
Abstract
Corticotropin-releasing hormone (CRH) and its receptor, CRH receptor-1 (CRHR1), have a key role in alcoholism. Especially, post-dependent and stress-induced alcohol intake involve CRH/CRHR1 signaling within extra-hypothalamic structures, but a contribution of the hypothalamic–pituitary–adrenal (HPA) axis activity might be involved as well. Here we examined the role of CRHR1 in various drinking conditions in relation to HPA and extra-HPA sites, and studied relapse-like drinking behavior in the alcohol deprivation model (ADE). To dissect CRH/CRHR1 extra-HPA and HPA signaling on a molecular level, a conditional brain-specific Crhr1-knockout (Crhr1NestinCre) and a global knockout mouse line were studied for basal alcohol drinking, stress-induced alcohol consumption, deprivation-induced intake, and escalated alcohol consumption in the post-dependent state. In a second set of experiments, we tested CRHR1 antagonists in the ADE model. Stress-induced augmentation of alcohol intake was lower in Crhr1NestinCre mice as compared with control animals. Crhr1NestinCre mice were also resistant to escalation of alcohol intake in the post-dependent state. Contrarily, global Crhr1 knockouts showed enhanced stress-induced alcohol consumption and a more pronounced escalation of intake in the post-dependent state than their control littermates. Basal intake and deprivation-induced intake were unaltered in both knockout models when compared with their respective controls. In line with these findings, CRHR1 antagonists did not affect relapse-like drinking after a deprivation period in rats. We conclude that CRH/CRHR1 extra-HPA and HPA signaling may have opposing effects on stress-related alcohol consumption. CRHR1 does not have a role in basal alcohol intake or relapse-like drinking situations with a low stress load.
Similar content being viewed by others


INTRODUCTION
Alcohol-related diseases, especially alcoholism, are the result of cumulative responses to alcohol exposure, the genetic make-up of an individual, and environmental perturbations over time (Spanagel, 2009). Hence, only an adverse combination of chronic alcohol consumption, genetic, and environmental risk factors favors the transition from alcohol drinking to its compulsive use (Vengeliene et al, 2009). It has been suggested that during the transition from recreational to compulsive alcohol use, a progressive recruitment of endogenous stress systems occurs. Specifically, this has been proposed to involve a pathological engagement of extra-hypothalamic corticotropin-releasing hormone (CRH) transmission, and CRH receptor-1 (CRHR1) signaling. These are thought to contribute to a chronic negative affective state and increased susceptibility to relapse behavior (Heilig and Koob, 2007; Koob and Le Moal, 2008; Heilig et al, 2010). A potential translation of this hypothesis may be offered by recent human genetic findings. Studies of several samples have shown that Crhr1 gene variation interacts with negative life events to determine risk for excessive alcohol consumption (Treutlein et al, 2006; Blomeyer et al, 2008; Barr, 2010; Nelson et al, 2010; Schmid et al, 2010).
The general role of extra-hypothalamic CRH transmission and CRHR1 activation is to mediate stress-induced behaviors, whereas stress-induced neuroendocrine effects are mediated by CRH/CRHR1 signaling within the hypothalamic–pituitary–adrenal (HPA) axis (De Kloet, 2004; McEwen, 2007). A pathological engagement of extra-HPA CRH/CRHR1 signaling in chronic alcohol effects, in particular in the amygdala, has been found in the post-dependent state (Funk et al, 2006; Koob and Le Moal, 2008; Sommer et al, 2008; Roberto et al, 2010). The term post-dependent state was introduced to describe a persistent neuroadaptive state after a prolonged history of dependence, such as, for example, observed after repeated intermittent alcohol vapor exposure. A core feature of the post-dependent state is persistent escalation of alcohol consumption, which mimics a key characteristic of addictive disorders (Roberts et al, 2000). Post-dependent escalation can be inhibited by systemic (Gehlert et al, 2007), central infusion (Valdez et al, 2002), or intra-amygdala application of CRH receptor antagonists (Funk et al, 2006; Finn et al, 2007). By systemic application of the non-peptide CRHR1 antagonist N,N-bis(2-methoxyethyl)-3-(4-methoxy-2-methylphenyl)-2,5-dimethyl-pyrazolo[1,5-a] pyrimidin-7-amine (MPZP)—a compound with high affinity and specificity for CRHR1—it could further be demonstrated that post-dependent escalation of alcohol drinking is diminished (Gilpin et al, 2008; Richardson et al, 2008). Furthermore, an upregulation of Crhr1 expression is consistently found within the amygdala in the post-dependent state (Sommer et al, 2008; Heilig et al, 2010; Contet et al, 2011). In conclusion, CRH/CRHR1 signaling within the amygdala mediates escalated alcohol intake in animals in a post-dependent state (Heilig and Koob, 2007).
CRH/CRHR1 signaling also has a role in mediating stress-related drinking situations. A variety of physical and psychological stressors are important risk factors for induction of craving and relapse (Uhart and Wand, 2009), and it has been shown that stress is a main contributor in reinstating alcohol-seeking behavior in animals (Lê et al, 2000; Lê and Shaham, 2002). Stress- but not cue-induced alcohol seeking responses are blocked by systemic administration of CRHR1 antagonists (Gehlert et al, 2007; Lê et al, 2000; Lê and Shaham, 2002; Liu and Weiss, 2002; Marinelli et al, 2007; Hansson et al, 2006), demonstrating selective involvement of CRH/CRHR1 signaling in stress-related drinking behaviors. Surprisingly, however, in mice lacking a functional CRHR1 (Timpl et al, 1998), augmented but delayed stress-induced alcohol consumption was observed (Sillaber et al, 2002), which is in stark contrast with the findings that a CRHR1 blockade should abolish stress-induced alcohol intake. We have hypothesized that CRH/CRHR1 signaling within the HPA axis might contribute to this unexpected effect of a constitutive and global Crhr1 inactivation (Spanagel, 2009).
To dissect on a molecular level the role of CRH/CRHR1 signaling in the HPA axis vs extra-HPA structures, conditional brain-specific Crhr1-knockout (Crhr1NestinCre) mice and global knockout (CRH1R−/−) mice were examined. Conditional Crhr1NestinCre-knockout mice lack the CRHR1 receptor in the entire central nervous system. However, CRHR1 expressed in the pituitary and adrenal glands of the HPA axis remain unaffected in this genetic mouse model (Müller et al, 2001; Lu et al, 2008). As global knockouts lack the receptor in all tissues they can be used in comparison with Crhr1NestinCre-knockout mice to differentiate between the HPA axis vs extra-HPA structures (note: the ideal comparison would be a conditional mouse model that lacks Crhr1 exclusively in the pituitary and adrenal glands; such a model however is not available). Both lines went through a long-term voluntary alcohol drinking procedure in their home cage and were then tested for stress-induced alcohol consumption or deprivation-induced intake (alcohol deprivation model (ADE)). After repeated intermittent vapor exposure, escalated alcohol consumption in the post-dependent state was also examined. As we unexpectedly found that relapse-like drinking was unaffected in both knockout models, we conducted a second set of experiments where we tested the CRHR1 antagonists, antalarmin and NBI-27914, on relapse-like drinking behavior in the rats in our long-term alcohol drinking model with repeated deprivation phases (Spanagel and Hölter, 1999). This model provides excellent face and construct validity (Vengeliene et al, 2009), and has shown predictive validity as well (Spanagel and Kiefer, 2008).
MATERIALS AND METHODS
Animals
Male mice (see below for background) and Wistar rats (from our own rat breeding colony at the CIMH, Mannheim, Germany) were housed individually in standard cages (Ehret, Emmendingen, Germany) under a 12-h artificial light–dark cycle (lights on at 0700 hours). Room temperature was kept constant (temperature: 22±1 °C, humidity: 55±5%). Standard laboratory rodent food (Sniff, Soest, Germany) and tap water were provided ad libitum throughout the experimental period. Body weights were measured weekly.
Global homozygous Crhr1 knockouts (Crhr1−/−) and control littermates were generated as described previously (Timpl et al, 1998) and were on a mixed 129 × 1/SvJ × CD1 background for more than 50 generations. Generation of mice carrying a loxP-flanked Crhr1 allele has been described by Müller et al (2003). For conditional inactivation of Crhr1 in the central nervous system, transgenic mice expressing the Cre recombinase under the control of the rat Nestin (Nes) promoter and enhancer (Tronche et al, 1999) were bred to homozygous Crhr1loxP/loxP mice. The resulting heterozygous F1 offspring (Crhr1+/loxP) were either positive or negative for Nes–Cre. From matings of Crhr1+/loxP with Crhr1+/loxP Nes–Cre mice we obtained F2 animals of the desired genotypes (Crhr1loxP/loxP Nes–Cre (Crhr1NestinCre) and Crhr1loxP/loxP), which were then inter-crossed to obtain sufficient numbers of F3 animals. Crhr1loxP/loxP mice were used as control littermates for conditional CRH1R mice. The mice used for this study were on a mixed 129S2/Sv × C57BL/6J × (C57BL/6 × SJL)F2 background, 129S2/Sv × C57BL/6J coming from the floxed line and (C57BL/6 × SJL)F2 coming from the Nes–Cre line. In Crhr1NestinCre-knockout mice Crhr1 expression is lost throughout the central nervous system (Romanowski et al, 2010). Of note, Cre recombinase driven by the Nestin promoter is not expressed in the anterior pituitary (Supplementary Figure S1). Therefore, Crhr1 in the anterior pituitary is undisturbed as has been demonstrated previously by a fully active HPA axis in Crhr1NestinCre-knockout mice (Schmidt et al, 2006).
In our detailed description of generation of our mutant mouse lines, we adhere to the Standards for the Publication of Mouse Mutant Studies (Crusio et al, 2009) and emphasize on maintaining the mutations in heterogeneous, segregating populations, that is, in a randomized genetic background as this procedure will over time gradually reduce the size of the flanking allele region (Gerlai, 1996; Crusio, 2004).
Twenty-five 2-month-old male Wistar rats (from our own breeding colony at the CIMH) were used for the ADE experiments. All rats were housed individually in standard rat cages (Ehret) under a 12-h artificial light–dark cycle (lights on at 0700 hours). Room temperature was kept constant (temperature: 22±1 °C, humidity: 55±5%). Standard laboratory rat food (Sniff) and tap water were provided ad libitum throughout the experimental period. Body weights were measured weekly.
All experimental procedures were approved by the Committee on Animal Care and Use (Regierungspräsidium Karlsruhe), and performed in accordance with the local Animal Welfare Act and the European Communities Council Directive of 24 November 1986 (86/609/EEC).
Drugs
All alcohol drinking solutions and alcohol for the ethanol vapor chamber system were prepared from 96% ethanol (Sigma-Aldrich, Taufkirchen, Germany) and diluted in tap water. Antalarmin (N-butyl-N-ethyl-2,5,6-trimethyl-7-(2,4,6-trimethylphenyl)-7H-pyrrolol[2,3-d]pyrimidin-4-amine) was provided by the NIAAA and NBI-27914 (5-chloro-N-(cyclopropylmethyl)-2-methyl-N-propyl-N′-(2,4,6-trichlorophenyl)-4,6-pyrimidinediamine) was a gift from Anton Bespalov (Abbott, Ludwigshafen). Antalarmin and NBI-27914 were suspended in 0.5% methylcellulose solution. The solutions were freshly prepared and injected in a volume of 3 ml/kg intraperitoneally (i.p.). Control experiments were performed after administration of 0.5% methylcellulose.
Free Choice, Two-Bottle Drinking Behavior in Mice
All mice (4 groups of animals with n=7–12 per genotype: Crhr1NestinCre and control littermates, and Crhr1−/− and controls; see figures for the exact number of animals used in each group) were single-housed and allowed to drink tap water from two bottles for 1 week. After this habituation, one of the bottles was changed to a 2% ethanol solution during 4 days. For the next 4 days the alcohol bottles contained a 4% ethanol solution. From then on all mice were kept on 8% ethanol vs water for approximately 5 months as described previously (Sillaber et al, 2002). Water intake (ml/day) and ethanol intake (ml/day) were measured daily (in connection with experimental interventions, for example, stress treatment) or weekly (between experiments). The position of the ethanol solution was changed once every week to avoid development of side preferences. From these data ethanol intake was calculated in grams of ethanol/kg body weight/day.
Stress-Induced Alcohol Drinking in Mice
After 2 months of home cage alcohol consumption at 8%, social defeat stress (SDS) was performed as described previously (Sillaber et al, 2002). Mice were confronted with an unfamiliar male mouse (resident) in a special cage (20 × 12 × 20 cm). The resident (6- to 7-month-old Swiss mice) had been housed in this cage for 2 days. In this paradigm, the resident attacked the intruder immediately. Directly after the attack, the two mice were separated by a wire mesh screen and the intruder was left in the smaller section of the cage (7 × 12 × 20 cm) for 15 min. The intruder was then returned to its home cage, where it had again free access to water and alcohol. The SDS procedure was applied for three consecutive days. Ethanol and water intake were measured daily for 5 days and then weekly for 4 weeks.
After these 4 weeks of weekly measurements, forced swim stress (FSS) was performed as described previously (Sillaber et al, 2002). Mice were placed in a water-filled glass cylinder (25 cm high, 14 cm wide) for 5 min (the water temperature was 21 °C). Afterward, the mice were gently dried and moved back to their home cages with free access to water and alcohol. The FSS procedure was applied for three consecutive days. Ethanol and water intake were measured daily for 5 days and then weekly for 4 weeks.
ADE in Mice
Approximately 5 weeks after the FSS a deprivation phase was introduced; the ethanol bottles were removed for a period of 2 weeks. At the end of the deprivation phase, the 8% ethanol bottles were reintroduced and ADE-induced alcohol (ml/day) and water intake (ml/day) were measured daily for 5 days.
Alcohol Vapor Exposure, Blood Sampling, and Post-Dependent Drinking in Mice
The vapor chamber system was purchased from La Jolla Alcohol Research (La Jolla, CA, USA) and was further modified for a more accurate use of mice. Briefly, four chambers are connected to a pump by a flask with four side-arms, the bottom of which sits in a heater. Alcohol flows from a large reservoir that contains 95% alcohol to a peristaltic pump, from which it is delivered to a side-arm flask at a flow rate that can be regulated. This round-bottom flask is placed inside the heater so that drops of alcohol on the bottom are vaporized. Airflow controlled by a pressure gauge is delivered to the flask and serves to carry the alcohol vapor to the individual chambers through the tubes connected to the four side-arms. Each tube also has its own pressure gauge that enables to adjust evenly the conditions for each chamber. The alcohol delivery rate was set to 56–60 ml/h.
Post-abstinence drinking was monitored in four additional groups of mice (n=7–12 per genotype). After 3 months of home cage, free choice, two-bottle drinking at 8% ethanol concentration, all four groups of mice were exposed to alcohol vapor. Prior to the start of the first exposure, all mice were injected i.p. with 2 g/kg ethanol and allowed to rest for approximately 20 min. The mice were then exposed to four cycles of 16 h of ethanol vapor separated by 8-h periods of withdrawal (Becker and Lopez, 2004). At the end of the last cycle each mouse was placed into a transparent plexiglass cylinder of approximately 2.5 cm diameter, the limb was gently stretched, and blood (∼20 μl) was sampled from the saphenous vein. Blood alcohol concentrations (BACs) were determined using a nicotinamide dinucleotide phosphate enzyme spectrophotometric method (Rolf Greiner BioChemica GmbH, Stuttgart, Germany) (Perreau-Lenz et al, 2009).
After exposure to repeated intermittent ethanol vapor and approximately 24 h into acute withdrawal, weekly alcohol and water intake were monitored for 4 weeks.
Long-Term Alcohol Consumption with Repeated Deprivation Phases in Rats
After 2 weeks of habituation to the animal room, rats were given ad libitum access to tap water and to 5, 10, and 20% ethanol solutions (v/v) as well. This procedure is described in great detail by Hölter et al (1998) and Vengeliene et al (2010). In brief, the first 2-week deprivation period was introduced after 8 weeks of continuous alcohol availability. After the deprivation period, rats were given access to alcohol again and four more deprivation periods were introduced in a random manner, that is, the duration of the following drinking and deprivation phases was irregular, that is, approximately 5±1 week and 2±1 week, respectively, in order to prevent adaptive behavioral mechanisms. The long-term voluntary alcohol drinking procedure, including all deprivation phases, lasted a total of 26 weeks.
The role of CRH on relapse-like drinking was assessed in long-term alcohol drinking rats. For this purpose, two different CRH receptor antagonists were tested during the onset of the fifth post-abstinence drinking period. Particularly, three separate groups of rats (n=8–9 per group) were matched in such way that the mean baseline total alcohol intake was approximately the same in each group (ie, ∼2.1 g/kg/day). Baseline drinking was measured daily for 1 week. After the last day of baseline measurement, the alcohol bottles were removed from the cages, leaving the rats with free access to food and water for 3 weeks. Thereafter, each rat was subjected to a total of 5 intraperitoneal injections (starting at 1900 hours, with 12-h intervals) of either vehicle, antalarmin (20 mg/kg), or NBI-27914 (10 mg/kg). The doses chosen were according to the literature the most effective yet still selective doses (Hansson et al, 2006; Marinelli et al, 2007; Hummel et al, 2010). The alcohol bottles were reintroduced after the second injection (at 0900 hours on the twenty-second day of alcohol deprivation) and the occurrence of an ADE was determined. Total ethanol (g/kg of body weight/day) and water intake (ml/kg of body weight/day) were measured daily at 009 hours for the subsequent week. Each rat's body weight was recorded 24 h before the first injection and 12 h after the last injection. The drug injection schedule was based on our previous studies using the same paradigm (eg, Vengeliene et al, 2010).
In order to test for any sedative effects resulting from the drug treatment, home-cage locomotor activity was monitored by use of a novel infrared sensor connected to a recording and data-storing system (Mouse-E-Motion by Infra-e-motion, Henstedt-Ulzburg, Germany) (Vengeliene et al, 2010). A Mouse-E-Motion device was placed above each cage (30 cm from the bottom) so that the rat could be detected at any position inside the cage. The device was sampling every second, whether the rat was moving or not. The sensor could detect body movement of the rat of at least 1.5 cm from one sample point to the successive one. The data measured by each Mouse-E-Motion device were downloaded into a personal computer and processed with Microsoft Excel. Monitoring of locomotor activity started 3 days before the drug treatment procedure and was continued for five more post-treatment days. The percentage of each rat's locomotor activity during and after treatment days was calculated by using the ‘before treatment’ activity data as a reference.
Statistics
Data derived from drinking (total alcohol intake, alcohol preference, water intake) and locomotor activity were analyzed by two-way analysis of variance (ANOVA) with repeated measures (factors were: genotype, or stress/deprivation/treatment, or ethanol concentration, or time (day, week)). Whenever significant differences were found, post-hoc Student's Newman–Keuls tests were performed. All data are presented as means±SEM and the chosen level of significance was p<0.05.
RESULTS
Baseline Alcohol Consumption, Stress-Induced, Alcohol Deprivation-Induced, and Alcohol Vapor-Induced Alcohol Intake in Crhr1NestinCre Mice
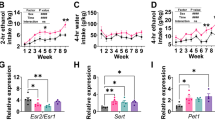
During the first days of exposure to increasing alcohol concentrations, Crhr1NestinCre mice and their control littermates consumed similar amounts of alcohol (factor genotype: p=0. 27) (Figure 1a). Alcohol consumption increased in both genotypes when higher concentrations of alcohol were offered (factor concentration: F(2, 44)=115.8, p<0.0001). SDS increased ethanol intake and preference as compared with baseline consumption levels in both Crhr1NestinCre and control mice (factor day: F(5, 110)=47.2, p<0.0001, and F(5, 110)=21.9, p<0.0001, for intake and preference, respectively). This increase was stronger in the control animals (factor genotype: F(1, 22)=4.6, p<0.05, and F(1, 22)=4.7, p<0.05, for intake and preference, respectively) (Figure 1b and Supplementary Figure S3A), with a trend-level statistical significance for the genotype × day interaction for alcohol intake (F(5, 110)=2.1, p=0.07). Three days after the last SDS exposure, alcohol intake returned to baseline levels for both genotypes and remained on baseline for the four subsequent weeks of measurement (Supplementary Figure S2A). During the FSS exposure both animal groups again significantly increased their alcohol intake (factor day: F(5, 110)=29.8, p<0.0001, and F(5, 110)=16.8, p<0.0001, for intake and preference, respectively). Alcohol consumption after FSS was significantly lower in Crhr1NestinCre mice compared with their control littermates (factor genotype: F(1, 22)=4.4, p<0.05, and F(1, 22)=3.8, p<0.05, for intake and preference, respectively) (Figure 1c and Supplementary Figure S3C). The behavioral parameters monitored during the FSS, that is, time spent struggling and floating, did not reveal any significant difference between genotypes (data not shown). Two days after the last FSS exposure, alcohol intake returned to baseline levels for both genotypes and remained on slightly fluctuating baseline levels for the four subsequent weeks of measurement (Supplementary Figure S2B). A 2-week alcohol deprivation phase was then introduced and after re-exposure alcohol consumption was different in both animal groups (factor day: F(5, 110)=8.4, p<0.0001, and F(5, 110)=3.3, p<0.01, for alcohol intake and preference, respectively) (Figure 1d and Supplementary Figure S3E). However, the genotype × day interaction effect for either alcohol intake (p=0.31) or preference (p=0.08) was not significant. There was also no significant difference in alcohol consumption between genotypes (factor genotype: p=0.45 and p=0.84, for alcohol intake and preference, respectively).

Baseline alcohol consumption (a), stress-induced (b, c), and alcohol deprivation-induced (d) alcohol intake (g/kg/day) in conditional brain-specific Crhr1-knockout (Crhr1NestinCre) mice as well as in their control counterpart mice. (a) Voluntary home-cage alcohol consumption during the first 4 days of concurrent exposure to water and either 2, 4, or 8% ethanol solution by Crhr1NestinCre (n=12) and control (n=12) mice. SDS (b) was performed during three subsequent days after 2 months of voluntary 8% ethanol consumption. After 1 more month of continuous alcohol drinking, all mice were subjected to a daily FSS for three subsequent days. The effect of FSS on alcohol consumption by Crhr1NestinCre and control mice is shown in panel c. An alcohol deprivation phase of 2 weeks was introduced 1 month after the last stress procedure. Pre- and post-abstinence alcohol consumption (alcohol deprivation effect) are shown in panel d. Alcohol intake measured during the last week before either stress or alcohol withdrawal is marked as baseline ‘B’. Data are presented as means±SEM. Although we report in the Results section significant genotype differences after stress procedures, we did not find significant interactions by time and therefore do not show post-hoc analyses here.
During the post-dependent drinking weeks, the Crhr1NestinCre mice and their control littermates tended to increase alcohol consumption as compared with that measured before vapor exposure (p=0.10). However, increased alcohol consumption during post-dependent state was seen only in control mice (3a and Supplementary Figure S3G). By contrast, Crhr1NestinCre mice had similar alcohol intake as compared with that before vapor exposure during the post-dependent drinking weeks, indicating lack of escalation of alcohol intake in the post-dependent state (factor genotype: F(1, 21)=3.9, p<0.05, and factor genotype × week interaction effect F(4, 84)=1.2, p=0.30, for alcohol intake, as well as factor genotype: F(1, 21)=7.5, p<0.05, and factor genotype × week interaction effect F(4, 84)=2.5, p<0.05, for alcohol preference). Post-hoc analysis of alcohol preference data showed that alcohol preference in Crhr1NestinCre mice was not significantly different during the first 4 weeks of post-dependent drinking as compared with that before vapor exposure. By contrast, a significant increase in alcohol preference during the first post-dependent drinking weeks was measured in their control littermate mice.
Baseline Alcohol Consumption, Stress-Induced, Alcohol Deprivation-Induced, and Alcohol Vapor-Induced Alcohol Intake in Crhr1−/− Mice
Crhr1−/− mice and their control littermates consumed comparable amounts of alcohol during the first days of exposure to increasing alcohol concentrations (factor genotype: p=0.52) (Figure 2a). Alcohol consumption increased in both genotypes when higher concentrations of alcohol were offered (factor concentration: F(2, 28)=16.1, p<0.0001). After SDS Crhr1−/− and control mice increased their ethanol intake as compared with baseline consumption levels (factor day: F(5, 70)=27.2, p<0.0001, and F(5, 70)=12.5, p<0.0001, for intake and preference, respectively). This SDS-induced increase in alcohol intake was significantly more pronounced in Crhr1−/− animals (factor genotype: F(1, 14)=5.6, p<0.05) (Figure 2b). The factor genotype × day interaction effect was found significant for both alcohol intake and preference: F(5, 70)=3.9, p<0.01 and F(5, 70)=3.1, p<0.05 (Figure 2b and Supplementary Figure S3B). Two days after the last SDS exposure, alcohol intake returned to baseline levels for both genotypes and remained on baseline for the four subsequent weeks of measurement (Supplementary Figure S2C). The subsequent FSS exposure significantly increased alcohol consumption in both animal groups (factor day: F(5, 70)=10.2, p<0.0001, and F(5, 70)=5.2, p<0.001, for intake and preference, respectively); however, alcohol consumption was not different between genotypes (factor genotype: p=0.98 and p=0.83 for intake and preference, respectively) (Figure 2c and Supplementary Figure S3D). The behavioral parameters monitored during the FSS, that is, time spent struggling and floating, did not reveal any significant difference between genotypes (data not shown). Two days after the last FSS exposure, alcohol intake returned to baseline levels for both genotypes and remained on baseline levels for the four subsequent weeks of measurement (Supplementary Figure S2D). Alcohol consumption after the deprivation phase was different in both animal groups (factor day: F(5, 70)=5.0, p<0.001, and F(5, 70)=1.6, p=0.17, for intake and preference, respectively) (Figure 2d and Supplementary Figure S3F). However, the genotype × day interaction effect for either alcohol intake (p=0.58) or preference (p=0.69) was not significant. There was also no significant difference in alcohol consumption between genotypes (factor genotype: p=0.63 and p=0.97 for intake and preference, respectively).

Baseline alcohol consumption (a), stress-induced (b, c), and alcohol deprivation-induced (d) alcohol intake (g/kg/day) in global Crhr1-knockout (CRH1R−/−) mice as well as in their control littermates. (a) Voluntary home-cage alcohol consumption during the first 4 days of concurrent exposure to water and either 2, 4, or 8% ethanol solution by CRH1R−/− (n=7) and control (n=9) mice. SDS (b) was performed during three subsequent days after 2 months of voluntary 8% ethanol consumption. After 1 more month of continuous alcohol drinking, all mice were subjected to a daily FSS for three subsequent days. The effect of FSS on alcohol consumption by CRH1R−/− and control mice is shown in panel c. An alcohol deprivation phase of 2 weeks was introduced 1 month after the last stress procedure. Pre- and post-abstinence alcohol consumption are shown in panel d. Alcohol intake measured during the last week before either stress or alcohol withdrawal is marked as baseline ‘B’. Data are presented as means±SEM. *indicates significant differences from the control mice group after post-hoc analysis (p<0.05).
During the post-dependent drinking weeks both Crhr1−/− mice and their control littermates had significantly increased alcohol consumption as compared with that measured before vapor exposure (factor week: F(4, 52)=9.7, p<0.0001, and F(4, 52)=3.6, p<0.05, for intake and preference, respectively) (Figure 3b and Supplementary Figure S3H). However, Crhr1−/− mice had significantly higher alcohol intake when compared with control mice, which remained increased for at least 4 weeks (factor genotype: F(1, 13)=13.1, p<0.01, and factor genotype × week interaction effect F(4, 52)=6.2, p<0.001, for alcohol intake, as well as factor genotype: F(1, 13)=9.0, p<0.05, and factor genotype × week interaction effect F(4, 52)=3.9, p<0.01, for alcohol preference). Post-hoc analysis has shown that increased alcohol intake in Crhr1−/− mice was significant during the first 2 weeks of post-dependent drinking, whereas this increase was significant only during the first post-dependent drinking week in their control littermates.

Average daily alcohol intake (g/kg/day) during the first weeks of vapor-induced drinking in (a) Crhr1NestinCre mice (n=11) and in their control counterpart mice (n=12), as well as in (b) CRH1R−/− mice (n=7) and in their control counterpart mice (n=9). All mice were exposed to voluntary 8% home-cage drinking for 3 months. Thereafter they received four cycles of 16-h ethanol vapor exposure separated by 8-h periods of withdrawal. Twenty-four hours after discontinuation of vapor exposure, all mice (except one CRH1R−/− mouse that died during the vapor exposure) were returned to voluntary home-cage drinking for 4 weeks. Alcohol intake measured during the last week before vapor exposure is marked as baseline ‘B’. Data are presented as means±SEM *indicates significant differences from the control mice group after post-hoc analysis (p<0.05). Although we report in the Results section significant genotype differences between Crhr1NestinCre and their control mice line during post-dependent drinking, we did not find significant interactions by time and therefore do not show post-hoc analyses here.
Blood Alcohol Concentration
BAC measured at the end of the last vapor exposure session in CRH1NestinCre mice and in their control littermates was 222±29 and 228±36 mg/dl, respectively. In the CRH1−/− mice and their control littermates 348±24 and 322±35 mg/dl respectively, indicating a high level of alcohol intoxication in all animals. Blood alcohol levels (BACs/grams) were found as significantly different among the genotypes (factor genotype; F(3, 30)=3.6, p<0.05). However, the post hoc analysis revealed that BACs between CRH1NestinCre and CRH1−/− mice as compared with their control littermates were not significantly different (p=0.89 and p=0.58, respectively).
Effects of CRHR1 Antagonists on ADE in Wistar Rats
After re-introduction of alcohol solutions after the fifth deprivation phase, the vehicle-treated group showed a typical increase in alcohol consumption indicating occurrence of an ADE (Figure 4). This increase was not different from that observed during the first four deprivation phases (data not shown). With respect to the antalarmin and NBI-27914 treatment, statistical analysis revealed a significantly different alcohol intake after a deprivation phase in all animal groups as compared with basal drinking (factor day: F(5, 110)=113.3, p<0.0001). Neither repeated administration of antalarmin or NBI-27914 did significantly reduce the expression of the ADE (factor treatment group: p=0.31 and treatment group × day interaction: p=0.15) (Figure 4). Similarly, water intake during the first five post-abstinence days was unaffected by both antalarmin and NBI-27914 treatments as compared with vehicle-treated rats (factor treatment group: p=0.71 and treatment group × day interaction: p=0.47) (Supplementary Figure S4). The small tendency of compounds to reduce alcohol intake was likely caused by the side effects of drug treatment recorded as change in the home-cage locomotor activity of an animal. Locomotor activity data were analyzed using recordings of 12-h post-injection intervals that corresponded to the rats’ active phase. Overall there was a general reduction in home-cage activity seen in all animal groups, which was likely caused by alcohol intoxication (Supplementary Figure S5). However, a significant change in the activity of both antalarmin- and NBI-27914-treated rats was seen when compared with vehicle-treated rats (treatment group × day interaction: F(10, 105)=4.2, p<0.0001). Post-hoc analysis showed that locomotor activity recovered to that of the vehicle-treated animal group immediately after treatment was stopped. Both antalarmin and NBI-27914 treatment also led to a small but significant loss of ∼1% of body weight, showing that food intake or metabolism was altered during the treatment days (factor treatment group: F(2, 22)=13.8, p<0.001).

Total ethanol intake (g/kg/day) before and after an alcohol deprivation period of 3 weeks in Wistar rats. The arrows indicate administration of either (a) vehicle (n=9) and 20 mg/kg antalarmin (n=8), or (b) vehicle (n=9) and 10 mg/kg NBI-27914 (n=8). The last-week measurements of ethanol intake are shown as baseline drinking ‘B’. Note: The same vehicle control group is shown in panels a and b. A separate display of antalarmin and NBI-27914 treatment has been chosen for better visualization. Data are presented as means±SEM.
DISCUSSION
Our alcohol drinking studies on global, constitutive Crhr1, and conditional neuronal knockout (Crhr1NestinCre) mice, and our pharmacological studies using CRHR1 antagonists, led to following key findings: (i) neuronal CRHR1 mediates stress-induced alcohol intake and is critically involved in the escalation of alcohol intake in the post-dependent state; (ii) neuronal CRHR1 is neither involved in mediating primary reinforcement, that is, baseline alcohol consumption nor in relapse-like drinking behavior after a period of abstinence; (iii) CRHR1 located within the HPA axis may mediate opposing effects on stress-induced and post-dependent alcohol consumption, that is, animals lacking Crhr1 show augmented stress-induced alcohol intake and enhanced escalation; and finally (iv) selective CRHR1 antagonists do not influence relapse-like drinking behavior in an animal model with good predictive validity. In conclusion, these findings are in line with the hypothesis that extra-hypothalamic CRH/CRHR1 systems are crucial for the transition into an excessive alcohol drinking state and for stress-induced augmentation of alcohol consumption (Heilig and Koob, 2007; Koob and Le Moal, 2008; Heilig et al, 2010). However, CRH/CRHR1 signaling seems to be of less importance for the early motivational stages of alcohol consumption and relapse-like drinking situation after abstinence with a low stress load (Heilig and Koob, 2007). This could also be seen in the results obtained from the stress experiments. Stress-induced differences in alcohol consumption between mutant mice and their control littermate mice were most apparent after exposure to a SDS. Contrarily, difference in alcohol consumption between groups of mice could hardly be measured after exposure to the FSS. Our earlier studies comparing the effect of different stressors on alcohol consumption have shown that response to swim stress with respect to alcohol consumption is very weak, suggesting that under the present experimental conditions swim stress act as a low-intensity stressor (Vengeliene et al, 2003). Furthermore, HPA axis-related CRHR1 has an opposing effect, leading to a further escalation in the post-dependent state and to augmented but delayed stress-induced alcohol consumption—an effect that has been described previously by us (Sillaber et al, 2002). The Koob laboratory also studied global Crhr1-knockout mice in the post-dependent state but did not find an escalation of alcohol reinforcement (Chu et al, 2007). The discrepancy between our data and the Chu et al report might be explained by non-operant and operant drinking procedures (Sanchis-Segura and Spanagel, 2006).
Taking our results into consideration one may call into question expectations for therapeutic efficacy of a pharmacological CRHR1 blockade in alcohol-dependent patients. A possible prediction based on the data presented here is that only patients who are suffering from a high acute stress load or who experienced chronic psychosocial adversity might be expected to benefit from CRHR1 blockade as treatment. This speculation is in line with previous human genetic studies describing an interaction between the hCrhr1 gene and stressful life events in mediating heavy alcohol use (Treutlein et al, 2006; Blomeyer et al, 2008; Schmid et al, 2010).
We used a conditional Crhr1-deficient mouse line, which lacks this receptor in all neurons (Crhr1NestinCre knockouts), while retaining Crhr1 expression in the anterior pituitary owing to a complete lack of Nestin expression in this region (Schmidt et al, 2006). Therefore, Crhr1NestinCre knockouts are an attractive tool to genetically dissect the functional role of extra-hypothalamic CRHR1-containing cell populations from that of HPA axis-related, CRHR1-expressing cells (Silberstein et al, 2009). The results we obtained with the Crhr1NestinCre knockouts in respect to stress-induced and post-dependent alcohol consumption are in line with the CRH hypothesis postulating a pathological engagement of extra-hypothalamic CRH transmission and CRHR1 signaling that contributes to a chronic negative affective state and subsequently to relapse behavior (Heilig and Koob, 2007; Koob and Le Moal, 2008; Heilig et al, 2010).
Global Crhr1-knockout mice (Timpl et al, 1998) show the opposite phenotype: augmented stress-induced alcohol consumption—a phenomenon described by us earlier (Sillaber et al, 2002)—and enhanced escalation of alcohol intake in the post-dependent state. These unexpected results merit additional discussion. The possibility that mutation of the Crhr1 gene affected the pharmacokinetics of alcohol can be excluded, as there are no differences in alcohol metabolism between these knockouts and control mice (Sillaber et al, 2002). Dysregulation of glucocorticoids, however, might contribute to the observed phenotype. In contrast to the Crhr1NestinCre mice, which have slightly elevated basal corticosterone levels (Jan Deussing, unpublished data; Schmidt et al, 2006), the global Crhr1 knockouts have reduced levels of basal corticosterone and a blunted endocrine stress response (Timpl et al, 1998). Corticosterone has usually a facilitatory role on voluntary alcohol consumption, demonstrated by the finding that adrenalectomy causes a decrease in alcohol drinking in both Wistar rats (Fahlke et al, 2004) and alcohol-preferring AA rats (Fahlke and Eriksson, 2000), whereas intra-cerebroventricular infusion of corticosterone increases voluntary alcohol intake in animals (Fahlke et al, 1996). Given that reduced corticosterone levels are found in the global knockouts, it is unlikely that corticosterone dysregulation contributes to the observed phenotype. Excluding these obvious explanations we can only speculate that CRH/CRHR1 signaling within the HPA axis per se shows a facilitatory role on stress-induced and post-dependent alcohol drinking.
One limitation of using these genetic mouse models is potential compensatory changes that might occur as a result of the gene deletion. In fact, compensatory changes in CRHR2 or the peptide itself might have contributed to the observed phenotypes. However, in global Crhr1-knockout mice no alterations in CRHR2 have been observed (Timpl et al, 1998) and therefore it seems unlikely that CRHR2 levels are altered in Crhr1NestinCre mice. On the other hand, CRH levels are increased in the paraventricular nucleus (PVN) in global knockouts and augmented levels of CRH also occur in forebrain-specific Crhr1-knockout mice (Müller et al, 2003). Therefore, it can be assumed that early loss of Crhr1 in Crhr1NestinCre mice is also compensated by an upregulation of CRH in the PVN. As these compensatory changes in CRH are aligned in both mouse models, it seems very unlikely that they have contributed to the opposing phenotypes. A further limitation is that both genetic mouse models derive from a different genetic background, which explains slight differences in baseline alcohol intake and potentially may also influence the other phenotypic observations.
If CRHR1 signaling within the HPA system shows an opposing role to the one in extra-hypothalamic sites, one could assume that CRHR1 blockade would not be efficient in relapse that is not triggered by stress. Indeed, we found that relapse-like drinking behavior was unaffected in both knockout models, and in our pharmacological experiments we did also not observe an effect on the ADE. By contrast, Sparta et al (2009) showed that systemic application of the CRHR1 antagonist CP-154,526 protected against the ADE. When comparing these two studies, one could argue that experimental design, such as operant vs non-operant drinking procedures, different compounds, time point for data collection (2 h vs daily), species, and short-term vs long-term alcohol exposure likely have contributed to the different outcomes. Usually, the ADE procedure involves only a minor stress component as shown in neuron-specific glucocorticoid receptor knockouts that express a normal ADE (Anna Molander and Michael Cowen, unpublished results). We therefore conclude that CRHR1 blockade might primarily be an attractive treatment mechanism for patients suffering from acute stress or having a strong psychosocial adversity load (Treutlein et al, 2006; Blomeyer et al, 2008; Schmid et al, 2010).
There is convincing evidence from preclinical and human genetic research that selective CRHR1 blockade may be a treatment option in alcohol dependent patients. In line with this, several pharmaceutical companies have developed new tools to be translated into the human situation. However, application of specific CRHR1 antagonists in the translatable human situation may face two limitations: First, only a relapse risk that is driven by a high stress load may be efficiently attenuated. Second, the actions of CRHR1 antagonists on HPA axis activity might counteract their desired therapeutic effects in alcohol-dependent patients.
References
Barr CS (2010). Commentary on Nelson et al (2010). Addict Biol 15: 12–14.
Becker HC, Lopez MF (2004). Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res 28: 1829–1838.
Blomeyer D, Treutlein J, Esser G, Schmidt MH, Schumann G, Laucht M (2008). Interaction between CRFR1 gene and stressful life events predicts adolescent heavy alcohol use. Biol Psychiatry 63: 146–151.
Chu K, Koob GF, Cole M, Zorrilla EP, Roberts AJ (2007). Dependence-induced increases in ethanol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacol Biochem Behav 86: 813–821.
Contet C, Gardon O, Filliol D, Becker JA, Koob GF, Kieffer BL (2011). Identification of genes regulated in the mouse extended amygdala by excessive ethanol drinking associated with dependence. Addict Biol 16: 615–619.
Crusio WE (2004). Flanking gene and genetic background problems in genetically manipulated mice. Biol Psychiatry 56: 381–385.
Crusio WE, Goldowitz D, Holmes A, Wolfer D (2009). Standards for the publication of mouse mutant studies. Genes Brain Behav 8: 1–4.
De Kloet ER (2004). Hormones and the stressed brain. Ann N Y Acad Sci 1018: 1–15.
Fahlke C, Engel JA, Eriksson CJ, Hard E, Soderpalm B (2004). Involvement of corticosterone in the modulation of ethanol consumption in the rat. Alcohol 11: 195–202.
Fahlke C, Eriksson CJ (2000). Effect of adrenalectomy and exposure to corticosterone on alcohol intake in alcohol-preferring and alcohol-avoiding rat lines. Alcohol Alcohol 35: 139–144.
Fahlke C, Hard E, Hansen S (1996). Facilitation of ethanol consumption by intracerebroventricular infusions of corticosterone. Psychopharmacology 127: 133–139.
Finn DA, Snelling C, Fretwell AM, Tanchuck MA, Underwood L, Cole M et al (2007). Increased drinking during withdrawal from intermittent ethanol exposure is blocked by the CRF receptor antagonist D-Phe-CRF(12-41). Alcohol Clin Exp Res 31: 939–949.
Funk CK, O’Dell LE, Crawford EF, Koob GF (2006). Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci 26: 11324–11332.
Gehlert DR, Cippitelli A, Thorsell A, Lê AD, Hipskind PA, Hamdouchi C et al (2007). 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci 27: 2718–2726.
Gerlai R (1996). Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci 19: 177–181.
Gilpin NW, Richardson HN, Koob GF (2008). Effects of CRF1-receptor and opioid-receptor antagonists on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res 32: 1535–1542.
Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Björk K, Soverchia L et al (2006). Variation at the rat CRFr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci USA 103: 15236–15241.
Heilig M, Egli M, Crabbe JC, Becker HC (2010). Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol 15: 169–184.
Heilig M, Koob GF (2007). A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci 30: 399–406.
Hölter SM, Engelmann M, Kirschke C, Liebsch G, Landgraf R, Spanagel R (1998). Long-term ethanol self-administration with repeated ethanol deprivation episodes changes ethanol drinking pattern and increases anxiety-related behaviour during ethanol deprivation in rats. Behav Pharmacol 9: 41–48.
Hummel M, Cummons T, Lu P, Mark L, Harrison JE, Kennedy JD et al (2010). Pain is a salient ‘stressor’ that is mediated by corticotropin-releasing factor-1 receptors. Neuropharmacology 59: 160–166.
Koob GF, Le Moal M (2008). Addiction and the brain antireward system. Annu Rev Psychol 59: 29–53.
Lê A, Shaham Y (2002). Neurobiology of relapse to alcohol in rats. Pharmacol Ther 94: 137–156.
Lê AD, Harding S, Juzytsch W, Watchus J, Shalev U, Shaham Y (2000). The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology 150: 317–324.
Liu X, Weiss F (2002). Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci 22: 7844–7849.
Lu A, Steiner MA, Whittle N, Vogl AM, Walser SM, Ableitner M et al (2008). Conditional mouse mutants highlight mechanisms of corticotropin-releasing hormone effects on stress-coping behavior. Mol Psychiatry 13: 1028–1042.
Marinelli PW, Funk D, Juzytsch W, Harding S, Rice KC, Shaham Y et al (2007). The CRF1 receptor antagonist antalarmin attenuates yohimbine-induced increases in operant alcohol self-administration and reinstatement of alcohol seeking in rats. Psychopharmacology 195: 345–355.
McEwen BS (2007). Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 87: 873–904.
Müller MB, Preil J, Renner U, Zimmermann S, Kresse AE, Stalla GK et al (2001). Expression of CRHR1 and CRHR2 in mouse pituitary and adrenal gland: implications for HPA system regulation. Endocrinology 142: 4150–4153.
Müller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P et al (2003). Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci 6: 1100–1107.
Nelson EC, Agrawal A, Pergadia ML, Wang JC, Whitfield JB, Saccone FS et al (2010). H2 haplotype at chromosome 17q21.31 protects against childhood sexual abuse-associated risk for alcohol consumption and dependence. Addict Biol 15: 1–11.
Perreau-Lenz S, Zghoul T, de Fonseca FR, Spanagel R, Bilbao A (2009). Circadian regulation of central ethanol sensitivity by the mPer2 gene. Addict Biol 14: 253–259.
Richardson HN, Zhao Y, Fekete EM, Funk CK, Wirsching P, Janda KD et al (2008). MPZP: a novel small molecule corticotropin-releasing factor type 1 receptor (CRF1) antagonist. Pharmacol Biochem Behav 88: 497–510.
Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M et al (2010). Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry 67: 831–839.
Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF (2000). Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology 22: 581–594.
Romanowski CP, Fenzl T, Flachskamm C, Wurst W, Holsboer F, Deussing JM et al (2010). Central deficiency of corticotropin-releasing hormone receptor type 1 (CRH-R1) abolishes effects of CRH on NREM but not on REM sleep in mice. Sleep 33: 427–436.
Sanchis-Segura C, Spanagel R (2006). Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol 11: 2–38.
Schmid B, Blomeyer D, Treutlein J, Zimmermann US, Buchmann AF, Schmidt MH et al (2010). Interacting effects of CRFR1 gene and stressful life events on drinking initiation and progression among 19-year-olds. Int J Neuropsychopharmacol 13: 703–714.
Schmidt MV, Deussing JM, Oitzl MS, Ohl F, Levine S, Wurst W et al (2006). Differential disinhibition of the neonatal hypothalamic–pituitary–adrenal axis in brain-specific CRF receptor 1-knockout mice. Eur J Neurosci 24: 2291–2298.
Silberstein S, Vogl AM, Bonfiglio JJ, Wurst W, Holsboer F, Arzt E et al (2009). Immunology, signal transduction, and behavior in hypothalamic–pituitary–adrenal axis-related genetic mouse models. Ann N Y Acad Sci 1153: 120–130.
Sillaber I, Rammes G, Zimmermann S, Mahal B, Zieglgänsberger W, Wurst W et al (2002). Enhanced and delayed stress-induced alcohol drinking in mice lacking functional CRF1 receptors. Science 296: 931–933.
Sommer WH, Rimondini R, Hansson AC, Hipskind PA, Gehlert DR, Barr CS et al (2008). Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala CRFr1 expression following a history of dependence. Biol Psychiatry 63: 139–145.
Spanagel R (2009). Alcoholism—a systems approach from molecular physiology to behavior. Physiol Rev 89: 649–705.
Spanagel R, Bartsch D, Brors B, Dahmen N, Deussing J, Eils R et al (2010). An integrated genome research network for studying the genetics of alcohol addiction. Addict Biol 15: 369–379.
Spanagel R, Hölter SM (1999). Long-term alcohol self-administration with repeated alcohol deprivation phases: an animal model of alcoholism? Alcohol Alcohol 34: 231–243.
Spanagel R, Kiefer F (2008). Drugs for relapse prevention of alcoholism—10 years of progress. Trends Pharmacol Sci 29: 109–115.
Sparta DR, Ferraro 3rd FM, Fee JR, Knapp DJ, Breese GR, Thiele TE (2009). The alcohol deprivation effect in C57BL/6J mice is observed using operant self-administration procedures and is modulated by CRF-1 receptor signaling. Alcohol Clin Exp Res 33: 31–42.
Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK et al (1998). Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet 19: 162–166.
Treutlein J, Kissling C, Frank J, Wiemann S, Dong L, Depner M et al (2006). Genetic association of the human corticotropin releasing hormone receptor 1 (CRFR1) with binge drinking and alcohol intake patterns in two independent samples. Mol Psychiatry 11: 594–602.
Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC et al (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99–103.
Uhart M, Wand GS (2009). Stress, alcohol and drug interaction: an update of human research. Addict Biol 14: 43–64.
Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP et al (2002). Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res 26: 1494–1501.
Vengeliene V, Celerier E, Chaskiel L, Penzo F, Spanagel R (2009). Compulsive drug and food taking behaviour in rodents. Addict Biol 14: 384–396.
Vengeliene V, Leonardi-Essmann F, Marston H, Sommer W, Spanagel R (2010). Glycine transporter-1 blockade leads to persistently reduced relapse-like alcohol drinking in rats. Biol Psychiatry 68: 704–711.
Vengeliene V, Siegmund S, Singer MV, Sinclair JD, Li TK, Spanagel R (2003). A comparative study on alcohol-preferring rat lines: effects of deprivation and stress phases on voluntary alcohol intake. Alcohol Clin Exp Res 27: 1048–1054.
Acknowledgements
We thank Sabrina Koch for technical assistance. This work was supported by the Bundesministerium für Bildung und Forschung (NGFN Plus; FKZ: 01GS08152, FKZ: 01GS08155 see under www.ngfn-alkohol.de and Spanagel et al, 2010; FKZ: 01GS08151) and Svenska Sällskapet för Medicinsk Forskning (SSMF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
All authors report having no conflict of interest, financial or otherwise. The authors declare that over the past 3 years RS has received compensations for research and consultant contracts from Abbott, GSK, Solvay, and Xenoport.
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article
Cite this article
Molander, A., Vengeliene, V., Heilig, M. et al. Brain-Specific Inactivation of the Crhr1 Gene Inhibits Post-Dependent and Stress-Induced Alcohol Intake, but Does Not Affect Relapse-Like Drinking. Neuropsychopharmacol 37, 1047–1056 (2012). https://doi.org/10.1038/npp.2011.297
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.297
Keywords
This article is cited by
-
GPCR and Alcohol-Related Behaviors in Genetically Modified Mice
Neurotherapeutics (2020)
-
Pituitary Adenylate Cyclase-Activating Peptide (PACAP) Signaling and the Dark Side of Addiction
Journal of Molecular Neuroscience (2019)
-
Genetic association of human Corticotropin-Releasing Hormone Receptor 1 (CRHR1) with Internet gaming addiction in Korean male adolescents
BMC Psychiatry (2018)
-
Social rank-associated stress vulnerability predisposes individuals to cocaine attraction
Scientific Reports (2018)
-
Targeted overexpression of CRH receptor subtype 1 in central amygdala neurons: effect on alcohol-seeking behavior
Psychopharmacology (2018)
