
Key Points
-
Duchenne muscular dystrophy (DMD) is characterized by a severe and progressive loss of muscle fibres, which is provoked by the dystrophin deficiency that results from frame-shifting mutations in the large dystrophin gene.
-
Conventional gene-therapy strategies (viral vectors, naked plasmids and cell transplantation) have been moderately efficient, but still have to overcome problems associated with low delivery efficiencies, immune responses, limited transgene persistence, poor cell survival and the physical barriers in muscle tissue itself.
-
Recently, relatively new and alternative strategies that are emerging from our increasing knowledge of the organization of the dystrophin gene and its role in muscle function are gaining credibility as therapeutic prospects.
-
Treatment of the mdx mouse model with DNA–RNA chimeric oligonucleotides to correct the nonsense mutation in exon 23, or with gentamicin to read through the resulting stop codon, has shown positive results. However, the efficiencies have been low and follow-up studies are required to further evaluate these methods.
-
The minimal requirements for the function of the dystrophin protein have been defined in more detail, which has allowed the construction of functional mini- and micro-dystrophins of only 6.2 kb and 3.6 kb, respectively. Now, using recombinant adeno-associated virus (rAAV) vectors, these small constructs have been efficiently delivered into the muscle of mdx mice and converted the dystrophic muscle morphology for more than six months post-injection.
-
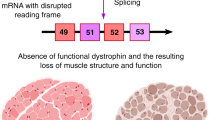
Antisense oligonucleotides have been successfully used to modify the splicing of the dystrophin pre-mRNA so that specific exons are skipped. Despite introducing a (larger) deletion, this frame-restoring treatment induced the synthesis of BMD-like dystrophins to therapeutic levels in cultured muscle cells from a series of DMD patients with different mutations, and from the mdx mouse both in vitro and in vivo.
-
Utrophin can compensate for dystrophin deficiency and its artificial upregulation in mdx mice has reduced dystrophic pathology and improved muscle performance. Several growth and transcription factors have been identified to enhance expression from the utrophin promoter A, but the challenge remains to find an effective small-molecule drug that will be sufficiently utrophin-specific to circumvent putative adverse effects.
-
In terms of safety and efficiency, non-pathogenic rAAV and synthetic small-molecule drugs for targeted exon skipping and utrophin upregulation are the most promising techniques, and they are progressing towards clinical applications in the near future.
Abstract
Since the initial characterization of the genetic defect for Duchenne muscular dystrophy, much effort has been expended in attempts to develop a therapy for this devastating childhood disease. Gene therapy was the obvious answer but, initially, the dystrophin gene and its product seemed too large and complex for this approach. However, our increasing knowledge of the organization of the gene and the role of dystrophin in muscle function has indicated ways to manipulate them both. Gene therapy for Duchenne muscular dystrophy now seems to be in reach.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
White, S. et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am. J. Hum. Genet. 71, 365–374 (2002).
van Essen, A. J., Busch, H. F., te Meerman, G. J. & ten Kate, L. P. Birth and population prevalence of Duchenne muscular dystrophy in The Netherlands. Hum. Genet. 88, 258–266 (1992).
Franco, A. & Lansman, J. B. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 344, 670–673 (1990).
Straub, V., Rafael, J. A., Chamberlain, J. S. & Campbell, K. P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 139, 375–385 (1997).
Hardiman, O. Dystrophin deficiency, altered cell signalling and fibre hypertrophy. Neuromuscul. Disord. 4, 305–315 (1994).
Nakamura, A., Harrod, G. V. & Davies, K. E. Activation of calcineurin and stress activated protein kinase/p38-mitogen activated protein kinase in hearts of utrophin–dystrophin knockout mice. Neuromuscul. Disord. 11, 251–259 (2001).
Kochanek, S. et al. A new adenoviral vector: replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and β-galactosidase. Proc. Natl Acad. Sci. USA 93, 5731–5736 (1996).
Chen, H. H. et al. Persistence in muscle of an adenoviral vector that lacks all viral genes. Proc. Natl Acad. Sci. USA 94, 1645–1650 (1997).
Havenga, M. J. et al. Exploiting the natural diversity in adenovirus tropism for therapy and prevention of disease. J. Virol. 76, 4612–4620 (2002).
Thirion, C. et al. Strategies for muscle-specific targeting of adenoviral gene transfer vectors. Neuromuscul. Disord. 12 (Suppl.), 30–39 (2002).
Akkaraju, G. R. et al. Herpes simplex virus vector-mediated dystrophin gene transfer and expression in mdx mouse skeletal muscle. J. Gene Med. 1, 280–289 (1999).
Huard, J. et al. Gene transfer to muscle using herpes simplex virus-based vectors. Neuromuscul. Disord. 7, 299–313 (1997).
Romero, N. B. et al. Current protocol of a research phase I clinical trial of full-length dystrophin plasmid DNA in Duchenne/Becker muscular dystrophies. Part II: clinical protocol. Neuromuscul. Disord. 12 (Suppl.), 45–48 (2002).
Murakami, T. et al. Full-length dystrophin cDNA transfer into skeletal muscle of adult mdx mice by electroporation. Muscle Nerve 27, 237–241 (2003).
Lu, Q. L., Liang, H. D., Partridge, T. & Blomley, M. J. Microbubble ultrasound improves the efficiency of gene transduction in skeletal muscle in vivo with reduced tissue damage. Gene Ther. 10, 396–405 (2003).
Gollins, H., McMahon, J., Wells, K. E. & Wells, D. J. High-efficiency plasmid gene transfer into dystrophic muscle. Gene Ther. 10, 504–512 (2003).
Tremblay, J. P. et al. Results of a triple blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Transplant. 2, 99–112 (1993).
Gussoni, E., Blau, H. M. & Kunkel, L. M. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nature Med. 3, 970–977 (1997).
Hodgetts, S. I., Spencer, M. J. & Grounds, M. D. A role for natural killer cells in the rapid death of cultured donor myoblasts after transplantation. Transplantation 75, 863–871 (2003).
Moisset, P. A., Gagnon, Y., Karpati, G. & Tremblay, J. P. Expression of human dystrophin following the transplantation of genetically modified mdx myoblasts. Gene Ther. 5, 1340–1346 (1998).
Qu, Z. et al. Development of approaches to improve cell survival in myoblast transfer therapy. J. Cell Biol. 142, 1257–1267 (1998).
Smythe, G. M., Fan, Y. & Grounds, M. D. Enhanced migration and fusion of donor myoblasts in dystrophic and normal host muscle. Muscle Nerve 23, 560–574 (2000).
Camirand, G., Caron, N. J., Asselin, I. & Tremblay, J. P. Combined immunosuppression of mycophenolate mofetil and FK506 for myoblast transplantation in mdx mice. Transplantation 72, 38–44 (2001).
Gussoni, E. et al. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 401, 390–394 (1999).
Sampaolesi, M. et al. Cell therapy of α-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 301, 487–492 (2003).
Liu, L., Parekh-Olmedo, H. & Kmiec, E. B. The development and regulation of gene repair. Nature Rev. Genet. 4, 679–689 (2003).
Bartlett, R. J. et al. In vivo targeted repair of a point mutation in the canine dystrophin gene by a chimeric RNA/DNA oligonucleotide. Nature Biotechnol. 18, 615–622 (2000).
Rando, T. A., Disatnik, M. H. & Zhou, L. Z. Rescue of dystrophin expression in mdx mouse muscle by RNA/DNA oligonucleotides. Proc. Natl Acad. Sci. USA 97, 5363–5368 (2000).
Bertoni, C., Lau, C. & Rando, T. A. Restoration of dystrophin expression in mdx muscle cells by chimeraplast-mediated exon skipping. Hum. Mol. Genet. 12, 1087–1099 (2003).
Barton-Davis, E. R., Cordier, L., Shoturma, D. I., Leland, S. E. & Sweeney, H. L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Invest. 104, 375–381 (1999).
Dunant, P., Walter, M. C., Karpati, G. & Lochmuller, H. Gentamicin fails to increase dystrophin expression in dystrophin-deficient muscle. Muscle Nerve 27, 624–627 (2003).
England, S. B. et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343, 180–182 (1990).
Corrado, K. et al. Transgenic mdx mice expressing dystrophin with a deletion in the actin-binding domain display a “mild Becker” phenotype. J. Cell Biol. 134, 873–884 (1996).
Warner, L. E. et al. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin–glycoprotein complex and partially prevents dystrophy. Hum. Mol. Genet. 11, 1095–1105 (2002).
Rafael, J. A. et al. Forced expression of dystrophin deletion constructs reveals structure–function correlations. J. Cell Biol. 134, 93–102 (1996).
Crawford, G. E. et al. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J. Cell Biol. 150, 1399–1410 (2000).
Yuasa, K. et al. Effective restoration of dystrophin-associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs. FEBS Lett. 425, 329–336 (1998).
Wang, B., Li, J. & Xiao, X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl Acad. Sci. USA 97, 13714–13719 (2000).
Harper, S. Q. et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nature Med. 8, 253–261 (2002). This study shows the functionality of mini- and micro-dystrophins and compares their therapeutic effect in mdx mice, following transgenic expression or rAAV-mediated transduction.
Sakamoto, M. et al. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem. Biophys. Res. Commun. 293, 1265–1272 (2002).
Fabb, S. A., Wells, D. J., Serpente, P. & Dickson, G. Adeno-associated virus vector gene transfer and sarcolemmal expression of a 144 kDa micro-dystrophin effectively restores the dystrophin-associated protein complex and inhibits myofibre degeneration in nude/mdx mice. Hum. Mol. Genet. 11, 733–741 (2002).
Watchko, J. et al. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther. 13, 1451–1460 (2002). This paper further shows the functionality of a mini-dystrophin gene that, besides ameliorating the dystrophic histopathology of mdx muscle, also increases its force and resistance to mechanical stress.
Cordier, L. et al. Muscle-specific promoters may be necessary for adeno-associated virus-mediated gene transfer in the treatment of muscular dystrophies. Hum. Gene Ther. 12, 205–215 (2001).
Yuasa, K. et al. Adeno-associated virus vector-mediated gene transfer into dystrophin-deficient skeletal muscles evokes enhanced immune response against the transgene product. Gene Ther. 9, 1576–1588 (2002).
Chao, H. et al. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol. Ther. 2, 619–623 (2000).
Hauck, B. & Xiao, W. Characterization of tissue tropism determinants of adeno-associated virus type 1. J. Virol. 77, 2768–2774 (2003).
Sherratt, T. G., Vulliamy, T., Dubowitz, V., Sewry, C. A. & Strong, P. N. Exon skipping and translation in patients with frameshift deletions in the dystrophin gene. Am J. Hum. Genet. 53, 1007–1015 (1993).
Nicholson, L. V. The “rescue” of dystrophin synthesis in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 3, 525–531 (1993).
Shiga, N. et al. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J. Clin. Invest. 100, 2204–2210 (1997).
Ginjaar, I. B. et al. Dystrophin nonsense mutation induces different levels of exon 29 skipping and leads to variable phenotypes within one BMD family. Eur J. Hum. Genet. 8, 793–796 (2000).
Fanin, M. et al. Dystrophin-positive fibers in Duchenne dystrophy: origin and correlation to clinical course. Muscle Nerve 18, 1115–1120 (1995).
Lu, Q. L. et al. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J. Cell Biol. 148, 985–996 (2000). An extensive study that shows that multiple exon skipping, rather than secondary deletion, is the most likely mechanism behind the presence of dystrophin-positive revertant fibres in mdx muscle.
Matsuo, M. et al. Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy kobe. J. Clin. Invest. 87, 2127–2131 (1991).
Tanaka, K., Watakabe, A. & Shimura, Y. Polypurine sequences within a downstream exon function as a splicing enhancer. Mol. Cell Biol. 14, 1347–1354 (1994).
Cartegni, L., Chew, S. L. & Krainer, A. R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nature Rev. Genet. 3, 285–298 (2002).
Pramono, Z. A. et al. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochem. Biophys. Res. Commun. 226, 445–449 (1996). This is the first report of antisense-induced exon skipping from the dystrophin transcript.
Dunckley, M. G., Manoharan, M., Villiet, P., Eperon, I. C. & Dickson, G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 7, 1083–1090 (1998).
Sicinski, P. et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244, 1578–1580 (1989).
Wilton, S. D. et al. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 9, 330–338 (1999).
Mann, C. J. et al. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl Acad. Sci. USA 98, 42–47 (2001).
Mann, C. J., Honeyman, K., McClorey, G., Fletcher, S. & Wilton, S. D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. 4, 644–654 (2002).
Wilton, S. D., Dye, D. E. & Laing, N. G. Dystrophin gene transcripts skipping the mdx mutation. Muscle Nerve 20, 728–734 (1997).
Aartsma-Rus, A. et al. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul. Disord. 12 (Suppl.), 71 (2002).
Lu, Q. L. et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nature Med. 9, 1009–1014 (2003). This study shows the in vivo applicability of therapeutic exon-23 skipping in mdx mice, which restores dystrophin synthesis to normal levels in large numbers of fibres and improves the functionality of the treated muscle.
van Deutekom, J. C. et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet. 10, 1547–1554 (2001).
Aartsma-Rus, A. et al. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 12, 907–914 (2003). In this study, BMD-like dystrophins were introduced into cultured muscle cells from DMD patients that were affected by different mutations, through the AON-induced skipping of different exons; the therapeutic potential and wide clinical applicability of this approach is discussed.
De Angelis, F. G. et al. Chimeric snRNA molecules carrying antisense sequences against the splice junctions of exon 51 of the dystrophin pre-mRNA induce exon skipping and restoration of a dystrophin synthesis in δ48–50 DMD cells. Proc. Natl Acad. Sci. USA 99, 9456–9461 (2002).
Errington, S. J., Mann, C. J., Fletcher, S. & Wilton, S. D. Target selection for antisense oligonucleotide induced exon skipping in the dystrophin gene. J. Gene Med. 5, 518–527 (2003).
Agrawal, S. et al. Toxicologic effects of an oligodeoxynucleotide phosphorothioate and its analogs following intravenous administration in rats. Antisense Nucleic Acid Drug Dev. 7, 575–584 (1997).
Agrawal, S. Importance of nucleotide sequence and chemical modifications of antisense oligonucleotides. Biochim Biophys. Acta 1489, 53–68 (1999).
Wahlestedt, C. et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl Acad. Sci. USA 97, 5633–5638 (2000).
Larsen, H. J., Bentin, T. & Nielsen, P. E. Antisense properties of peptide nucleic acid. Biochim Biophys. Acta 1489, 159–166 (1999).
Summerton, J. & Weller, D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 7, 187–195 (1997).
Gebski, B. L., Mann, C. J., Fletcher, S. & Wilton, S. D. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 12, 1801–1811 (2003).
Fluiter, K. et al. Tumor genotype-specific growth inhibition in vivo by antisense oligonucleotides against a polymorphic site of the large subunit of human RNA polymerase II. Cancer Res. 62, 2024–2028 (2002).
Bijsterbosch, M. K. et al. In vivo fate of phosphorothioate antisense oligodeoxynucleotides: predominant uptake by scavenger receptors on endothelial liver cells. Nucleic Acids Res. 25, 3290–3296 (1997).
Tinsley, J. M. et al. Primary structure of dystrophin-related protein. Nature 360, 591–593 (1992).
Pozzoli, U. et al. Comparative analysis of the human dystrophin and utrophin gene structures. Genetics 160, 793–798 (2002).
Pearce, M. et al. The utrophin and dystrophin genes share similarities in genomic structure. Hum. Mol. Genet. 2, 1765–1772 (1993).
Matsumura, K., Ervasti, J. M., Ohlendieck, K., Kahl, S. D. & Campbell, K. P. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature 360, 588–591 (1992).
Winder, S. J., Gibson, T. J. & Kendrick-Jones, J. Dystrophin and utrophin: the missing links! FEBS Lett. 369, 27–33 (1995).
Takemitsu, M. et al. Dystrophin-related protein in the fetal and denervated skeletal muscles of normal and mdx mice. Biochem. Biophys. Res. Commun. 180, 1179–1186 (1991).
Nguyen, T. M. et al. Localization of the DMDL gene-encoded dystrophin-related protein using a panel of nineteen monoclonal antibodies: presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J. Cell Biol. 115, 1695–1700 (1991).
Campanelli, J. T., Roberds, S. L., Campbell, K. P. & Scheller, R. H. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell 77, 663–674 (1994).
Grady, R. M., Merlie, J. P. & Sanes, J. R. Subtle neuromuscular defects in utrophin-deficient mice. J. Cell Biol. 136, 871–882 (1997).
Karpati, G. et al. Localization and quantitation of the chromosome 6-encoded dystrophin-related protein in normal and pathological human muscle. J. Neuropathol. Exp. Neurol. 52, 119–128 (1993).
Mizuno, Y., Nonaka, I., Hirai, S. & Ozawa, E. Reciprocal expression of dystrophin and utrophin in muscles of Duchenne muscular dystrophy patients, female DMD-carriers and control subjects. J. Neurol. Sci. 119, 43–52 (1993). This analysis of muscle biopsies shows that utrophin compensates, to some extent, for the dystrophin efficiency in muscles from DMD patients, which hinted at its therapeutic potential in dystrophic muscle.
Galvagni, F., Cantini, M. & Oliviero, S. The utrophin gene is transcriptionally up-regulated in regenerating muscle. J. Biol. Chem. 277, 19106–19113 (2002).
Deconinck, A. E. et al. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90, 717–727 (1997).
Grady, R. M. et al. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell 90, 729–738 (1997).
Tinsley, J. M. et al. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 384, 349–353 (1996).
Deconinck, N. et al. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nature Med. 3, 1216–1221 (1997).
Gilbert, R. et al. Efficient utrophin expression following adenovirus gene transfer in dystrophic muscle. Biochem. Biophys. Res. Commun. 242, 244–247 (1998).
Gilbert, R. et al. Adenovirus-mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum. Gene Ther. 10, 1299–1310 (1999).
Rafael, J. A., Tinsley, J. M., Potter, A. C., Deconinck, A. E. & Davies, K. E. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nature Genet. 19, 79–82 (1998).
Wakefield, P. M. et al. Prevention of the dystrophic phenotype in dystrophin/utrophin-deficient muscle following adenovirus-mediated transfer of a utrophin minigene. Gene Ther. 7, 201–204 (2000).
Cerletti, M. et al. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 10, 750–757 (2003).
Tinsley, J. et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nature Med. 4, 1441–1444 (1998). This study shows that relatively low levels of expression of a full-length utrophin are sufficient to prevent the development of muscular dystrophy in transgenic mdx mice.
Gillis, J. M. Multivariate evaluation of the functional recovery obtained by the overexpression of utrophin in skeletal muscles of the mdx mouse. Neuromuscul. Disord. 12 (Suppl.), 90–94 (2002).
Fisher, R. et al. Non-toxic ubiquitous over-expression of utrophin in the mdx mouse. Neuromuscul. Disord. 11, 713–721 (2001).
Dennis, C. L., Tinsley, J. M., Deconinck, A. E. & Davies, K. E. Molecular and functional analysis of the utrophin promoter. Nucleic Acids Res. 24, 1646–1652 (1996).
Perkins, K. J., Burton, E. A. & Davies, K. E. The role of basal and myogenic factors in the transcriptional activation of utrophin promoter A: implications for therapeutic up-regulation in Duchenne muscular dystrophy. Nucleic Acids Res. 29, 4843–4850 (2001). This is one of a series of basic studies on the regulatory mechanisms that control utrophin expression, which aims to identify regulatory elements that might provide targets for reagents to effect the therapeutic upregulation of utrophin.
Burton, E. A. et al. A second promoter provides an alternative target for therapeutic up-regulation of utrophin in Duchenne muscular dystrophy. Proc. Natl Acad. Sci. USA 96, 14025–14030 (1999).
Funk, W. D., Ouellette, M. & Wright, W. E. Molecular biology of myogenic regulatory factors. Mol. Biol. Med. 8, 185–195 (1991).
Santoro, I. M., Yi, T. M. & Walsh, K. Identification of single-stranded-DNA-binding proteins that interact with muscle gene elements. Mol. Cell Biol. 11, 1944–1953 (1991).
Koike, S., Schaeffer, L. & Changeux, J. P. Identification of a DNA element determining synaptic expression of the mouse acetylcholine receptor δ-subunit gene. Proc. Natl Acad. Sci. USA 92, 10624–10628 (1995).
Gramolini, A. O. et al. Local transcriptional control of utrophin expression at the neuromuscular synapse. J. Biol. Chem. 272, 8117–8120 (1997).
Gramolini, A. O. et al. Muscle and neural isoforms of agrin increase utrophin expression in cultured myotubes via a transcriptional regulatory mechanism. J. Biol. Chem. 273, 736–743 (1998). Following an earlier study that showed that nerve-derived trophic factors activate utrophin transcription, this paper reports that agrin doubled the amount of utrophin mRNAs and increased utrophin levels through interaction with the N-box in promoter A.
Gramolini, A. O. et al. Induction of utrophin gene expression by heregulin in skeletal muscle cells: role of the N-box motif and GA binding protein. Proc. Natl Acad. Sci. USA 96, 3223–3227 (1999).
Khurana, T. S. et al. Activation of utrophin promoter by heregulin via the ets-related transcription factor complex GA-binding protein α/β. Mol. Biol. Cell 10, 2075–2086 (1999).
Galvagni, F., Capo, S. & Oliviero, S. Sp1 and Sp3 physically interact and co-operate with GABP for the activation of the utrophin promoter. J. Mol. Biol. 306, 985–996 (2001).
Chaubourt, E. et al. Nitric oxide and l-arginine cause an accumulation of utrophin at the sarcolemma: a possible compensation for dystrophin loss in Duchenne muscular dystrophy. Neurobiol. Dis. 6, 499–507 (1999).
Courdier-Fruh, I., Barman, L., Briguet, A. & Meier, T. Glucocorticoid-mediated regulation of utrophin levels in human muscle fibers. Neuromuscul. Disord. 12 (Suppl.), 95–104 (2002).
Vater, R. et al. Utrophin mRNA expression in muscle is not restricted to the neuromuscular junction. Mol. Cell Neurosci. 10, 229–242 (1998).
Weir, A. P., Burton, E. A., Harrod, G. & Davies, K. E. A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J. Biol. Chem. 277, 45285–45290 (2002).
Briguet, A., Bleckmann, D., Bettan, M., Mermod, N. & Meier, T. Transcriptional activation of the utrophin promoter B by a constitutively active Ets-transcription factor. Neuromuscul. Disord. 13, 143–150 (2003).
Perkins, K. J. & Davies, K. E. Ets, Ap-1 and GATA factor families regulate the utrophin B promoter: potential regulatory mechanisms for endothelial-specific expression. FEBS Lett. 538, 168–172 (2003).
Galvagni, F. & Oliviero, S. Utrophin transcription is activated by an intronic enhancer. J. Biol. Chem. 275, 3168–3172 (2000).
Yang, J. et al. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J. Virol. 73, 9468–9477 (1999).
Schnepp, B. C., Clark, K. R., Klemanski, D. L., Pacak, C. A. & Johnson, P. R. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J. Virol. 77, 3495–3504 (2003).
Xiao, X., Li, J. & Samulski, R. J. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol. 70, 8098–8108 (1996). This is the first report of efficient rAAV-mediated lacZ gene transduction in muscle, which persisted for more than 1.5 years without evidence of a cellular immune response.
Greelish, J. P. et al. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nature Med. 5, 439–443 (1999).
Herzog, R. W. et al. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc. Natl Acad. Sci. USA 94, 5804–5809 (1997).
Chao, H. et al. Persistent expression of canine factor IX in hemophilia B canines. Gene Ther. 6, 1695–1704 (1999).
Manno, C. S. et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 101, 2963–2972 (2003).
Fraites, T. J. et al. Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol. Ther. 5, 571–578 (2002).
Goudy, K. et al. Adeno-associated virus vector-mediated IL-10 gene delivery prevents type 1 diabetes in NOD mice. Proc. Natl Acad. Sci. USA 98, 13913–13918 (2001).
Koenig, M., Monaco, A. P. & Kunkel, L. M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53, 219–226 (1988).
Hoffman, E. P., Brown, R. H. & Kunkel, L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 (1987).
Ervasti, J. M. & Campbell, K. P. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122, 809–823 (1993).
Chapman, V. M., Miller, D. R., Armstrong, D. & Caskey, C. T. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl Acad. Sci. USA 86, 1292–1296 (1989).
Acknowledgements
Work in the authors' laboratories is supported by grants from the Dutch Duchenne Parent Project, the Princess Beatrix Fund (The Netherlands), the European Union, the Muscular Dystrophy Campaign (United Kingdom) and the Muscular Dystrophy Association (United States).
Author information
Authors and Affiliations
Corresponding author
Related links
Related links
DATABASES
LocusLink
OMIM
FURTHER INFORMATION
Glossary
- EXTRACELLULAR MATRIX
-
In muscle, this is a thin layer (basal lamina) that contains collagen, elastin and fibronectin, which surrounds each muscle fibre. This might act as a semipermeable filter or a selective cellular barrier and is important in regeneration after damage.
- TRANSDUCTION
-
The transfer of genetic material into a cell using a viral vector.
- IMMUNOGENICITY
-
The properties of a virus, transgene, vector, compound or molecule that provoke an immune response.
- CYTOTOXICITY
-
The properties of a virus, transgene, vector, compound or molecule that are toxic for cells.
- ELECTROPORATION
-
The application of an electric current to the plasma membrane of a cell, to temporarily open pores or channels through which DNA might pass.
- PRESSURIZED ISOLATED-LIMB PERFUSION
-
The introduction of therapeutic agents under pressure in a limb after isolation of the blood circulation by clamping.
- MICROBUBBLES
-
Encapsulated gas microbubbles that can be used as drug or gene carriers, which are able to penetrate into the smallest membranes. When exposed to sufficiently high-amplitude ultrasound, the microbubbles rupture and release the drugs and genes that are contained in their encapsulating layer.
- TRANSFECTION
-
The transfer of exogenous DNA into a cell.
- MYOBLAST TRANSPLANTATION
-
The implantation of exogenous muscle-progenitor cells into muscle to generate new myofibres or to support existing myofibres.
- AMINOGLYCOSIDES
-
A group of antibiotics (such as gentamicin) that inhibit bacterial protein synthesis and are particularly active against Gram-negative bacteria.
- F-ACTIN
-
A protein that is involved in the contractile apparatus and the maintenance of the cytoskeleton of myofibres.
- β-DYSTROGLYCAN
-
The α- and β-dystroglycans are the laminin-binding components of the dystrophin–glycoprotein complex, which provides a linkage between the subsarcolemmal cytoskeleton and the extracellular matrix.
- DYSTROBREVINS
-
The components of the dystrophin–glycoprotein complex that bind to syntrophin and (indirectly) to the C-terminal of dystrophin. Dystrobrevin-α recruits signalling proteins, such as neuronal nitric oxide synthase.
- SYNTROPHINS
-
Peripheral membrane proteins that bind to the C-terminal of dystrophin, which might have a role in the process of synaptogenesis.
- SPECTRIN
-
A large contractile submembrane protein that, similar to dystrophin, contains an actin-binding domain and a long repeat domain.
- DEPENDOVIRUS
-
A single-stranded DNA virus from the family parvoviridae (subfamily parvovirinae), which is dependent on a co-infection with helper adenoviruses or herpes viruses for efficient replication.
- EPISOMES
-
DNA that can replicate autonomously in the cytoplasm of host cells.
- HEK-293 CELLS
-
Host cells that generate viral particles following transfection with the rAAV plasmid and the helper plasmid.
- NEO-ANTIGEN
-
A foreign (transgene) product that is able to stimulate an immune response.
- PRE-mRNA SPLICING
-
The removal of introns from the precursor mRNA molecule; the remaining exons are spliced together.
- PRIMARY MUSCLE-CELL CULTURES
-
Cells that are taken into culture directly from a tissue biopsy. In contrast to cell lines that only contain immortalized cells, these cultures contain heterogeneous cell populations.
- SPLICEOSOMAL COMPLEX
-
A large dynamic complex that consists of small nuclear RNA molecules and protein components. It mediates the two catalytic steps of the splicing reaction: the excision of introns from the pre-mRNA and the ligation of the two exon termini.
- RNaseH
-
Ribonuclease H. An enzyme that cleaves RNA/DNA complexes.
- PHARMACOKINETIC PROFILE
-
The characteristics of a drug that determine its absorption, distribution and elimination in the body.
- SARCOLEMMA
-
The membrane that encloses a striated muscle fibre.
- ACETYLCHOLINE
-
A neurotransmitter (C7H17NO3) that is released at autonomic synapses and neuromuscular junctions. It is active in the transmission of nerve impulses and is formed enzymatically in tissues from choline.
- CpG ISLAND
-
Genomic regions that are rich in the CpG pattern, are resistant to methylation and are often associated with promoter activity.
Rights and permissions
About this article
Cite this article
van Deutekom, J., van Ommen, GJ. Advances in Duchenne muscular dystrophy gene therapy. Nat Rev Genet 4, 774–783 (2003). https://doi.org/10.1038/nrg1180
Issue Date:
DOI: https://doi.org/10.1038/nrg1180
This article is cited by
-
Fully automated fast-flow synthesis of antisense phosphorodiamidate morpholino oligomers
Nature Communications (2021)
-
Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future
Pharmacological Reports (2020)
-
sPIF promotes myoblast differentiation and utrophin expression while inhibiting fibrosis in Duchenne muscular dystrophy via the H19/miR-675/let-7 and miR-21 pathways
Cell Death & Disease (2019)
-
Molecular characterization of exonic rearrangements and frame shifts in the dystrophin gene in Duchenne muscular dystrophy patients in a Saudi community
Human Genomics (2018)
-
Antisense oligonucleotides: the next frontier for treatment of neurological disorders
Nature Reviews Neurology (2018)
