
Key Points
-
Hepatitis C virus (HCV) represents an important global healthcare burden. At least 3% of the world's population is chronically infected and consequently at risk of developing liver cirrhosis and/or hepatocellular carcinoma.
-
Morbidity and mortality rates associated with HCV are predicted to rise in the coming years, creating an urgent need for more efficacious and tolerable therapies. This is particularly important for those patients who are refractory to the current standard of care, pegylated IFN-α and ribavirin.
-
To date, no prophylactic vaccine is available. Attempts to develop peptide-based therapeutic vaccines have been undertaken with various rates of success.
-
Increased understanding of the HCV lifecycle has led to the identification of novel therapeutic targets. A number of innovative agents are in development, most notably protease and polymerase inhibitors.
-
The rapid mutation rate of HCV increases the likelihood of the emergence of viruses with reduced sensitivity to therapy, necessitating the development of treatment strategies that minimize the development of resistance.
-
Development of combination therapy strategies using agents with different modes of action has the potential to improve treatment success rates while minimizing the emergence of resistant viruses.
Abstract
Infection with the hepatitis C virus (HCV) represents an important health-care problem worldwide. The prevalence of HCV-related disease is increasing, and no vaccine is yet available. Since the identification of HCV as the causative agent of non-A, non-B hepatitis, treatment has progressed rapidly, but morbidity and mortality rates are still predicted to rise. Novel, more efficacious and tolerable therapies are urgently needed, and a greater understanding of the viral life cycle has led to an increase in the number of possible targets for antiviral intervention. Here we review the specific challenges posed by HCV, and recent developments in the design of vaccines and novel antiviral agents.
Similar content being viewed by others

Main
Hepatitis C virus (HCV) infection represents an important global health-care burden, which is likely to increase over the coming years. There are approximately 3–4 million new cases of HCV infection each year, and current estimates suggest that a minimum of 3% of the world's population (approximately 170 million people) are chronically infected, and are at risk of developing liver cirrhosis and/or hepatocellular carcinoma1. Today, in developed countries, most cases are acquired through the sharing of infected needles whilst injecting drugs or, to a much lesser extent, via sexual and perinatal transmission2. However, in a significant number of patients the route of infection remains unknown. Before the routine screening of blood for HCV, many patients were infected by blood transfusions or treatment with infected blood products. At present, most new cases of HCV infection occur in the developing world3, and it is believed that immigration will impact on HCV prevalence and subsequent disease burden in the developed world. In the developed world, infection with HCV is responsible for 50–76% of all cases of liver cancer and accounts for two-thirds of all liver transplants4.
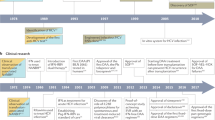
Since the discovery of the virus in 1989 (Ref. 5), the development of HCV therapy has progressed significantly (Fig. 1). With the introduction of IFN monotherapy, and the current recommendation of pegIFN-a and ribavirin, the proportion of patients achieving sustained antiviral response (SVR) has increased significantly6,7,8,9,10,11,12,13,14,15. The mechanism of action of IFN-α and ribavirin is still incompletely understood. IFN has a direct antiviral effect and acts on the immune system of the host, and ribavirin alone does not inhibit HCV replication significantly but augments the antiviral action of IFN. Importantly, ribavirin prevents relapse after the end of antiviral treatment. Despite this, the morbidity and mortality rates associated with HCV are predicted to rise in the coming years, and more efficacious and tolerable therapies are urgently required, particularly for the increasing proportion of patients who are refractory to treatment with IFN-α and ribavirin. Numerous studies have estimated the extent to which the burden of the disease will increase, but these projections may prove to be an underestimate. Consequently, HCV-related annual mortality is set to increase in most Western countries over the next two decades. In France, for example, the likely future mortality of HCV has been examined using the back-calculation method; this predicted a rise from 3,000 in 1998 to 4,500 in 2022 (Ref. 16). This is unlikely to change unless at least 50% of the HCV-infected population is treated effectively. For this, HCV carriers have to be readily identified. Projections in the United States suggest that if half of all patients infected with HCV are identified, even with the most aggressive treatment at optimal doses and durations, the best possible outcome is a 24% reduction in the incidence of decompensated cirrhosis after 20 years17. By 2020, the proportion of all US HCV cases with liver cirrhosis is estimated to increase from 16% to 32%, and decompensation will increase by 106% over current levels17 resulting in an increased need for liver transplantation.

The proportion of patients achieving a sustained virological response (SVR) has increased with advances in the treatment of hepatitis C virus (HCV) infection, from interferon (IFN) monotherapy to the current standard of care. The numbers above the columns, and the paler shaded area of the columns, represent the ranges of SVR reported in the literature for each treatment or patient population6,7,8,9,10,11,12,13,14. PegIFN, pegylated-interferon.
This Review is based on a satellite meeting held at the 12th International Symposium on Viral Hepatitis and Liver Disease in Paris, 2006, and is updated to include the current knowledge and recent developments in the field of HCV therapy at the time of publication. Given the shortcomings of current HCV treatment, we examine emerging new therapies for HCV, the impact of viral resistance, and key lessons from HIV management, in particular the potential of combination treatment strategies.
Obstacles in current HCV management
Recent studies have highlighted the barriers and challenges that exist in ensuring patients newly diagnosed with HCV receive appropriate treatment18,19. In a US study of patients infected with HCV in primary care, obstacles to receiving appropriate treatment included negative views of patients regarding treatment, inadequate patient follow-up, a tendency for providers not to consider treatment of past drug abusers, and delays in obtaining specialist input18. An observational study in the UK found that among all patients diagnosed with HCV over a 2-year period, only about half of all patients were appropriately referred for further management and only 10% began treatment19. Conversely, in France, the Ministry of Health has implemented a nationwide viral hepatitis prevention and control programme aimed at increasing both detection of seropositive individuals and provision of antiviral treatment20. By 2002, it was estimated that 60% of new HCV patients had been diagnosed through improved HCV screening programmes, and the number of patient referrals to hepatology reference centres had more than doubled from 2,063 in 2000 to 4,259 in 2002. Despite this success, the programme recommended that additional efforts and new strategies were needed to improve treatment compliance and for treating non-responders20. Nationwide screening for HCV began in 2002 in Japan, and as a consequence, a reduction in hepatocellular carcinoma and in the number of candidates requiring liver transplantation is anticipated21.
Limitations of current treatment options
Long-term studies have shown that SVR indicates clearance of virus and cure of the disease22,23. However, the response to therapy is dependent on several factors, including viral genotype (Fig. 1) and patient characteristics. There are six different genotypes of HCV, with numerous subtypes. Genotype 1 is the most prevalent and most difficult to treat viral strain in Europe and North America, and represents the greatest unmet treatment need24. Genotypes 2 and 3 appear to be more prevalent in the Far East. Of the other genotypes, genotype 4 is common in Africa and the Middle East, whereas genotypes 5 and 6 are predominant in South Africa and South-East Asia, respectively3.
Certain patient populations are difficult to treat; these include non-responders to prior treatment with IFN-based therapies, patients with severe liver fibrosis or cirrhosis, those of African–American ethnicity, individuals co-infected with HIV, and patients with comorbidities, such as alcohol consumption, fatty liver or insulin resistance25,26,27,28,29,30,31,32. For example, response rates in African–American patients with genotype 1 HCV have been shown to be as low as 6–26%, and 50% in those with genotypes 2 or 3 (Refs 29,33). This is compared with overall cure rates of 40–50% for genotype 1 and more than 75% in patient groups with genotypes 2 and 3 (Refs 8,11–13).
There are no approved treatment options available for patients who have failed to respond to previous treatments. Studies suggest that in non-responders to IFN monotherapy, re-treatment with pegIFN and ribavirin can achieve SVR rates of 25–40%; and in non-responders to IFN and ribavirin, re-treatment can achieve SVR rates of up to 10%28,34. It has also been shown that extending the treatment duration in slow responders infected with HCV genotype 1 might increase the rate of SVR to the current standard of care for this patient population105,106. Trials investigating re-treatment of non-responders with current standard of care are ongoing, but the results available so far are not promising.
Studies in patients co-infected with HIV have shown SVR rates of 17–62% (17–29% for genotypes 1 or 4 and 44–62% for genotypes 2, 3 and 5)31,32. These responses may, in part, be explained by viral kinetics — the response to therapy generally being delayed in patients with co-infection31,32. For example, Torriani et al. state that although patients who are mono-infected with HCV genotype 2 or 3 require 24 weeks of pegIFN-α plus ribavirin therapy, those co-infected with HCV and HIV probably need 48 weeks of treatment31. This could be due to the higher viral load in co-infected patients, as well as host immune deficiency. It should be noted that in initial trials for HIV–HCV co-infected patients, lower ribavirin dosages were used than dosages commonly recommended for treatment of HCV infection. Subsequent studies were able to demonstrate significantly higher SVR rates in HIV–HCV co-infected patients with genotype 1 infection if standard weight-adapted ribavirin dosing was used35.
In addition to inadequate response rates, current therapies are associated with numerous side effects, including flu-like illness, fever, fatigue, haematological disease, anaemia, leucopaenia, thrombocytopaenia, alopecia and depression. Treatment-associated side effects are an important consideration in the management of patients with HCV. A review of current treatments indicates that side effects may reduce adherence to therapy, resulting in 10–20% of premature treatment discontinuations36. Consequently, improvements in tolerability and the addition of supportive strategies, such as patient-focused treatment education, may drive overall success rates.
As the ultimate goal of HCV therapy is the complete elimination of the virus in all patients, new strategies for treatment are needed. Prophylactic and therapeutic vaccines are in development, and new approaches include the development of innovative new agents targeting different stages of the viral life cycle, as well as improvements to current strategies. Furthermore, a combination of complementary approaches and individualization based on genotype, viral load and early virological response will improve outcomes.
HCV vaccine development
As yet, no prophylactic vaccine is available for HCV, but extensive studies of a recombinant vaccine in chimpanzees showed encouraging results. Based on the viral envelope proteins E1/E2 (see Fig. 2a), it protected more than 80% of the animals from developing chronic infection following the experimental challenge with either homologous or heterologous HCV-1a viral strains107. A T-cell vaccine eliciting broad cellular responses to HCV-1b non-structural proteins 3, 4 and 5, was also shown to exhibit prophylactic activity in chimpanzees after heterologous HCV-1a challenge108.

Hepatitis C virus (HCV) is a single-stranded RNA virus belonging to the Flaviviridae family75. a | Genomic organization of proteins encoded by HCV, comprising the structural proteins core (C), envelope 1 (E1), envelope 2 (E2), and P7 (presumed to be an ion channel) and the non-structural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B), which are mainly enzymes essential to the viral life cycle. b | The nucleocapsid of the HCV genome is surrounded by an envelope that facilitates attachment and penetration into host cells. Upon enty into the host cell by endocytosis, the virus undergoes a fusion and uncoating step. Its RNA genome is translated into a polyprotein of approximately 3,000 amino acids5, that is processed by cellular and viral proteases (including NS3) to yield four structural and six non-structural proteins50. The non-structural protein NS5B, a RNA-dependent RNA polymerase, catalyses the replication of the viral genome; negative-strand RNA intermediates are formed, which, in turn, serve as templates for the synthesis of new positive-strand RNAs. These are either encapsulated to form new viruses or used as mRNA for viral protein synthesis. The newly formed viral particles are released by exocytosis97. Each HCV structure represents a potential antiviral target for drug and vaccine development46,98. For example, protease inhibitors target the NS3/4 protease, which is essential for viral polyprotein processing; polymerase inhibitors target the NS5B RNA-dependent RNA polymerase, which is essential for viral RNA replication; cyclophilin inhibitors block cyclophilin-induced stimulation of RNA-binding activity of NS5B; and α-glucosidase inhibitors block the action of a host enzyme required for viral assembly, release and infectivity. Examples of drugs that are or have been in clinical development are included. Fig. 1b modified with permission from Nature Ref. 99 © (2005) Macmillan Publishers Ltd.
Several approaches are also being taken to develop therapeutic vaccines. For example, a clinical-grade HCV E1 protein produced and purified from mammalian cells (InnoVac-C) has been evaluated in clinical trials37,38. In a Phase IIa study involving 35 patients with chronic HCV infection, cellular immune responses were boosted with a recombinant E1 vaccine, including a significant T-cell response. However, these cellular immune responses were not accompanied by any significant reductions in serum HCV RNA37. Another peptide-based therapeutic HCV vaccine, IC-41, also induced significant T-cell responses, but HCV decay was not more than 1 log10 in individual patients39. The only parameter that was shown to correlate with RNA response to IC-41 was an increase in HCV-specific IFN-γ secreting CD8+ cytotoxic T cells above a critical threshold. A clinical trial was initiated with the aim to increase T cell responses, and an optimized schedule increased responder rates, caused a fivefold stronger CD8+ response (sustained for at least 20 weeks), and a broader induction of cytotoxic T-cell responses40. The optimized regimen is currently being tested in a clinical trial of treatment-naive HCV patients. Such immune boosting in HCV carriers is likely to be most effective when used as an adjunct therapy with standard-of-care antiviral drugs. Other approaches to therapeutic HCV vaccines include the use of the recombinant core protein adjuvanted with Iscomatrix. This combination elicited an unusually strong T-helper and cytotoxic T-cell response to HCV in rhesus macaques109, and a clinical trial in HCV patients who previously failed IFN therapy is underway.
Innovative agents in clinical development
For the development of new, specific anti-HCV drugs, an understanding of the HCV life cycle (Fig. 2b), in particular the genomic organization and polyprotein processing, is essential. It has resulted in the development of several agents that target specific stages of the life cycle, the so-called specifically targeted antiviral therapy for HCV (STAT-C) drugs. Potential processes for viral inhibition include virus entry into the host cell, proteolytic processing, RNA replication, and the assembly and release of the new virions. Among the most promising new agents in development are the protease and polymerase inhibitors, as discussed below. RNA-targeted therapies, such as antisense oligonucleotides41, ribozymes42 and small interfering RNA (siRNA)-targeting structures43, have shown substantial success at inhibiting the HCV life cycle in vitro, but not in vivo. The structural viral envelope proteins E1 and E2, as well as their assembly, represent other potential antiviral targets44,110. Analogous to a recently developed HIV cell fusion inhibitor, detailed understanding of HCV cell fusion and cell entry could permit the development of specific HCV entry inhibitors.
Protease inhibitors. The non-structural protein NS3 possesses a protease domain that is responsible for polyprotein processing and is a potential target for antiviral intervention. Despite the catalytic site being a shallow and largely hydrophobic groove, making it difficult to target, several compound inhibitors of the NS3 protease have been successfully designed and are currently in preclinical and clinical development (for example, telaprevir (VX-950), boceprevir (SCH503034) BI12202, MK-7009, TMC435350 and ITMN-191). The proof-of-principle for this class of compounds was provided by BILN 2061, an NS3 protease inhibitor that provides at least a 2−3 log10 decrease in HCV load within 48 hours45. However, the clinical development of BILN 2061 was stopped owing to significant side effects.
Protease inhibitors have been associated with substantial reductions in serum HCV RNA in clinical studies when given alone or in combination with pegIFN-α46,47,48,49 (see also clinical trials section below). NS3 possesses a helicase domain that has multiple functions, including RNA-stimulated nucleoside 5′-triphosphate hydrolase (NTPase) activity, RNA binding and unwinding of RNA regions with extensive secondary structure. Other potential targets include the NTP binding site and the binding site for single-stranded RNA50.
Polymerase inhibitors. The protein NS5B is cleaved from the HCV polyprotein by the NS3 serine protease, and functions as a RNA-dependent RNA polymerase. It is the key enzyme for synthesis of a complementary minus-strand RNA, using the genome as a template, and the subsequent synthesis of genomic plus-strand RNA from this minus-strand RNA template. Several compound inhibitors of the NS5B polymerase are, or have been, in clinical development. Two separate classes of compounds have shown inhibitory effects on the NS5B through two distinct mechanisms: first, nucleoside polymerase inhibitors, which directly inhibit the active site causing chain termination (for example, valopicitabine (NM-283), MK-0608, R1626, PSI-6130 and its prodrug R7128), and second, non-nucleoside polymerase inhibitors, which cause allosteric inhibition resulting in a conformational change of the protein (for example, BILB 1941 and HCV-796)50. Preclinical studies have shown that agents targeting the HCV RNA polymerase are associated with significant reductions in serum HCV RNA51 and clinical studies have demonstrated the promising antiviral effects of NS5B inhibitors when used either as monotherapy or in combination with pegIFN-α (Refs 52–54). However, due to safety concerns and unfavourable risk-benefit profiles, the development of several polymerase inhibitors, including HCV-796, BILB 1941 and valopicitabine, is on hold.
Immune modulators. Other mechanisms that are under investigation include immune modulators targeting the cellular immune response, which plays a major role in HCV infection. Examples include agents that generate and/or promote an effective immune response by inducing or modulating cytokine responses, such as the toll-like receptor (TLR) agonists (for example, CPG 10101 and ANA 975), which have shown antiviral efficacy in initial clinical studies55. CPG 10101 (Coley Pharmaceuticals) is a synthetic oligodeoxynucleotide TLR9 agonist that also induces T-helper type 1 cytokine responses, resulting in high levels of type 1 IFN, natural killer (NK) cell stimulation and other viral-specific immunomodulatory responses. In a Phase 1b clinical trial, patients with HCV genotype 1 who received at least 1 mg CPG 10101 twice a week for 4 weeks experienced increases in IFN-α and other markers of immune response along with a mean 1 log10 decline in HCV RNA levels55,111. However, improved SVR results have not been reported so far. The clinical development of the TLR7 and TLR9 agonists is currently on hold — Coley Pharmaceuticals has stopped further development of CPG 10101 for viral hepatitis and are concentrating their efforts towards the more promising use of CPG 10101 as an anticancer drug. The development of the TLR7 agonist ANA 975 (Anadys Pharmaceuticals) was stopped owing to preclinical safety issues, as it was found to induce a general inflammatory response in animals.
Further novel investigational agents. The effectiveness of inhibitors of cyclophilin B (for example, NIM-811 and DEBIO-025), a host factor involved in viral replication, is being evaluated in patients with HCV. NIM-811, a cyclosporin A analogue, suppresses HCV genome replication in a cell culture system and may provide a novel strategy for anti-HCV treatment56,57. DEBIO-025 has demonstrated strong antiviral activity in vitro against HCV genotype 1 and HIV-1. In a Phase Ib study of HCV–HIV co-infected patients, those receiving treatment with DEBIO-025 achieved a mean HCV viral load reduction of 3.6 log10 after 15 days compared with 0.7 log10 for patients receiving placebo58.
Recently, it has also been reported that NS4A, a cofactor for the NS3 protease, is a valid therapeutic target for chronic HCV infection. ACH-806 (GS-9132) binds to HCV NS4A, inhibiting the correct proteolytic processing of the HCV polyprotein and thereby the formation of a functional replication complex, consequently decreasing viral RNA synthesis. Results of a randomized, double-blind, placebo-controlled, dose-escalation study demonstrated clinical proof-of-concept, although reversible nephrotoxicity precludes further development of ACH-806 (Ref. 59). Furthermore, glucosidase inhibitors have been in development for many years albeit with slow progress.
Improvements to current therapies. Longer-acting IFNs and IFN-inducing molecules are in development. One example is albinterferon-α2b (albIFN-α2b), a fusion protein comprising albumin and IFN-α2b, which has been shown to have antiviral activity in a clinical trial setting, with a less frequent dosing regimen than current pegIFNs60. A recent Phase IIb, active-controlled study evaluated the efficacy and safety of three therapeutic dosage regimens of albIFN-α2b (900 μg or 1200 μg every 2 weeks or 1200 μg every 4 weeks) compared with pegIFN-α2a (180 μg once a week) in treatment-naive patients with genotype 1 chronic HCV infection. All treatments were in combination with ribavirin 1,000–1,200 mg per day (based on body weight). SVR rates for the albIFN-α2b arms were 58.5% for 900 μg every 2 weeks, 55.5% for 1,200 μg every 2 weeks and 50.9% for 1200 μg every 4 weeks, compared with 57.9% for the weekly pegIFN-α2a arm61. In addition, patients who received albIFN-α2b 900 μg every 2 weeks reported less impairment of quality of life (measured using the SF-36 Health Survey62) than those who received weekly pegIFN-α2a. These data suggest that albIFN-α2b given every 2 weeks may offer comparable efficacy to pegIFN-α2a, with an improved dosing schedule and the potential for less impairment of quality of life.
Other strategies to improve IFN efficacy include gene shuffle (this compound was developed by Maxygen and was in development together with Roche), IFN variants112 and the development of long-lasting IFNs, like albIFN-α2b (Human Genome Sciences and Novartis Pharma), locteron (OctoPlus) and omega IFN with a subcutaneous delivery device (Intarcia Therapeutics) lasting 12 weeks. Furthermore, ribavirin derivatives have been developed to improve efficacy and tolerability — these include levovirin and viramidine (taribavirin). However, combination of levovirin and pegIFN-α2a fails to generate virological responses comparable with ribavirin–pegIFN-α2a combination therapy in patients with chronic HCV63. Fixed-dose viramidine was shown to be less efficacious than ribavirin in two Phase III clinical studies, although anaemia rates were significantly lower in patients treated with viramidine compared with those treated with ribavirin64,65. Weight-based dosing of viramidine is currently being evaluated in a Phase IIb study of treatment-naive patients with HCV genotype 1.
Clinical trials of NS3 and NS5B inhibitors
The two novel innovative agents furthest in clinical development (late Phase II) (Fig. 3) are the protease inhibitors telaprevir (VX-950) and boceprevir (SCH503034). Valopicitabine (NM-283) was the first polymerase inhibitor to reach Phase IIb clinical testing, but was recently placed on clinical hold in the United States following a review by the Food and Drug Administration (FDA)66. These three agents have been shown to have significant antiviral activity in patients with HCV genotype 1, including treatment-naive patients and those not responding to other therapies54,67,68,69,70.

New antivirals for the treatment of HCV and present stage of development.
A Phase II clinical study in treatment-naive patients with genotype 1 evaluated valopicitabine 200–800 mg once a day with pegIFN-α2a for 12 weeks compared with pegIFN-α2a alone for the first 4 weeks, followed by pegIFN-α2a and valopicitabine (400–800 mg) from week 5 onwards71. At week 4, all combination therapy groups demonstrated greater reductions in HCV RNA than the pegIFN-α2a monotherapy group, and end-of-treatment data indicated that valopicitabine maintained antiviral activity for up to 48 weeks. In a Phase IIb study in non-responders to pegIFN-α2a and ribavirin, SVR data demonstrated comparable results for valopicitabine plus pegIFN-α2a versus re-treatment with pegIFN-α and ribavirin; SVR was not achieved by any patient in the valopicitabine plus pegIFN-α2a arm and one patient (3%) in the pegIFN-α2a and ribavirin arm72. The clinical hold imposed by the FDA was based on the agency's overall assessment of the risk–benefit profile observed to date. R1626, a prodrug of R1479, is a polymerase inhibitor currently in Phase II development, which has shown a maximum mean (median) HCV RNA reduction of 3.7 (4.1) log10 in treatment-naive patients at a dose of 4,500 mg twice daily for 14 days73.
Phase Ib studies have evaluated telaprevir as monotherapy69 and in combination with pegIFN-α2a and ribavirin in treatment-naive patients with HCV genotype 1 (Ref. 46). Telaprevir was well tolerated as monotherapy (750 mg every 8 hours) for 14 days and in combination with pegIFN-α2a and ribavirin, and patients receiving telaprevir plus pegIFN-α2a and ribavirin demonstrated the largest reduction in plasma HCV RNA levels46. Telaprevir is currently being evaluated in three Phase II studies. An interim analysis of one of these studies, PROVE 1, showed that 70% of patients who received telaprevir (750 mg every 8 hours) plus pegIFN-α2a and ribavirin had HCV RNA below 10 IU per ml after 12 weeks of treatment compared with 39% of patients who were treated with pegIFN-α2a and ribavirin alone (intention-to-treat analysis)74. According to the study protocol, patients in one of the study treatment arms (telaprevir plus pegIFN-α2a plus ribavirin) were eligible to stop all treatment at week 12 if they met certain on-treatment criteria, including a rapid virological response (RVR, defined as less than 10 IU per ml HCV RNA at week 4) and maintenance of this response at week 10. Nine out of 17 patients achieved week-4 RVR and discontinued therapy at 12 weeks; six of these patients continued to have undetectable HCV RNA 20 weeks post-treatment. Of the remaining eight patients in this study arm, four discontinued owing to adverse events before week 12 and four did not achieve RVR. The first SVR data of the PROVE 1 study114, as well as first results from PROVE 2, another Phase II trial of telaprevir with treatment naive patients, have just been reported115.
A dose-ranging study of boceprevir (100–400 mg twice a day) in patients with HCV genotype 1 that had previously failed pegIFN-α2a therapy indicates that this protease inhibitor has dose-related antiviral activity as monotherapy70. A Phase Ib 14-day study of boceprevir (200 or 400 mg three times daily) administered in combination with pegIFN-α2a (1.5 μg per kg weekly) demonstrated a dose–response relationship in non-responder patients with HCV genotype 1. Mean maximum log10 reductions in HCV RNA were 2.45 and 2.88 for 200 and 400 mg boceprevir plus pegIFN-α2a, respectively49, and the combination of agents provided greater antiviral activity than either drug as monotherapy. Boceprevir 800 mg three times a day is currently being evaluated in combination with pegIFN-α2a and ribavirin in a Phase II trial of non-responders. A further Phase II trial of boceprevir 800 mg three times a day in combination with pegIFN-α2a and ribavirin has also been initiated in treatment-naive patients. Recent preliminary results from this so-called SPRINT (Serine Protease Inhibitor Therapy) study are comparable to the two telapravir Phase II studies in treatment naive patients113,114,115.
Many of the studies with novel agents conducted so far have focused on the response in patients infected with genotype 1. Studies of the agents in patients infected with other genotypes and in non-responder populations with refractory disease are also required as clinical programmes progress. For example, clinical studies with the now discontinued protease inhibitor BILN-2061 highlighted that antiviral activity may be less pronounced in patients infected with genotypes 2 or 3 compared with those infected with genotype 1 (Refs 48,75).
Resistance to new HCV antivirals
Response to therapy is dependent on several factors including treatment-related factors, host characteristics (including the ability of host cells to respond to IFN, induce antiviral defences and clear infected cells), viral-related factors and disease-related factors76,78,79. In addition, the genetic heterogeneity or quasispecies nature of HCV has important therapeutic implications, as the generation and selection of resistant variants can allow the virus to escape the antiviral pressure exerted by treatment77. Indeed, mutations in both the polymerase and protease enzymes have already been identified (Table 1). In addition, the overall prevalence of individual mutations changes over time, indicating that the relative fitness (that is, the ability to replicate) of a resistant variant will have a role in viral dynamics during treatment.
As previously discussed, many emerging HCV treatments are targeted against specific HCV enzymes; among the most promising are the NS3 serine protease inhibitors and the NS5B RNA-dependent RNA polymerase inhibitors. As the active site for protease inhibitors is a long shallow groove, a single-point mutation in this enzyme might be sufficient to hinder the binding of these antivirals, with different mutations conferring low-level or high-level resistance (Fig. 4). For example, sequencing studies using samples from patients treated with telaprevir have identified several mutations that confer low-level and high-level resistance80. Resistant isolates are selected rapidly and therefore combination therapy with pegIFN-α2a or other antiviral agents will be required to limit the development of resistance to telaprevir. As far as we know, telaprevir-resistant mutants are sensitive to IFN-α. The T54A mutation will confer resistance to both telaprevir and boceprevir, whereas the A156S mutation leads to resistance to telaprevir, but not boceprevir81,82. There have been several other reports of the selection of HCV-resistant mutants against various protease inhibitors using the in vitro replicon system70,83,84,85,86.

NS3 serine protease (green) and central domain of NS4A (red) showing sites of resistance mutations (D168, A156, R155). The residues that constitute the enzyme catalytic triad (H57, D81 and S139) are shown as yellow (stick representation), and the structural zinc atom is indicated in purple. The protease inhibitor BILN 2061 (discontinued) is modelled in the active site. Figure reproduced with permission from Nature Ref. 83 © (2005) Macmillan Publishers Ltd.
The active site of the NS5B RNA-dependent RNA polymerase is a highly conserved region in all HCV genotypes and any amino-acid mutations in this region may inhibit the ability of the virus to replicate (Fig. 5). This suggests that resistance to nucleoside polymerase inhibitors by mutation in the enzyme may not readily develop. Although selection of replicons resistant to 2′-C-methyl-nucleosides has shown that HCV is rapidly able to discriminate between antiviral agents and natural nucleosides81, in vitro studies have shown that replicons carrying these mutations showed decreased replication fitness83,87,88,89. There are several binding sites for non-nucleoside analogues within the NS5B polymerase (Fig. 5). Several mutations have been identified as determinants for resistance to non-nucleosides. For example, it has been demonstrated in vitro that replacement of P495 with alanine or leucine strongly reduces affinity for non-nucleoside inhibitors83,89. Such a mutation decreases the efficiency of viral replication, but viral fitness can be restored by mutations elsewhere in the NS5B coding region83.

NS5B RNA-dependent RNA polymerase (thumb, palm and finger domains are blue, green and red, respectively) showing sites of resistance mutations to nucleoside and non-nucleoside polymerase inhibitors. Figure reproduced with permission from Nature Ref. 83 © (2005) Macmillan Publishers Ltd.
In vitro data suggest a low probability of cross-resistance between some of the different nucleoside polymerase inhibitors or between nucleoside and non-nucleoside inhibitors (see also Table 1). For example, production of mutant viral strains by an amino-acid substitution at S96T alone or in combination with N142T confers resistance to R1479 (for which R1626 is the pro-drug), but not valopicitabine87,90, and the S282T substitution confers resistance to valopicitabine but not to R1479. Furthermore, molecular biology suggests no cross-resistance between protease and polymerase inhibitors83. There was also no cross-resistance observed between the cyclophilin B inhibitor NIM-811 and NM-107, the active moiety of valopicitabine91. These data suggest that NIM-811, an agent that targets host–viral interactions, provides another option for combination therapy with other antiviral agents, which would reduce the emergence of resistance82,87.
From the results of in vitro studies we can anticipate drug resistance in vivo and consider options to reduce it, such as the use of agents with a low probability of cross-resistance in combination. For example, telaprevir monotherapy in treatment-naive patients with HCV genotype 1 produced subsets of patients that had a plateau in HCV RNA decline or breakthrough response during 14 days of dosing92. Sequencing assays of the viral RNA in these patients detected that these responses correlated with the selection of viruses containing one or two mutations in the NS3 protease region. In vitro analysis demonstrated that specific mutations correlated with the level of resistance; viruses with mutations at A156V/T conferred a high level of resistance to telaprevir, whereas T54A conferred a low level of resistance. In the absence of drug-selective pressure, high-level resistant variants rapidly became undetectable and replaced with wild-type variants92. Moreover, administration of telaprevir in combination with pegIFN-α2a alone or with ribavirin appeared to prevent the selection of inhibitor-resistant variants and, hence, viral rebound46,69.
Lessons from HIV combination therapy?
The HIV epidemic had a major impact on drug development, and antiretrovirals now encompass a number of drug classes of which many have already been developed beyond first and second generation. Drug combinations have significantly changed the face of HIV management, enabling significant viral load suppression, thus preventing or delaying the development of drug-resistant mutations and thereby prolonging patient benefit by slowing disease progression.
HIV and HCV have important differences: HIV is a retrovirus that integrates into the host DNA and establishes persistent infection, whereas HCV does not integrate into the host DNA, and about 15–50% of exposed individuals clear the infection spontaneously. The viruses also differ with regard to response to therapies: HIV therapy can only suppress virus replication below the limit of detection, whereas viral clearance with HCV therapy can be achieved in a high proportion of patients. Despite these obvious differences, there are many similarities between the two diseases, including high levels of viral replication, viral heterogeneity, the importance of patient management, the use of combination therapy, the challenge of resistance to treatment and the lack of an effective vaccine93,94.
The lessons learned from the treatment of HIV may influence the approach to the future treatment of chronic HCV infections. Combinations of drugs with different mechanisms of action should allow clinicians to improve efficacy and reduce viral resistance. Analogous to HIV therapy, the success of future HCV antiviral agents will be influenced by their resistance profiles; that is, their ability to inhibit viral variants and prevent the emergence of resistance mutants. Agents with complementary, but different modes of action have the potential to be used in combination and have exhibited limited cross-resistance82,83,87,89,95. Thus, combination therapy using multiple small molecules designed to inhibit different virus-specific targets and producing diverse resistance patterns may improve response rates; for example, protease inhibitors with polymerase inhibitors or nucleoside inhibitors with non-nucleoside inhibitors96. Development of new combination strategies and the use of short-term therapy will potentially allow improved treatment success while minimizing the potential for developing resistance to any single agent. Ideal antiviral regimens should be based on potency as well as tolerability and convenience, thereby promoting adherence and minimizing the risk of treatment failure.
Outlook
There is an urgent need for a prophylactic vaccine, and for improved strategies for HCV management that achieve the ultimate goal of HCV therapy: a complete cure for all infected patients. In addition to efficacy of treatment, the duration of therapy, viral kinetics, side effects and treatment of patient populations with refractory disease are all factors that need to be addressed. Here, the lessons learned from the development of treatment regimens for HIV could prove valuable. In particular, further improvements in patient outcomes might be gained from the addition of one or more of the new small-molecule antivirals to existing regimens to improve SVR rates and/or reduce treatment duration. As well as potentially improving success rates, the advancement of combination therapies will be vital in the prevention and management of resistance to any single agent. Furthermore, as many patients cannot tolerate IFN-α or ribavirin, there also needs to be a shift toward treatment regimens that are associated with less serious side effects, which might be achieved by the use of all-oral combination therapy regimens.
Demonstrating these possibilities for one or more of the new anti-HCV STAT-C drugs in treatment-naive patients, patients who have relapsed from previous treatment, and non-responders to current treatment regimes is the next step in anti-HCV drug development. The full release of clinical trial information on novel drugs that have been or are being evaluated, whether successful or not, would also considerably enhance efforts to develop more effective therapies that could achieve the ultimate goal of curing all patients infected with HCV.
References
World Health Organization (WHO). Hepatitis C. Fact Sheet No. 164. Revised October 2000. WHO web site [online], (2000).
McHutchison, J. G. Understanding hepatitis C. Am. J. Manag. Care 10, S21–S29 (2004).
Sy, T. & Jamal, M. M. Epidemiology of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 3, 41–46 (2006).
World Health Organization (WHO). Initiative for Vaccine Research. Hepatitis C. WHO web site [online], (2007).
Choo, Q. L. et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244, 359–362 (1989). This paper describes the first isolation of the hepatitis C genome from a patient diagnosed with non-A, non-B hepatitis.
Davis, G. L. et al. Treatment of chronic hepatitis C with recombinant interferon α. A multicenter randomized, controlled trial. Hepatitis InterventionalTherapy Group. N. Engl. J. Med. 321, 1501–1506 (1989). This paper describes one of the first clinical trials to investigate the use of IFN-α in the treatment of chronic hepatitis C.
Poynard, T. et al. A comparison of three interferon α-2b regimens for the long-term treatment of chronic non-A, non-B hepatitis. Multicenter Study Group. N. Engl. J. Med. 332, 1457–1462 (1995).
McHutchison, J. G. et al. Interferon α-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N. Engl. J. Med. 339, 1485–1492 (1998).
Poynard, T. et al. Randomised trial of interferon α2b plus ribavirin for 48 weeks or for 24 weeks versus interferon α2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT). Lancet 352, 1426–1432 (1998). References 8 and 9 describe the combined use of IFN-α and ribavirin to treat chronic hepatitis C, showing combination therapy to be more effective than IFN-α alone.
Zeuzem, S. et al. Peginterferon α-2a in patients with chronic hepatitis C. N. Engl. J. Med. 343, 1666–1672 (2000).
Lindsay, K. L. et al. A randomized, double-blind trial comparing pegylated interferon α-2b to interferon α-2b as initial treatment for chronic hepatitis C. Hepatology 34, 395–403 (2001).
Manns, M. P. et al. Peginterferon α-2b plus ribavirin compared with interferon α-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358, 958–965 (2001). This paper describes the first pivotal trial to show the improved efficacy of pegylated IFN-α plus ribavirin over (non-pegylated) IFN-α plus ribavirin in the treatment of chronic hepatitis C as the new standard of care.
Fried, M. W. et al. Peginterferon α-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347, 975–982 (2002). The second paper to describe the beneficial role of pegylated IFN-α plus ribavirin as the new standard of care.
Pockros, P. J. et al. Efficacy and safety of two-dose regimens of peginterferon α-2a compared with interferon α-2a in chronic hepatitis C: a multicenter, randomized controlled trial. Am. J. Gastroenterol. 99, 1298–1305 (2004).
Zic, I. Peginterferon α/ribavirin combination therapy for the treatment of hepatitis C infection. Gastroenterol. Nurs. 28, 317–328 (2005).
Deuffic-Burban, S. et al. Comparing the public health burden of chronic hepatitis C and HIV infection in France. J. Hepatol. 40, 319–326 (2004).
Davis, G. L., Albright, J. E., Cook, S. F. & Rosenberg, D. M. Projecting future complications of chronic hepatitis C in the United States. Liver Transpl. 9, 331–338 (2003).
Morrill, J. A., Shrestha, M. & Grant, R. W. Barriers to the treatment of hepatitis C. Patient, provider, and system factors. J. Gen. Intern. Med. 20, 754–758 (2005).
Irving, W. L. et al. Clinical pathways for patients with newly diagnosed hepatitis C — what actually happens. J. Viral. Hepat. 13, 264–271 (2006).
Guerin, N. & Roudot-Thoraval, F. Hepatitis C in France. Viral Hepatitis 13, 9–12 (2005).
Kiyosawa, K. et al. Hepatocellular carcinoma: recent trends in Japan. Gastroenterology 127, S17–S26 (2004).
McHutchison, J. G. et al. Sustained virologic response (SVR) to interferon-α-2b +/− ribavirin therapy at 6 months reliably predicts long-term clearance of HCV at 5-year follow up. J. Hepatol. 44 (Suppl. 2), S275 (2006).
Swain, M. G. et al. Durable sustained virological response after treatment with peginterferon a-2a (Pegasys®) alone or in combination with ribavirin (Copegus®): 5-year follow-up and the criteria of a cure. J. Hepatol. 46 (Suppl. 1), S3 (2007).
WHO. Global surveillance and control of hepatitis C. Report of a WHO consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J. Viral Hepat. 6, 35–47 (1999).
Hoefs, J. & Aulakh, V. S. Treatment of chronic HCV infection in special populations. Int. J. Med. Sci. 3, 69–74 (2006).
Dienstag, J. L. & McHutchison, J. G. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology 130, 231–264 (2006).
Herrine, S. K., Rossi, S. & Navarro, V. J. Management of patients with chronic hepatitis C infection. Clin. Exp. Med. 6, 20–26 (2006).
Shiffman, M. L. et al. Peginterferon α-2a and ribavirin in patients with chronic hepatitis C who have failed prior treatment. Gastroenterology 126, 1015–1023 (2004).
Jeffers, L. J., Cassidy, W., Howell, C. D., Hu, S. & Reddy, K. R. Peginterferon α-2a (40 kd) and ribavirin for black American patients with chronic HCV genotype 1. Hepatology 39, 1702–1708 (2004).
Muir, A. J., Bornstein, J. D. & Killenberg, P. G. Peginterferon α-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites. N. Engl. J. Med. 350, 2265–2271 (2004).
Torriani, F. J. et al. Peginterferon α-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N. Engl. J. Med. 351, 438–450 (2004).
Carrat, F. et al. Pegylated interferon α-2b vs standard interferon α-2b, plus ribavirin, for chronic hepatitis C in HIV-infected patients: a randomized controlled trial. JAMA 292, 2839–2848 (2004).
Veldt, B. J. et al. Retreatment of hepatitis C non-responsive to interferon. A placebo controlled randomized trial of ribavirin monotherapy versus combination therapy with Ribavirin and Interferon in 121 patients in the Benelux [ISRCTN53821378]. BMC Gastroenterol. 3, 24 (2003).
Brau, N. et al. Black patients with chronic hepatitis C have a lower sustained viral response rate than non-Blacks with genotype 1, but the same with genotypes 2/3, and this is not explained by more frequent dose reductions of interferon and ribavirin. J. Viral. Hepat. 13, 242–249 (2006).
Nunez, M. et al. The PRESCO Study: role of extended therapy and/or optimal doses of ribavirin in the treatment of chronic hepatitis C in HIV-infected patients. National AIDS Treatment Advocacy Project web site [online], (2006).
Manns, M. P., Wedemeyer, H. & Cornberg, M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut 55, 1350–1359 (2006).
Nevens, F. et al. A pilot study of therapeutic vaccination with envelope protein E1 in 35 patients with chronic hepatitis C. Hepatology 38, 1289–1296 (2003).
Leroux-Roels, G. et al. Immunogenicity and tolerability of intradermal administration of an HCV E1-based vaccine candidate in healthy volunteers and patients with resolved or ongoing chronic HCV infection. Hum. Vaccin. 1, 61–65 (2005).
Manns, M. P. et al. Immunization with the therapeutic hepatits C virus peptide vaccine IC41 in 66 chronic hepatits C non-responder patients. Hepatology 40 (Suppl. 1), 251A (2004).
Klade, C. et al. Therapeutic peptide vaccination against chronic hepatitis C virus infection. J. Hepatol. 46 (Suppl. 1), S229 (2007).
McHutchison, J. G. et al. A phase I trial of an antisense inhibitor of hepatitis C virus (ISIS 14803), administered to chronic hepatitis C patients. J. Hepatol. 44, 88–96 (2006).
Jarczak, D., Korf, M., Beger, C., Manns, M. P. & Kruger, M. Hairpin ribozymes in combination with siRNAs against highly conserved hepatitis C virus sequence inhibit RNA replication and protein translation from hepatitis C virus subgenomic replicons. FEBS J. 272, 5910–5922 (2005).
Kanda, T., Steele, R., Ray, R. & Ray, R. B. Small interfering RNA targeted to hepatitis C virus 5′ nontranslated region exerts potent antiviral effect. J. Virol. 81, 669–676 (2007).
Li, Y. P., Kang, H. N., Babiuk, L. A. & Liu, Q. Elicitation of strong immune responses by a DNA vaccine expressing a secreted form of hepatitis C virus envelope protein E2 in murine and porcine animal models. World J. Gastroenterol. 12, 7126–7135 (2006).
Lamarre, D. et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426, 186–189 (2003). This paper describes the study that established the proof-of-principle for the use of protease inhibitors in the treatment of HCV infection.
Lawitz, E. J. et al. 28 Days of the hepatitis C protease inhibitor VX-950, in combination with PEG-Interferon-α-2a and ribavirin, is well-tolerated and demonstrates robust antiviral effects. Gastroenterology 131, 950–951 (2006).
Hinrichsen, H. et al. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 127, 1347–1355 (2004).
Reiser, M. et al. Antiviral efficacy of NS3-serine protease inhibitor BILN-2061 in patients with chronic genotype 2 and 3 hepatitis C. Hepatology 41, 832–835 (2005).
Sarrazin, C. et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon α-2b for genotype 1 nonresponders. Gastroenterology 132, 1270–1278 (2007). Beneficial role of adding interferon to the new direct antiviral hepatitis C agents (so called STAT-C drugs).
Pawlotsky, J. M. & McHutchison, J. G. Hepatitis C. Development of new drugs and clinical trials: promises and pitfalls. Summary of an AASLD hepatitis single topic conference, Chicago, IL, February 27-March 1, 2003. Hepatology 39, 554–567 (2004).
Olsen, D. B. et al. HCV antiviral activity and resistance analysis in chronically infected chimpanzees treated with NS3/4A protease and NS5B polymerase inhibitors. J. Hepatol. 46 (Suppl. 1), S298 (2007).
Roberts, S. et al. Interim results of a multiple ascending dose study of R1626, a novel nucleoside analog targeting HCV polymerase in chronic HCV patients. J. Hepatol. 44 (Suppl. 2), S269 (2006).
Afdhal, N. et al. Valopicitabine (NM283), alone or with peg-interferon, compared to peg-interferon/ribavirin (pegIFN/RBV) retreatment in hepatitis C patients with prior non-response to pegIFN/RBV: week 24 results. J. Hepatol. 44 (Suppl. 2), S19 (2006).
Dieterich, D. et al. Early clearance of HCV RNA with valopicitabine (NM283) plus peg-interferon in treatment-naive patients with HCV-1 infection: first results from a Phase IIb trial. J. Hepatol. 44 (Suppl. 2), S271 (2006).
McHutchison, J. G., Bartenschlager, R., Patel, K. & Pawlotsky, J. M. The face of future hepatitis C antiviral drug development: recent biological and virologic advances and their translation to drug development and clinical practice. J. Hepatol. 44, 411–421 (2006).
Goto, K. et al. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 343, 879–884 (2006).
Ma, S. et al. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with α interferon. Antimicrob. Agents Chemother. 50, 2976–2982 (2006).
Flisiak, R. et al. The cyclophilin inhibitor DEBIO-025 has a potent dual anti-HIV and anti-HCV activity in treatment-naive HIV/HCV co-infected subjects. National AIDS Treatment Advocacy Project web site [online], (2006).
Pottage Jr JC et al. Short-term antiviral activity and safety of ACH-806 (GS-9132), an NS4A antagonist, in HCV genotype 1 infected individuals. J. Hepatol. 46 (Suppl. 1), S294 (2007).
Balan, V. et al. A Phase I/II study evaluating escalating doses of recombinant human albumin-interferon-α fusion protein in chronic hepatitis C patients who have failed previous interferon-α-based therapy. Antivir. Ther. 11, 35–45 (2006).
Zeuzem S et al. Sustained virologic response rates with albinterferon α-2b plus ribavirin treatment in IFN-naive, chronic hepatitis C genotype I patients. Hepatology 46 (Suppl. 1), 317A (2007).
Ware JE et al. The SF-36® Health Survey: development and use in mental health research and the IQOLA Project. Int. J. Ment. Health 23, 49–73 (1994).
Pockros, P. J. et al. Combination of levovirin (LVV) and peginterferon α-2a (40KD) (Pegasys) fails to generate a virological response comparable to ribavirin (RBV, Copegus) and peginterferon α-2a (40KD) in patients with chronic hepatitis C. Hepatology 40, (Suppl. 1), 391A (2004).
Benhamou, Y. et al. The safety and efficacy of viramidine® plus pegylated interferon α-2b versus ribavirin plus pegylated interferon α-2b in therapy-naive patients infected with HCV: Phase 3 results. J. Hepatol. 44 (Suppl. 2), S273 (2006).
Marcellin P. et al. The safety and efficacy of taribavirin plus pegylated interferon α-2a versus ribavirin plus pegylated interferon α-2a in therapy-naive patients infected with HCV: Phase 3 results. J. Hepatol. 46 (Suppl. 1), S7 (2007).
Idenix Pharmaceuticals. News release 13 July 2007. Valopicitabine Development Program Placed on Clinical Hold in the United States. Idenix Pharmaceuticals web site [online], (2007).
Carroll, S. S. & Olsen, D. B. Nucleoside analog inhibitors of hepatitis C virus replication. Infect. Disord. Drug Targets 6, 17–29 (2006). A comprehensive review of nucleoside analogue polymerase inhibitors covering their development and progress in clinical trials.
Lin, C., Kwong, A. D. & Perni, R. B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets 6, 3–16 (2006).
Reesink, H. W. et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: a phase Ib, placebo-controlled, randomized study. Gastroenterology 131, 997–1002 (2006).
Zeuzem, S. et al. Antiviral activity of SCH 503034, a HCV protease inhibitor, admisitered as monotherapy in hepatitis C genotype 1 (HCV-1) patients refractory to pegylated interferon (PEG-IFN-alpha). Hepatology 42 (Suppl. 1), 233A (2005).
Lawitz, E. et al. Clearance of HCV RNA with valopicitabine (NM283) plus peg-interferon in treatment-naive patients with HCV-1 infection: results at 24 and 48 weeks. J. Hepatol. 46 (Suppl. 1), S9 (2007).
Afdhal, N. et al. Valopicitabine (NM283) alone or with peg-Interferon, compared to peg-Interferon/ribavirin (PEGIFN/RBV) retreatment in patients with HCV-1 infection and prior non-response to PEGIFN/RBV: one-year results. J. Hepatol. 46 (Suppl. 1), S5 (2007).
Roberts, S. et al. Results of a Phase 1B, multiple dose study of R1626, a novel nucleoside analog targeting HCV polymerase in chronic HCV genotype 1 patients. Hepatology 44, 692A (2006).
Vertex Pharmaceuticals Inc. Press Release 14 April 2007. Interim Results Presented at EASL from PROVE 1 Clinical Trial of Investigational Drug Telaprevir in Patients with Genotype 1 Hepatitis C. Vertex Pharmaceuticals web site [online], (2007).
Pawlotsky, J. M. & Gish, R. G. Future therapies for hepatitis C. Antivir. Ther. 11, 397–408 (2006).
Pawlotsky, J. M. The nature of interferon-α resistance in hepatitis C virus infection. Curr. Opin. Infect. Dis. 16, 587–592 (2003).
Martell, M. et al. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol. 66, 3225–3229 (1992).
Chen, L. et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology 128, 1437–1444 (2005).
Puig-Basagoiti, F. et al. Dynamics of hepatitis C virus NS5A quasispecies during interferon and ribavirin therapy in responder and non-responder patients with genotype 1b chronic hepatitis C. J. Gen. Virol. 86, 1067–1075 (2005).
Sarrazin, C. et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132, 1767–1777 (2007).
Lin, C. et al. In vitro resistance studies of hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061: structural analysis indicates different resistance mechanisms. J. Biol. Chem. 279, 17508–17514 (2004).
Tong, X. et al. Identification and analysis of fitness of resistance mutations against the HCV protease inhibitor SCH 503034. Antiviral. Res. 70, 28–38 (2006).
De Francesco, R. & Migliaccio, G. Challenges and successes in developing new therapies for hepatitis C. Nature 436, 953–960 (2005). A review article describing the novel antiviral molecules being evaluated for the treatment of hepatitis C and the development of resistance to these antivirals.
Lu, L. et al. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 48, 2260–2266 (2004).
Seiwert, S., Andrews, S. W. & Tan, H. Generation and characterization of HCV replicons with reduced sensitivity to ITMN 191, a macrocyclic inhibitor of NS3/4A. National AIDS Treatment Advocacy Project web site [online], (2006).
Trozzi, C. et al. In vitro selection and characterization of hepatitis C virus serine protease variants resistant to an active-site peptide inhibitor. J. Virol. 77, 3669–3679 (2003).
Le Pogam, S. et al. In vitro selected Con1 subgenomic replicons resistant to 2′-C-methyl-cytidine or to R1479 show lack of cross resistance. Virology 351, 349–359 (2006).
Migliaccio, G. et al. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 278, 49164–49170 (2003).
Tomei, L., Altamura, S., Paonessa, G., De Francesco, R. & Migliaccio, G. HCV antiviral resistance: the impact of in vitro studies on the development of antiviral agents targeting the viral NS5B polymerase. Antivir. Chem. Chemother. 16, 225–245 (2005).
Najera, I. et al. Resistance profile for 4′-azido-cytidine (R1479), reveals lack of cross resistance with 2′-C-methyl-cytidine, interferon-α and ribavirin. International Conference on Antimicrobial Agents and Chemotherapy web site [online], (2006).
Lin, K., Boerner, J., Ma, S. & Compton, T. NIM811, a cyclophilin inhibitor, and NM107, an HCV polymerase inhibitor, synergistically inhibits HCV replication and suppresses the emergence of resistance in vitro. J. Hepatol. 46, (Suppl. 1), S230 (2007).
Kieffer, T. et al. Wild-type HCV NS3 protease re-emerges during follow-up after 14 days of dosing with VX-950 in patients with genotype 1 HCV. J. Hepatol. 44 (Suppl. 2), S7 (2006).
Sepkowitz, K. A. AIDS — the first 20 years. N. Engl. J. Med. 344, 1764–1772 (2001).
Tan, S. L., Pause, A., Shi, Y. & Sonenberg, N. Hepatitis C therapeutics: current status and emerging strategies. Nature Rev. Drug Discov. 1, 867–881 (2002).
Lin, K., Kwong, A. D. & Lin, C. Combination of a hepatitis C virus NS3-NS4A protease inhibitor and alpha interferon synergistically inhibits viral RNA replication and facilitates viral RNA clearance in replicon cells. Antimicrob. Agents Chemother. 48, 4784–4792 (2004).
Mo, H. et al. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an HCV serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 49, 4305–4314 (2005). This paper describes the development of resistance mutations in the HCV replicon system under selective pressure from different antiviral compounds.
Penin, F., Dubuisson, J., Rey, F. A., Moradpour, D. & Pawlotsky, J. M. Structural biology of hepatitis C virus. Hepatology 39, 5–19 (2004).
Bartenschlager, R. The hepatitis C virus replicon system: from basic research to clinical application. J. Hepatol. 43, 210–216 (2005).
Lindenbach, B. D. & Rice, C. M. Unravelling hepatitis C virus replication from genome to function. Nature 436, 933–938 (2005). A detailed review of the progress that has been made in understanding HCV replication, with special emphasis on recent developments.
Villano, S. Analysis of HCV NS5B genetic variants following monotherapy with HCV-796, a non-nucleoside polymerase inhibitor, in treatment naive HCV-infected patients. Hepatology 44, 607A (2006).
Lin, C. et al. In vitro studies of cross-resistance mutations against two hepatitis C virus serine protease inhibitors, VX-950 and BILN 2061. J. Biol. Chem. 280, 36784–36791 (2005).
Koev G. et al. Antiviral interactions of an HCV polymerase inhibitor with an HCV protease inhibitor or interferon in vitro. Antiviral Res. 73, 78–83 (2007).
Le Pogam S et al. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 80, 6146–6154 (2006).
Tomei L et al. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77, 13225–13231 (2003).
Sanchez Tapias, J. M. et al. Peginterferon-alfa2a plus ribavirin for 48 versus 72 weeks in patients with detectable hepatitis C virus RNA at week 4 of treatment. Gastroenterology 131, 451–460 (2006).
Berg, T. et al. Extended treatment duration for hepatitis C virus type 1: comparing 48 versus 72 weeks of peginterferon-alfa-2a plus ribavirin. Gastroenterology 130, 1086–1097 (2006).
Houghton, M. & Abrignanis, S. Prospects for a vaccine against the hepatitis C virus Nature 436, 961–966 (2005).
Folgori, A. et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nature Med. 12, 190–197 (2006).
Polakos, N.K., et al. Characterization of hepatitis C virus core-specific immune responses primed in rhesus macaques by a nonclassical ISCOM vaccine. J. Immunol. 166, 3589–3598 (2001).
Chapel, C. et al. Reduction of the infectivity of hepatitis C virus pseudoparticles by incorporation of misfolded glycoproteins induced by glucosidase inhibitors. J. Gen Virol. 88, 1133–1143 (2007).
McHutchison, J.G. et al. Phase 1B, randomized, double-blind, dose-escalation trial of CPG 10101 in patients with chronic hepatitis C virus. Hepatology 46, 1341–1349 (2007).
Escuret, V. et al. Novel alpha interferon (IFN-alpha) variant with improved inhibitory activity against hepatitis C virus genotype 1 replication compared to IFN-alpha2b therapy in a subgenomic replicon system. Antimicrob Agents Chemother. 50, 3984–3991 (2006).
Schering–Plough. Press release 18 Oct. Initial results of Phase II Study with HCV protease inhibitor boceprevir in treatment-naive hepatitis C patients show a high rate of early virologic response. Schering-Plough web site [online], (2007).
Jacobson, I. M. et al. Interim analysis results from a Phase 2 study of telaprevir with Peginterferon alfa-2A and ribavirin in treatment-naïve subjects with hepatitis C. Hepatology 46, S1 315a–316a (2007).
Hezode, C. et al. PROVE2: Phase II study of VX950 (TELAPREVIR) in combination with Peginterferon ALFA2A with or without ribavirin in subjects with chronic Hepatitis C, first interim analysis. Hepatology 46, S1 71a (2007).
Acknowledgements
The authors would like to thank R. Douglas and L. O'Mahony for help in preparing the manuscript; this support was funded by Novartis. M.P.M., J.K.R., S.Z. acknowledge the support of Hep-Net, a network sponsored by BMBF, and M.P.M., S.Z., F.Z. are supported by VIRGIL, an EU6 Framework Project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Manns is an investigator/consultant/speaker at Novartis, Roche, Schering-Plough, Gilead Sciences Inc., Tibotec, Vertex, GlaxoSmithKline, Boehringer Ingelheim, Bristol-Myers Squibb, Idenix and Merck.
Dr Foster has received funding and payment for lectures and consultancy work from companies who are developing and/or manufacturing drugs for the treatment of hepatitis C, including Novartis, Roche, Human Genome Sciences, Idenix and Gilead.
Dr Rockstroh has received lecture honorarium or consultancy fees from Abbott, Bristol-Myers Squibb, GlaxoSmithKline, Novartis, Roche, Schering-Plough, Boehringer Ingelheim and Vertex.
Dr Zeuzem is a clinical advisor and investigator for Gilead, Human Genome Sciences, Novartis, Roche, Schering-Plough, Tibotec and Vertex.
Dr Houghton is a consultant to Novartis Vaccines and owns shares in Vertex and Pharmasset.
Related links
Related links
DATABASES
OMIM
FURTHER INFORMATION
Glossary
- Sustained antiviral response
-
HCV RNA below the limit of detection at week 24 following treatment completion.
Rights and permissions
About this article
Cite this article
Manns, M., Foster, G., Rockstroh, J. et al. The way forward in HCV treatment — finding the right path. Nat Rev Drug Discov 6, 991–1000 (2007). https://doi.org/10.1038/nrd2411
Issue Date:
DOI: https://doi.org/10.1038/nrd2411
This article is cited by
-
Alkaloid and benzopyran compounds of Melicope latifolia fruit exhibit anti-hepatitis C virus activities
BMC Complementary Medicine and Therapies (2021)
-
Investigation of the Immunomodulatory effect of Berberis vulgaris on core-pulsed dendritic cell vaccine
BMC Complementary and Alternative Medicine (2016)
-
Direkter Angriff auf das Hepatitis-C-Virus
MMW - Fortschritte der Medizin (2015)
-
Hepatitis C — Therapie-Update
CME (2015)
-
Treatment of HCV in Patients who Failed First-Generation PI Therapy: a Review of Current Literature
Current Gastroenterology Reports (2015)
