibitor was rationally designed to bind to the second PDZ domain of MAGI (Fujii et al, 2003). The compound dose-dependently (IC50 ∼10 to 30 μM) inhibited the binding of a peptide corresponding to the PDZ ligand of the lipid phosphatase PTEN to the membrane-associated MAGI protein in a fluorescence polarization assay. It also increased the activity of PKB (or Akt) in cells, consistent with increased phosphatidyl inositol 3,4,5-trisphosphate levels due to reduced PTEN recruitment to the membrane (Fujii et al, 2003). Eventually, this indole scaffold was developed into a reversible, albeit weak (IC50 ∼1 mM), inhibitor of the second PDZ domain of MAGI (Fujii et al, 2007a). A similar indole scaffold was used to develop an inhibitor of the disheveled PDZ domain, an important scaffold in the Wnt/β-catenin pathway (Fujii et al, 2007c). This compound, named FJ9, blocked the interaction between the PDZ ligand at the C-terminal of the 7TM receptor Frizzled 7 with the disheveled PDZ domain both in vitro and in cells (in vitro IC50 30–60 μM). It also suppressed the growth of tumor cells in a β-catenin-dependent manner (Fujii et al, 2007c). Another inhibitor of the disheveled PDZ domain has been described. This relatively weak (∼200 μM IC50) inhibitor was identified in a virtual screen against the disheveled PDZ domain and was capable of inhibiting Wnt signaling in a zebrafish embryo model of Wnt signaling (Shan et al, 2005). HTS attempts to develop PDZ inhibitors of the MINT1 PDZ domain have been published, but to date there have been no published structures of a selective MINT1 PDZ inhibitor (Chen et al, 2007). Inhibition of PDZ domains has the potential to provide very useful pharmacologic tools for the study of protein trafficking, synaptic function, and other scaffolding-dependent processes. Although current compounds still have only modest affinities, a selective inhibitor of some particular PDZ domains may also provide useful therapeutic agents, although this hypothesis needs to be tested.
CONCLUSIONS
The examples described in this chapter provide a view of current approaches to the development of PPIIs. This process often starts with the characterization of the protein interaction interface, either through crystallographic methods or by alanine scanning to identify interaction ‘hot spots.’ Then, peptide inhibitors of the interaction are developed using either a rational or screening approach. With the information and assay tools obtained from the peptide studies, small molecule peptidomimetics can be designed or high-throughput random chemical screening can be performed to identify small, ‘drug-like’ inhibitors. This general method has provided the most consistent chance of success in the development of PPIIs. In some cases (eg the Gβγ inhibitors) there is sufficient potency and cellular activity to demonstrate CNS actions, however, even in that case, blood–brain barrier penetration was not achieved.
FUTURE RESEARCH DIRECTIONS
It is clear that substantial challenges lie ahead in the development of PPI modulators for CNS therapeutics. Continued progress is being made in the cancer arena where the requirements of oral absorption and/or blood–brain barrier penetration are less critical. Indeed, the affinity of PPIIs in the cancer field has increased (to IC50 values <10 nM in vitro and 10–100 s of nM in cells) and molecular weight has decreased over the past 10 years. That work and continued academic efforts to develop tool compounds blocking PPIs of a range of CNS targets should advance this field substantially. The need to maintain small size and appropriate physicochemical properties of compounds to access the CNS may require novel approaches. Rather than targeting the immediate PPI site, the identifying and targeting of allosteric sites on the target protein may permit the use of more suitable chemical structures. In the cancer drug design arena, fragment-based design of PPIIs has proven useful so applications to CNS targets should be pursued. Also, identifying compounds that could make use of neuronal uptake mechanisms could permit compounds to achieve higher intracellular concentrations and result in additional cell-type specificity of action.
There are a large number of potential CNS PPI targets that could provide subtle modulation of neural signaling if a successful drug could be developed. As with the history of protein kinase inhibitors, overcoming initial reluctance to embrace the concept will likely require a success story. At present, it is hard to predict a major breakthrough in this field but continued refinement of existing approaches and further development of existing targets is likely to reach a threshold of success in the not too distant future. In the meantime, substantial genetic and biological studies will continue to define novel PPI targets. With the rapid advances in target identification, an increased pace of chemical discovery related to PPIs will be critical to exploit the potential of this novel field.
References
Aisen PS (2005). The development of anti-amyloid therapy for Alzheimer's disease: from secretase modulators to polymerisation inhibitors. CNS Drugs 19: 989–996.
Aisen PS, Gauthier S, Vellas B, Briand R, Saumier D, Laurin J et al (2007). Alzhemed: a potential treatment for Alzheimer's disease. Curr Alzheimer Res 4: 473–478.
Amer DA, Irvine GB, El-Agnaf OM (2006). Inhibitors of alpha-synuclein oligomerization and toxicity: a future therapeutic strategy for Parkinson's disease and related disorders. Exp Brain Res 173: 223–233.
Arkin MR, Wells JA (2004). Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat Rev Drug Discov 3: 301–317.
Bakker SC, Hoogendoorn ML, Hendriks J, Verzijlbergen K, Caron S, Verduijn W et al (2007). The PIP5K2A and RGS4 genes are differentially associated with deficit and non-deficit schizophrenia. Genes Brain Behav 6: 113–119.
Berman DM, Kozasa T, Gilman AG (1996). The GTPase-activating protein RGS4 stabilizes the transition state for nucleotide hydrolysis. J Biol Chem 271: 27209–27212.
Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI et al (2004). RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem 279: 21248–21256.
Bilen J, Bonini NM (2005). Drosophila as a model for human neurodegenerative diseases. Annu Rev Genet 39: 153–171.
Bodenstein J, Sunahara RK, Neubig RR (2007). N-terminal residues control proteasomal degradation of RGS2, RGS4, and RGS5 in human embryonic kidney 293 cells. Mol Pharmacol 71: 1040–1050.
Bodles AM, El-Agnaf OM, Greer B, Guthrie DJ, Irvine GB (2004). Inhibition of fibril formation and toxicity of a fragment of alpha-synuclein by an N-methylated peptide analogue. Neurosci Lett 359: 89–93.
Bogan AA, Thorn KS (1998). Anatomy of hot spots in protein interfaces. J Mol Biol 280: 1–9. This paper describes the “hot spot” theory in protein-protein interactions.
Bolognesi ML, Andrisano V, Bartolini M, Banzi R, Melchiorre C (2005). Propidium-based polyamine ligands as potent inhibitors of acetylcholinesterase and acetylcholinesterase-induced amyloid-beta aggregation. J Med Chem 48: 24–27.
Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D et al (2006). Differential targeting of Gbetagamma-subunit signaling with small molecules. Science 312: 443–446. This paper outlines the development of small molecule Gβγ inhibitors. It shows a complete development pathway from virtual screening based on protein structure to in vivo data demonstrating analgesic actions upon i.c.v. administration. It also reveals the capability of small molecule inhibitors to selectively disrupt specific signaling outputs from a protein messenger.
Bowden NA, Scott RJ, Tooney PA (2007). Altered expression of regulator of G-protein signalling 4 (RGS4) mRNA in the superior temporal gyrus in schizophrenia. Schizophr Res 89: 165–168.
Bragin V, Chemodanova M, Dzhafarova N, Bragin I, Czerniawski JL, Aliev G (2005). Integrated treatment approach improves cognitive function in demented and clinically depressed patients. Am J Alzheimers Dis Other Demen 20: 21–26.
Brunton LL, Lazo JS, Parker KL (eds) (2006). Goodman & Gilman's: The Pharmacological Basis of Therapeutics. McGraw-Hill Companies, Inc: New York.
Burchett SA (2005). Psychostimulants, madness, memory … and RGS proteins? Neuromolecular Med 7: 101–127.
Campbell DB, Ebert PJ, Skelly T, Stroup TS, Lieberman J, Levitt P et al (2008). Ethnic stratification of the association of RGS4 variants with antipsychotic treatment response in schizophrenia. Biol Psychiatry 63: 32–41.
Capsoni S, Cattaneo A (2006). On the molecular basis linking Nerve Growth Factor (NGF) to Alzheimer's disease. Cell Mol Neurobiol 26: 619–633.
Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A (2000). Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci USA 97: 6826–6831.
Cardone RA, Bellizzi A, Busco G, Weinman EJ, Dell’Aquila ME, Casavola V et al (2007). The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol Biol Cell 18: 1768–1780.
Cavalli A, Bolognesi ML, Capsoni S, Andrisano V, Bartolini M, Margotti E et al (2007). A small molecule targeting the multifactorial nature of Alzheimer's disease. Angew Chem Int Ed Engl 46: 3689–3692. This article describes the effects of memoquin in the treatment of Alzheimer's disease.
Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M et al (2008). Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 51: 347–372.
Chen X, Longgood JC, Michnoff C, Wei S, Frantz DE, Bezprozvanny L (2007). High-throughput screen for small molecule inhibitors of Mint1-PDZ domains. Assay Drug Dev Technol 5: 769–783.
Chowdari KV, Bamne M, Wood J, Talkowski ME, Mirnics K, Levitt P et al (2008). Linkage disequilibrium patterns and functional analysis of RGS4 polymorphisms in relation to schizophrenia. Schizophr Bull 34: 118–126.
Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T et al (2002). Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet 11: 1373–1380.
Clark MJ, Harrison C, Zhong H, Neubig RR, Traynor JR (2003). Endogenous RGS protein action modulates mu-opioid signaling through Galphao. Effects on adenylyl cyclase, extracellular signal-regulated kinases, and intracellular calcium pathways. J Biol Chem 278: 9418–9425.
Collins MO, Grant SG (2007). Supramolecular signalling complexes in the nervous system. Subcell Biochem 43: 185–207.
Conway KA, Rochet JC, Bieganski RM, Lansbury Jr PT (2001). Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294: 1346–1349. This paper describes a series of of catechol and non-catechol compounds that inhibit α-synuclein aggregation.
Darnell SJ, LeGault L, Mitchell JC (2008). KFC Server: interactive forecasting of protein interaction hot spots. Nucleic Acids Res 36 (Web Server issue): W265–W269.
Dev KK, van der Putten H, Sommer B, Rovelli G (2003). Part I: parkin-associated proteins and Parkinson's disease. Neuropharmacology 45: 1–13.
Dickson DW, Rogers J (1992). Neuroimmunology of Alzheimer's disease: a conference report. Neurobiol Aging 13: 793–798.
Doupnik CA, Xu T, Shinaman JM (2001). Profile of RGS expression in single rat atrial myocytes. Biochim Biophys Acta 1522: 97–107.
Duda JE, Lee VM, Trojanowski JQ (2000). Neuropathology of synuclein aggregates. J Neurosci Res 61: 121–127.
El-Agnaf OM, Paleologou KE, Greer B, Abogrein AM, King JE, Salem SA et al (2004). A strategy for designing inhibitors of alpha-synuclein aggregation and toxicity as a novel treatment for Parkinson's disease and related disorders. FASEB J 18: 1315–1317.
Elbaz A, Tranchant C (2007). Epidemiologic studies of environmental exposures in Parkinson's disease. J Neurol Sci 262: 37–44.
Feany MB (2004). New genetic insights into Parkinson's disease. N Engl J Med 351: 1937–1940.
Fischer TB, Arunachalam KV, Bailey D, Mangual V, Bakhru S, Russo R et al (2003). The binding interface database (BID): a compilation of amino acid hot spots in protein interfaces. Bioinformatics 19: 1453–1454.
Fotouhi N, Graves B (2005). Small molecule inhibitors of p53/MDM2 interaction. Curr Top Med Chem 5: 159–165.
Fu Y, Huang X, Zhong H, Mortensen RM, D’Alecy LG, Neubig RR (2006). Endogenous RGS proteins and Galpha subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ Res 98: 659–666.
Fu Y, Zhong H, Nanamori M, Mortensen RM, Huang X, Lan K et al (2004). RGS-insensitive G-protein mutations to study the role of endogenous RGS proteins. Methods Enzymol 389: 229–243.
Fujii N, Haresco JJ, Novak KA, Gage RM, Pedemonte N, Stokoe D et al (2007a). Rational design of a nonpeptide general chemical scaffold for reversible inhibition of PDZ domain interactions. Bioorg Med Chem Lett 17: 549–552.
Fujii N, Haresco JJ, Novak KA, Stokoe D, Kuntz ID, Guy RK (2003). A selective irreversible inhibitor targeting a PDZ protein interaction domain. J Am Chem Soc 125: 12074–12075. This paper, and subsequent papers by Fujii, et al. , provide an example of a rationally developed series of PDZ domain inhibitors.
Fujii N, Shelat A, Hall RA, Guy RK (2007b). Design of a selective chemical probe for class I PDZ domains. Bioorg Med Chem Lett 17: 546–548.
Fujii N, You L, Xu Z, Uematsu K, Shan J, He B et al (2007c). An antagonist of dishevelled protein–protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res 67: 573–579.
Garner CC, Nash J, Huganir RL (2000). PDZ domains in synapse assembly and signalling. Trends Cell Biol 10: 274–280.
Garzon J, Rodriguez-Munoz M, Lopez-Fando A, Garcia-Espana A, Sanchez-Blazquez P (2004). RGSZ1 and GAIP regulate mu- but not delta-opioid receptors in mouse CNS: role in tachyphylaxis and acute tolerance. Neuropsychopharmacology 29: 1091–1104.
Gestwicki JE, Crabtree GR, Graef IA (2004). Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science 306: 865–869. This article provides a novel approach to modulating protein-protein interactions that takes advantage of intracellular proteins to improve the activity of the small molecule.
Gestwicki JE, Marinec PS (2007). Chemical control over protein–protein interactions: beyond inhibitors. Comb Chem High Throughput Screen 10: 667–675.
Giasson BI, Lee VM-Y, Trojanowski JQ (2004). Animal models of neurodegenerative dementing disorders other than Alzheimer's disease. Clin Neurosci Res 3: 427–436.
Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E et al (2003). ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19: 163–164.
Grafstein-Dunn E, Young KH, Cockett MI, Khawaja XZ (2001). Regional distribution of regulators of G-protein signaling (RGS) 1, 2, 13, 14, 16, and GAIP messenger ribonucleic acids by in situ hybridization in rat brain. Brain Res Mol Brain Res 88: 113–123.
Gu Z, Jiang Q, Yan Z (2007). RGS4 modulates serotonin signaling in prefrontal cortex and links to serotonin dysfunction in a rat model of schizophrenia. Mol Pharmacol 71: 1030–1039.
Guo S, Tang W, Shi Y, Huang K, Xi Z, Xu Y et al (2006). RGS4 polymorphisms and risk of schizophrenia: an association study in Han Chinese plus meta-analysis. Neurosci Lett 406: 122–127.
Hague C, Bernstein LS, Ramineni S, Chen Z, Minneman KP, Hepler JR (2005). Selective inhibition of alpha1A-adrenergic receptor signaling by RGS2 association with the receptor third intracellular loop. J Biol Chem 280: 27289–27295.
Hajduk PJ, Burns DJ (2002). Integration of NMR and high-throughput screening. Comb Chem High Throughput Screen 5: 613–621.
Hammond MC, Harris BZ, Lim WA, Bartlett PA (2006). Beta strand peptidomimetics as potent PDZ domain ligands. Chem Biol 13: 1247–1251.
Hébert L (2007). Neurochem Announces Results from Tramiprosate (ALZHEMED(TM)) North American Phase III Clinical Trial. Neurochem Inc..
Heiser V, Engemann S, Brocker W, Dunkel I, Boeddrich A, Waelter S et al (2002). Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington's disease by using an automated filter retardation assay. Proc Natl Acad Sci USA 99 (Suppl 4): 16400–16406.
Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N et al (2000). Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington's disease therapy. Proc Natl Acad Sci USA 97: 6739–6744.
Hollinger S, Hepler JR (2002). Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev 54: 527–559.
Huang X, Charbeneau RA, Fu Y, Kaur K, Gerin I, MacDougald OA et al (2008). Resistance to diet-induced obesity and improved insulin sensitivity in mice with a regulator of G protein signaling-insensitive G184S Gnai2 allele. Diabetes 57: 77–85.
Huang X, Fu Y, Charbeneau RA, Saunders TL, Taylor DK, Hankenson KD et al (2006). Pleiotropic phenotype of a genomic knock-in of an RGS-insensitive G184S Gnai2 allele. Mol Cell Biol 26: 6870–6879.
Ishiguro H, Horiuchi Y, Koga M, Inada T, Iwata N, Ozaki N et al (2007). RGS4 is not a susceptibility gene for schizophrenia in Japanese: association study in a large case–control population. Schizophr Res 89: 161–164.
Ishii M, Fujita S, Yamada M, Hosaka Y, Kurachi Y (2005a). Phosphatidylinositol 3,4,5-trisphosphate and Ca2+/calmodulin competitively bind to the regulators of G-protein-signalling (RGS) domain of RGS4 and reciprocally regulate its action. Biochem J 385 (Part 1): 65–73.
Ishii M, Ikushima M, Kurachi Y (2005b). In vivo interaction between RGS4 and calmodulin visualized with FRET techniques: possible involvement of lipid raft. Biochem Biophys Res Commun 338: 839–846.
Iwata A, Maruyama M, Akagi T, Hashikawa T, Kanazawa I, Tsuji S et al (2003). Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum Mol Genet 12: 2625–2635.
Jelen F, Oleksy A, Smietana K, Otlewski J (2003). PDZ domains—common players in the cell signaling. Acta Biochim Pol 50: 985–1017.
Jin Y, Zhong H, Omnaas JR, Neubig RR, Mosberg HI (2004). Structure-based design, synthesis, and activity of peptide inhibitors of RGS4 GAP activity. Methods Enzymol 389: 266–277.
Johnston TH, Brotchie JM (2004). Drugs in development for Parkinson's disease. Curr Opin Investig Drugs 5: 720–726.
Johnston TH, Brotchie JM (2006). Drugs in development for Parkinson's disease: an update. Curr Opin Investig Drugs 7: 25–32.
Kennedy MB (1998). Signal transduction molecules at the glutamatergic postsynaptic membrane. Brain Res Brain Res Rev 26: 243–257.
Kimple AJ, Willard FS, Giguere PM, Johnston CA, Mocanu V, Siderovski DP (2007). The RGS protein inhibitor CCG-4986 is a covalent modifier of the RGS4 Galpha-interaction face. Biochim Biophys Acta 1774: 1213–1220.
Kortemme T, Kim DE, Baker D (2004). Computational alanine scanning of protein–protein interfaces. Sci STKE 2004: pl2.
Kreimann EL, Morales FC, de Orbeta-Cruz J, Takahashi Y, Adams H, Liu TJ et al (2007). Cortical stabilization of beta-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene 26: 5290–5299.
Lamprecht G, Seidler U (2006). The emerging role of PDZ adapter proteins for regulation of intestinal ion transport. Am J Physiol Gastrointest Liver Physiol 291: G766–G777.
Lan KL, Sarvazyan NA, Taussig R, Mackenzie RG, DiBello PR, Dohlman HG et al (1998). A point mutation in Galphao and Galphai1 blocks interaction with regulator of G protein signaling proteins. J Biol Chem 273: 12794–12797.
Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T et al (2005). ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33 (Web Server issue): W299–W302.
Lane HY, Liu YC, Huang CL, Chang YC, Wu PL, Huang CH et al (2008). RGS4 polymorphisms predict clinical manifestations and responses to risperidone treatment in patients with schizophrenia. J Clin Psychopharmacol 28: 64–68.
Lang UE, Puls I, Muller DJ, Strutz-Seebohm N, Gallinat J (2007). Molecular mechanisms of schizophrenia. Cell Physiol Biochem 20: 687–702.
Lansbury PT, Lashuel HA (2006). A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443: 774–779.
Laurie NA, Schin-Shih C, Dyer MA (2007). Targeting MDM2 and MDMX in retinoblastoma. Curr Cancer Drug Targets 7: 689–695.
Lee VM, Trojanowski JQ (2006). Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52: 33–38.
Lehmann DM, Seneviratne AM, Smrcka AV (2008). Small molecule disruption of G protein beta gamma subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol 73: 410–418.
Levitt P, Ebert P, Mirnics K, Nimgaonkar VL, Lewis DA (2006). Making the case for a candidate vulnerability gene in schizophrenia: convergent evidence for regulator of G-protein signaling 4 (RGS4). Biol Psychiatry 60: 534–537.
Li D, He L (2006). Association study of the G-protein signaling 4 (RGS4) and proline dehydrogenase (PRODH) genes with schizophrenia: a meta-analysis. Eur J Hum Genet 14: 1130–1135.
Li J, Zhu M, Manning-Bog AB, Di Monte DA, Fink AL (2004a). Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson's and Alzheimer's disease. FASEB J 18: 962–964.
Li J, Zhu M, Rajamani S, Uversky VN, Fink AL (2004b). Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem Biol 11: 1513–1521.
Mayasundari A, Ferreira AM, He L, Mahindroo N, Bashford D, Fujii N (2008). Rational design of the first small-molecule antagonists of NHERF1/EBP50 PDZ domains. Bioorg Med Chem Lett 18: 942–945.
Melchiorre C, Andrisano V, Bolognesi ML, Budriesi R, Cavalli A, Cavrini V et al (1998). Acetylcholinesterase noncovalent inhibitors based on a polyamine backbone for potential use against Alzheimer's disease. J Med Chem 41: 4186–4189.
Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P (2001). Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry 6: 293–301.
Morris DW, Rodgers A, McGhee KA, Schwaiger S, Scully P, Quinn J et al (2004). Confirming RGS4 as a susceptibility gene for schizophrenia. Am J Med Genet B Neuropsychiatr Genet 125B: 50–53.
Morrison RS, Kinoshita Y, Johnson MD, Ghatan S, Ho JT, Garden G (2002). Neuronal survival and cell death signaling pathways. Adv Exp Med Biol 513: 41–86.
Nagano T, Jourdi H, Nawa H (1998). Emerging roles of Dlg-like PDZ proteins in the organization of the NMDA-type glutamatergic synapse. J Biochem 124: 869–875.
Natochin M, Granovsky AE, Artemyev NO (1997). Regulation of transducin GTPase activity by human retinal RGS. J Biol Chem 272: 17444–17449.
Necula M, Chirita CN, Kuret J (2005). Cyanine dye N744 inhibits tau fibrillization by blocking filament extension: implications for the treatment of tauopathic neurodegenerative diseases. Biochemistry 44: 10227–10237.
Necula M, Kayed R, Milton S, Glabe CG (2007). Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282: 10311–10324.
Nekrasova ER, Berman DM, Rustandi RR, Hamm HE, Gilman AG, Arshavsky VY (1997). Activation of transducin guanosine triphosphatase by two proteins of the RGS family. Biochemistry 36: 7638–7643.
Neubig RR (2002). Regulators of G protein signaling (RGS proteins): novel central nervous system drug targets. J Pept Res 60: 312–316.
Neubig RR, Siderovski DP (2002). Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov 1: 187–197.
Nicodemus KK, Kolachana BS, Vakkalanka R, Straub RE, Giegling I, Egan MF et al (2007). Evidence for statistical epistasis between catechol-O-methyltransferase (COMT) and polymorphisms in RGS4, G72 (DAOA), GRM3, and DISC1: influence on risk of schizophrenia. Hum Genet 120: 889–906.
Nicodemus KK, Marenco S, Batten AJ, Vakkalanka R, Egan MF, Straub RE et al (2008). Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol Psychiatry 13: 873–877.
Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H et al (2005). Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem 280: 21212–21219.
Norris EH, Giasson BI, Lee VM (2004). Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol 60: 17–54.
Nunn C, Mao H, Chidiac P, Albert PR (2006). RGS17/RGSZ2 and the RZ/A family of regulators of G-protein signaling. Semin Cell Dev Biol 17: 390–399.
O’Brien RJ, Lau LF, Huganir RL (1998). Molecular mechanisms of glutamate receptor clustering at excitatory synapses. Curr Opin Neurobiol 8: 364–369.
O’Tuathaigh CM, Babovic D, O’Meara G, Clifford JJ, Croke DT, Waddington JL (2007). Susceptibility genes for schizophrenia: characterisation of mutant mouse models at the level of phenotypic behaviour. Neurosci Biobehav Rev 31: 60–78.
Ono K, Hasegawa K, Naiki H, Yamada M (2005). Preformed beta-amyloid fibrils are destabilized by coenzyme Q10 in vitro. Biochem Biophys Res Commun 330: 111–116.
Pagliaro L, Felding J, Audouze K, Nielsen SJ, Terry RB, Krog-Jensen C et al (2004). Emerging classes of protein–protein interaction inhibitors and new tools for their development. Curr Opin Chem Biol 8: 442–449.
Pan Y, Wang L, Dai JL (2006). Suppression of breast cancer cell growth by Na+/H+ exchanger regulatory factor 1 (NHERF1). Breast Cancer Res 8: R63.
Piazzi L, Rampa A, Bisi A, Gobbi S, Belluti F, Cavalli A et al (2003). 3-(4-[[Benzyl(methyl)amino]methyl]phenyl)-6, 7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation: a dual function lead for Alzheimer's disease therapy. J Med Chem 46: 2279–2282.
Planas AM, Gorina R, Chamorro A (2006). Signalling pathways mediating inflammatory responses in brain ischaemia. Biochem Soc Trans 34 (Part 6): 1267–1270.
Popov SG, Krishna UM, Falck JR, Wilkie TM (2000). Ca2+/Calmodulin reverses phosphatidylinositol 3,4,5-trisphosphate-dependent inhibition of regulators of G protein-signaling GTPase-activating protein activity. J Biol Chem 275: 18962–18968.
Porat Y, Abramowitz A, Gazit E (2006). Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des 67: 27–37.
Redies C (1997). Cadherins and the formation of neural circuitry in the vertebrate CNS. Cell Tissue Res 290: 405–413.
Riddle EL, Schwartzman RA, Bond M, Insel PA (2005). Multi-tasking RGS proteins in the heart: the next therapeutic target? Circ Res 96: 401–411.
Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA et al (2004). Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson's disease. J Mol Neurosci 23: 23–34.
Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig RR (2007). Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol Pharmacol 71: 169–175. This paper details the development of CCG-4986, the first small molecule inhibitor of an RGS protein.
Roof RA, Jin Y, Roman DL, Sunahara RK, Ishii M, Mosberg HI et al (2006). Mechanism of action and structural requirements of constrained peptide inhibitors of RGS proteins. Chem Biol Drug Des 67: 266–274.
Roof RA, Sobczyk-Kojiro K, Turbiak AJ, Roman DL, Pogozheva ID, Blazer LL et al (2008). Novel peptide ligands of RGS4 from a focused one-bead, one-compound library. Chem Biol Drug Des 72: 111–119.
Rosini M, Andrisano V, Bartolini M, Bolognesi ML, Hrelia P, Minarini A et al (2005). Rational approach to discover multipotent anti-Alzheimer drugs. J Med Chem 48: 360–363.
Sanders AR, Duan J, Levinson DF, Shi J, He D, Hou C et al (2008). No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am J Psychiatry 165: 497–506.
Sarek G, Ojala PM (2007). p53 reactivation kills KSHV lymphomas efficiently in vitro and in vivo: new hope for treating aggressive viral lymphomas. Cell Cycle 6: 2205–2209.
Sarrouilhe D, di Tommaso A, Metaye T, Ladeveze V (2006). Spinophilin: from partners to functions. Biochimie 88: 1099–1113.
Savitt JM, Dawson VL, Dawson TM (2006). Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest 116: 1744–1754.
Scott JK, Huang SF, Gangadhar BP, Samoriski GM, Clapp P, Gross RA et al (2001). Evidence that a protein–protein interaction ‘hot spot’ on heterotrimeric G protein betagamma subunits is used for recognition of a subclass of effectors. EMBO J 20: 767–776.
Shan J, Shi DL, Wang J, Zheng J (2005). Identification of a specific inhibitor of the dishevelled PDZ domain. Biochemistry 44: 15495–15503.
Shenolikar S, Weinman EJ (2001). NHERF: targeting and trafficking membrane proteins. Am J Physiol Renal Physiol 280: F389–F395.
Shibata T, Chuma M, Kokubu A, Sakamoto M, Hirohashi S (2003). EBP50, a beta-catenin-associating protein, enhances Wnt signaling and is over-expressed in hepatocellular carcinoma. Hepatology 38: 178–186.
Shulman-Peleg A, Shatsky M, Nussinov R, Wolfson HJ (2008). MultiBind and MAPPIS: webservers for multiple alignment of protein 3D-binding sites and their interactions. Nucleic Acids Res 36 (Web Server issue): W260–W264.
Siderovski DP, Willard FS (2005). The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci 1: 51–66.
Skovronsky DM, Lee VM, Trojanowski JQ (2006). Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu Rev Pathol 1: 151–170.
Slep KC, Kercher MA, He W, Cowan CW, Wensel TG, Sigler PB (2001). Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature 409: 1071–1077.
Smrcka AV, Lehmann DM, Dessal AL (2008). G Protein betagamma subunits as targets for small molecule therapeutic development. Comb Chem High Throughput Screen 11: 382–395.
So HC, Chen RY, Chen EY, Cheung EF, Li T, Sham PC (2008). An association study of RGS4 polymorphisms with clinical phenotypes of schizophrenia in a Chinese population. Am J Med Genet B Neuropsychiatr Genet 147B: 77–85.
Sutrala SR, Norton N, Williams NM, Buckland PR (2007). Gene copy number variation in schizophrenia. Am J Med Genet B Neuropsychiatr Genet 147B: 606–611.
Talkowski ME, Seltman H, Bassett AS, Brzustowicz LM, Chen X, Chowdari KV et al (2006). Evaluation of a susceptibility gene for schizophrenia: genotype based meta-analysis of RGS4 polymorphisms from thirteen independent samples. Biol Psychiatry 60: 152–162.
Tesmer JJ, Berman DM, Gilman AG, Sprang SR (1997). Structure of RGS4 bound to AlF4—activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell 89: 251–261.
Thorn KS, Bogan AA (2001). ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics 17: 284–285.
Tomiyama T, Asano S, Suwa Y, Morita T, Kataoka K, Mori H et al (1994). Rifampicin prevents the aggregation and neurotoxicity of amyloid beta protein in vitro. Biochem Biophys Res Commun 204: 76–83.
Tomiyama T, Shoji A, Kataoka K, Suwa Y, Asano S, Kaneko H et al (1996). Inhibition of amyloid beta protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. J Biol Chem 271: 6839–6844.
Tosetti P (2004). Evaluating chick Galpha-interacting protein selectivity. Methods Enzymol 390: 3–17.
Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P et al (2001). Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev 21: 499–512.
Traynor JR, Neubig RR (2005). Regulators of G protein signaling & drugs of abuse. Mol Interv 5: 30–41.
Vilella E, Costas J, Sanjuan J, Guitart M, De Diego Y, Carracedo A et al (2008). Association of schizophrenia with DTNBP1 but not with DAO, DAOA, NRG1 and RGS4 nor their genetic interaction. J Psychiatr Res 42: 278–288.
Voltz JW, Weinman EJ, Shenolikar S (2001). Expanding the role of NHERF, a PDZ-domain containing protein adapter, to growth regulation. Oncogene 20: 6309–6314.
Wang Y, Lee Y, Zhang J, Young KH (2008). Identification of Peptides That Inhibit Regulator of G Protein Signaling 4 Function. Pharmacology 82: 97–104.
Weintraub D, Comella CL, Horn S (2008). Parkinson's disease—part 1: pathophysiology, symptoms, burden, diagnosis, and assessment. Am J Manag Care 14 (2 Suppl): S40–S48.
Wells JA (1991). Systematic mutational analyses of protein–protein interfaces. Methods Enzymol 202: 390–411. This is one of the seminal papers on alanine scanning, a technique that led to the appreciation of protein-protein interaction “hot spots.”
Whitty A, Kumaravel G (2006). Between a rock and a hard place? Nat Chem Biol 2: 112–118.
Wieland T, Chen CK, Simon MI (1997). The retinal specific protein RGS-r competes with the gamma subunit of cGMP phosphodiesterase for the alpha subunit of transducin and facilitates signal termination. J Biol Chem 272: 8853–8856.
Williams NM, Preece A, Spurlock G, Norton N, Williams HJ, McCreadie RG et al (2004). Support for RGS4 as a susceptibility gene for schizophrenia. Biol Psychiatry 55: 192–195.
Wood LS, Pickering EH, Dechairo BM (2007). Significant support for DAO as a schizophrenia susceptibility locus: examination of five genes putatively associated with schizophrenia. Biol Psychiatry 61: 1195–1199.
Woulfe DS, Stadel JM (1999). Structural basis for the selectivity of the RGS protein, GAIP, for Galphai family members. Identification of a single amino acid determinant for selective interaction of Galphai subunits with GAIP. J Biol Chem 274: 17718–17724.
Wright TM (2006). Tramiprosate. Drugs Today (Barc) 42: 291–298.
Xie W, Samoriski GM, McLaughlin JP, Romoser VA, Smrcka A, Hinkle PM et al (1999). Genetic alteration of phospholipase C beta3 expression modulates behavioral and cellular responses to mu opioids. Proc Natl Acad Sci USA 96: 10385–10390.
Zhong H, Neubig RR (2001). Regulator of G protein signaling proteins: novel multifunctional drug targets. J Pharmacol Exp Ther 297: 837–845.
Zhong H, Wade SM, Woolf PJ, Linderman JJ, Traynor JR, Neubig RR (2003). A spatial focusing model for G protein signals. Regulator of G protein signaling (RGS) protien-mediated kinetic scaffolding. J Biol Chem 278: 7278–7284.
Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL (2004). The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J Biol Chem 279: 26846–26857.
Acknowledgements
This study was supported by NIH R01GM39561 and NIH RO1DA23252 to RRN and by a fellowship awarded to LLB by the CBI Training Program T32-GM008597.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
Dr Neubig has received a gift of equipment and research supplies from Luminex Corporation, consulting fees from Berlex/Bayer, and lecture fees from Glaxo-Smith-Kline, Abbott Laboratories, Merck Research Labs, and Epix Pharmaceuticals. Levi Blazer has no conflicts to disclose.
Rights and permissions
About this article
Cite this article
Blazer, L., Neubig, R. Small Molecule Protein–Protein Interaction Inhibitors as CNS Therapeutic Agents: Current Progress and Future Hurdles. Neuropsychopharmacol 34, 126–141 (2009). https://doi.org/10.1038/npp.2008.151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.151
Keywords
This article is cited by
-
Detection, visualization and quantification of protein complexes in human Alzheimer’s disease brains using proximity ligation assay
Scientific Reports (2023)
-
Develop quantitative FRET (qFRET) technology as a high-throughput universal assay platform for basic quantitative biomedical and translational research and development
Med-X (2023)
-
Site-directed modification of multifunctional lignocellulose-degrading enzymes of straw based on homologous modeling
World Journal of Microbiology and Biotechnology (2023)
-
Defective repair of topoisomerase I induced chromosomal damage in Huntington’s disease
Cellular and Molecular Life Sciences (2022)
-
Recent advances in the development of protein–protein interactions modulators: mechanisms and clinical trials
Signal Transduction and Targeted Therapy (2020)
