
Key Points
-
CD8+ T cells have a central role in immune control of HIV. However, the recent failure of a T-cell-based vaccine trial prompts an urgent refocus on what constitutes an effective CD8+ T-cell response.
-
CD8+ T cells can recognize and then kill HIV-infected cells following the presentation of fragments of viral proteins (epitopes) in the peptide-binding groove of MHC class I molecules on the infected cell surface. The genes encoding the MHC molecules are the most polymorphic of the entire human genome. MHC class I polymorphisms define the characteristic viral peptides that can be presented to CD8+ T cells.
-
The outcome of HIV infection is strongly influenced by the particular HLA genes expressed by the infected individual. Certain HLA alleles, such as HLA-B57 and HLA-B27, are strongly associated with effective control of HIV, whereas other alleles are associated with poor control of HIV.
-
HLA alleles associated with effective control of HIV typically present multiple epitopes from the Gag protein. HIV-infected individuals who mount CD8+ T-cell responses to multiple Gag epitopes tend to have lower viral loads; CD8+ T-cell targeting of multiple non-Gag-specific epitopes is not associated with low viral loads.
-
The reasons why Gag is a successful target for CD8+ T-cell responses might be related to the fact that, first, the capsid protein sequence is highly conserved, which means that viral escape mutations within epitopes derived from this protein might attenuate the virus; and, second, Gag epitopes can be presented very early following infection of a target cell.
-
The path forward for a T-cell-based vaccine against HIV remains unclear, with problems such as vaccine immunogenicity, vector-specific immunity, HIV sequence diversity and viral adaptation to effective CD8+ T-cell responses being among the major obstacles that need to be overcome.
Abstract
The recent failure of the T-cell-based HIV vaccine trial led by Merck & Co., Inc. prompts the urgent need to refocus on the question of which T-cell responses are required to control HIV replication. The well-described association between the expression of particular MHC class I molecules and successful containment of HIV or, in the macaque model, SIV replication provide a valuable starting point from which to evaluate more precisely what might constitute effective CD8+ T-cell responses. Here, we review recent studies of T-cell-mediated control of HIV and SIV infection, and offer insight for the design of a successful T-cell-based HIV vaccine in the future.
Similar content being viewed by others

Main
The HIV epidemic continues unabated. In 2007, there were an estimated 2.5 million new reported cases of infection and 2.1 million HIV-related deaths (see the UNAIDS WHO AIDS epidemic update). This brings the estimated total number of people who have been infected with HIV since 1981 to 58 million, of whom 25 million have died. Although antiretroviral therapy has, since the mid-1990s, transformed the management and prognosis of HIV infection in resource-rich countries, only a small fraction — approximately a quarter (see the AVERT website) — of HIV-infected people in resource-poor countries have access to needed antiretroviral therapy. The demand for an effective HIV vaccine has never been more urgent, and yet in 2007, the first advanced trials of a T-cell-based vaccine were abruptly halted because of lack of efficacy1. So, it is an opportune moment to reflect on what is currently known about the immune responses that can successfully control HIV replication, the extent to which data from studies of non-human primates infected with simian immunodeficiency virus (SIV) can be extrapolated to infection of humans with HIV, and what the prospects are for a successful HIV vaccine in the future.
This Review focuses on the central role of MHC class I alleles in the control of immunodeficiency virus replication. It is striking that, in a recent whole-genome association study of key determinants for host control of HIV-1, the six most highly significant protective determinants were within the MHC region2. We review the central role of CD8+ T cells in the control of HIV and SIV replication, the evidence that the impact of MHC molecules on HIV and SIV disease is substantially mediated by the particular epitope peptides presented by different MHC class I molecules, and describe the pre-eminent role of MHC-B alleles and CD8+ T-cell activity directed against the viral protein Gag in the control of HIV and SIV replication. We examine aspects of the effectiveness or dysfunctional nature of CD8+ T-cell activity in HIV infection that are unrelated or indirectly related to specificity of the response. Finally, we address the question of whether HIV is able to adapt to HLA alleles that are currently associated with control of the virus, and the prospects for a successful T-cell-based vaccine against HIV in the future.
Role of CD8 + T cells in control of HIV replication
During acute HIV infection in adults, the increase in viraemia to a peak of ∼107 HIV RNA copies per ml of plasma3 is followed by a marked decline over a few weeks to a median viral setpoint of 30,000 HIV RNA copies per ml of plasma4. The temporal association observed between this 102–103-fold decrease in viral load and the appearance of HIV-specific CD8+ cytotoxic T lymphocyte (CTL) responses5,6 suggests that CTLs may be responsible for reducing the levels of virus at this stage of infection. More direct evidence for the role of CTLs in mediating the decline in viraemia during acute infection has come from studies of the SIV-macaque model in which administration of CD8-specific monoclonal antibodies abrogated the decline in viraemia from its peak level7,8,9. Short-term infusions of CD8-specific monoclonal antibodies in animals with chronic SIV infection led to increased viral load in those animals in which SIV-associated disease progressed rapidly ('progressors') and in those that controlled the disease ('controllers'; also known as long-term non-progressors) and also simultaneously reduced the numbers of SIV-specific CD8+ T cells being monitored using peptide–MHC class I tetramers7,10,11.
Three additional lines of evidence support the central role of CD8+ T cells in the control of HIV infection. First, specific HLA class I molecules are consistently associated with particular HIV disease outcomes (see later). Second, more rapid disease progression is observed in individuals with HLA class I homozygosity12,13. Third, the selection of particular viral mutants that escape CD8+ T-cell recognition are temporally associated with loss of immune control of infection. Set against these findings are the observations that high-frequency HIV-specific CD8+ T-cell responses are usually detectable in HIV-infected individuals who have developed AIDS14,15, that the majority of HLA class I alleles seem to have little impact on the disease outcome of HIV infection16 and that most selected viral escape mutants do not significantly affect viraemia17. Taken together, these studies lead to the conclusion that there are differences among CD8+ T-cell specificities and among HLA class I molecules that are crucial for effective control of HIV replication in different individuals. This gives rise to the hypothesis that the HLA class I associations with characteristic disease outcomes from infectious diseases such as HIV largely result from the CD8+ T-cell responses against the particular pathogen-specific peptides that are presented by the different HLA class I alleles. Support for this hypothesis in relation to HIV and SIV is presented in the next sections.
The significance of MHC class I polymorphism
The human MHC region, located on the short arm of chromosome 6, is central to the immune response, as it encodes proteins that help to distinguish between self and non-self protein components. Of the three MHC class I loci in humans (HLA-A, HLA-B and HLA-C), HLA-B is the most polymorphic, with 817 different HLA-B molecules described, compared to 486 distinct HLA-A and 263 distinct HLA-C molecules (see the IMGT/HLA database). Indeed, HLA-B is the most polymorphic locus on chromosome 6 and in the entire human genome18. Differences in the frequency of various HLA-B alleles in selected populations worldwide are shown in Fig. 1 to illustrate the extraordinary diversity of distinct human populations with respect to HLA allele expression.

HLA class I diversity is illustrated by the prevalence of nine HLA-B molecules in eight selected populations (see the allele frequencies website). For simplicity, HLA class I alleles are shown by two digits only.
Almost all of this extensive HLA class I polymorphism is restricted to those residues that line the peptide-binding groove of these molecules19, thereby defining the peptides that bind to each HLA molecule. The significance of HLA class I polymorphism is that the differences among the various HLA molecules and the peptides they present are of sufficient functional importance to be subject to Darwinian natural selection: in short, these differences are a matter of life and death. Seemingly insignificant, single amino-acid differences between closely related HLA alleles may have crucial consequences in terms of outcome from particular infections. In the setting of HIV infection, examples include the association of HLA-B*3502 and HLA-B*3503 with rapid disease progression, and a lack of any such association for HLA-B*3501, which differs from HLA-B*3502 and HLA-B*3503 by only three and one amino acid, respectively20,21,22. These small changes would be expected to affect the binding of the C-terminal amino acid of the peptide to the F pocket of the HLA molecule: the preferred residue for binding to the F pocket of HLA-B*3501 is tyrosine23, whereas a smaller hydrophobic residue is preferred for binding to the F pocket of HLA-B*3502 and of HLA-B*3503 (Refs 19, 22). This therefore affects the binding of peptides such as the HLA-B*3501-restricted epitope of Gag p24, PPIPVGEIY (Gag residues 254–262), to HLA-B*3502 and HLA-B*3503. The observed differences in Gag-specific CD8+ T-cell responses between HLA-B*3501-, HLA-B*3502- and HLA-B*3503- positive study subjects24 are therefore not unexpected, and if Gag is an important target for immune control of HIV17, such HLA differences may be of crucial importance. In a more extreme example, a study of HIV infection in Durban, South Africa, found a strong association between HLA-B*5801 and low viral load, whereas HLA-B*5802, which differs from HLA-B*5801 by three amino acids, was associated with high viral load16. Again, these few differences between apparently closely related HLA class I molecules translate into substantial differences in peptide-binding specificity19.
Scrutiny of the peptides presented by superficially similar HLA class I molecules invariably reveals subtle but important differences. One study of five closely-related HLA alleles within the HLA-B7 supertype (HLA-B*0702, HLA-B*3910, HLA-B*4201, HLA-B*4202 and HLA-B*8101) demonstrated that, even when an identical peptide is presented by distinct alleles, different selection pressure can be imposed on the virus25. As described above, this result should not be unexpected because the very existence of HLA class I polymorphism is strong evidence that meaningful differences in disease outcome result from it.
The HLA class I alleles that have been identified consistently to have a significant impact on the rate of disease progression, or on viral setpoint (which is strongly predictive of the rate of disease progression16,26,27,28,29,30,31,32,33,34) are shown in Table 1. MHC class I molecules that are associated with successful control of SIV infection, as shown in studies of the SIV-macaque model of HIV infection, are also included35,36,37,38,39,40,41,42,43,44,45,46,47,48. It is evident that in HIV infection, HLA-B molecules have the strongest impact on viral setpoint compared with HLA-A and HLA-C molecules16 for reasons that remain unclear. A more diverse selection of peptide-binding motifs is offered by HLA-B alleles compared with HLA-A alleles, as illustrated by the residues at position 2 (P2) of the 100 optimally defined HIV epitopes that were most strongly targeted by CD8+ T cells in a recent large population-based study17 (Fig. 2). In particular, proline is frequently an anchor-binding residue at P2 in HLA-B-binding peptides, which also include peptides with charged residues at P2.

Amino acids have been grouped into the following categories: amino acids with hydroxyl-containing side chains (Thr, Ser); small-to-medium sized hydrophobic residues (Gly, Ala, Val, Leu, Ile, Met); amino acids with a cyclic side chain (Pro); amino acids with aromatic side chains (Phe, Tyr, Trp); amino acids with acidic side chains (Asp, Glu); amino acids with basic side chains (His, Lys, Arg); amino acids with amide side chains (Gln). Peptides that bind to HLA-A alleles are almost exclusively restricted to those that have hydrophobic or hydroxyl-containing residues at position 2, whereas HLA-B-binding peptides include these and peptides with proline, charged residues or glutamine at position 2. HLA-C-binding peptides are less diverse than HLA-B-binding peptides, but they are more closely related to HLA-B than HLA-A in their peptide-binding motif. Data taken from Refs 17,117.
It is noteworthy that Mamu-B*08 and Mamu-B*17, two rhesus macaque MHC class I alleles that are associated with the control of SIV replication36,37,40, have peptide-binding motifs that in some respects resemble those of the two HLA class I molecules most consistently associated with control of HIV infection, HLA-B*27 (which binds peptides with an arginine at P2) and HLA-B*57 (which binds peptides with a tryptophan at the C terminus), respectively19. Indeed, the eight currently known Mamu-B*08-restricted CD8+ T-cell epitopes fit the peptide-binding requirements defined for HLA-B*27 (Ref. 48), and peptides that bind to HLA-B*27 also bind to Mamu-B*08 with high affinity (J. T. Loffredo, unpublished observations). Interestingly, most of the HLA-B*27- and Mamu-B*08-restricted T-cell responses are directed towards peptides with two arginine residues at the N terminus (that is, at P1 and P2). It has recently been suggested that peptides with di-basic N termini are resistant to peptidase degradation, thereby increasing their intracellular half-lives. So, fewer molecules of the target antigen may be required to generate the necessary HLA-B*27- and Mamu-B*08-bound peptides to trigger CTL recognition and responses.
Despite the homology between SIV and HIV, there are no viral epitopes that are common targets of the CTL response in both HLA-B*27-positive humans and Mamu-B*08-positive macaques. However, the Gag epitope TSTLQEQIAW (which is abbreviated as TW10 and corresponds to the Gag amino acids 240–249) is the main target of the acute CD8+ T-cell response in HIV-infected subjects who have HLA-B*57 or HLA-B*5801 and is very similar to the SIV homologue (SSVDEQIQW, Gag 241–249) presented by the Mamu-A*90120-5 class I molecule, which is also associated with control of SIV in Burmese macaques42. The Gag epitope KAFSPEVIPMF (KF11, Gag 162–172), which is the dominant focus of the CD8+ T-cell response in HLA-B*57-positive subjects with chronic HIV infection17 overlaps with the SIV epitope KKFGAEVVP (KP9, Gag 162–170) presented by Mane-A*10, an MHC class I molecule that is associated with the control of SIV infection in pigtail macaques46,47. So, certain MHC class I molecules associated with the control of HIV replication in humans and SIV replication in macaque models can have similar or dissimilar peptide-binding motifs that nonetheless enable peptides from the same regions of particular viral proteins to be targeted by CD8+ T cells.
HLA-B * 27/57 control of HIV replication
The first evidence that specific HLA class I alleles could influence the outcome of HIV infection came from studies of HIV-infected HLA-B*27-positive individuals who mounted strong CTL responses against the immunodominant epitope, KRWIILGLNK (KK10, Gag 263–272)34,35,49,50. HIV-infected individuals expressing HLA-B*27 who responded to this epitope showed control of viraemia, whereas those not responding to this epitope, or whose response declined following the selection of viral escape mutations within the epitope, typically had a high level of viraemia34,35,50. Similarly, in the SIV macaque model, Mamu-A*01-positive animals show some measure of control of SIV replication after immunization with Gag-containing vaccines51. The dominant CD8+ T-cell response against the capsid protein epitope CTPYDINQM (CM9, Gag 181–189) in Mamu-A*01-positive macaques after infection with SIV might have a role, albeit minor, in the control of viral replication in naive Mamu-A*01-positive macaques41,52,53,54,55. Mamu-A*01-positive animals 'primed' with a DNA vaccine and 'boosted' with a recombinant-adenovirus-based vaccine encoding Gag (DNA/Ad5Gag) typically show lower viral loads following infection than unvaccinated, infected Mamu-A*01-positive macaques51. In one Mamu-A*01-positive animal infected with SHIV89.6P, the CM9-specific CTL response disappeared in association with the emergence of viral escape mutations in the CM9 epitope and resulted in rapid progression to disease56. Furthermore, the emergence of viral escape mutations in this epitope correlated with disease progression in SIVsmE660-infected macaques57. However, the picture is more complex than this: many Mamu-A*01-positive macaques experience high viral loads after SIV challenge but show no evidence of escape mutations in the CM9 epitope; and virus breakthrough after the transient control in DNA/Ad5Gag-vaccinated Mamu-A*01-positive macaques is not related to escape from the CM9-specific response58. These data suggest that CD8+ T-cell responses to the KK10 and CM9 epitopes can make important contributions to the control of HIV and SIV replication under certain conditions.
The striking feature of the emergence of viral escape mutations in the KK10 and CM9 epitopes is that it occurs late in the course of infection34,50,56. The hypothesis that crucial escape mutations might be costly in terms of viral replicative capacity is supported by studies of viral escape in individuals with HLA-B*57. In these individuals, in contrast to what had been observed following escape mutations in the KK10 or CM9 epitopes, escape mutations in the TW10 epitope that is targeted in acute infection do not result in rapid progression to AIDS. In general, HLA-B*57- and HLA-B*5801-positive subjects infected with a virus that has the common T242N mutation in TW10 tend to maintain low viral loads59. The observation that the virus with this T242N mutation was transmitted to HLA-B*57- and HLA-B*5801-negative individuals and then reverted to the wild-type sequence indicated that this mutation confers a cost to viral fitness in vivo in the absence of the selecting HLA alleles59. In vitro competition assays confirmed the negative effect of the T242N mutation on viral replicative capacity60. Similar observations were made with respect to the T182A escape mutation in the CM9 epitope, in that this mutation abrogated in vitro viral replication when engineered into cloned SIVmac239 in the absence of other mutations that compensate for the cost to viral fitness61,62. Similar to the R264K escape mutation in the HLA-B*27-restricted epitope KK10, which seems to precipitate progression to AIDS34,50, the T182A mutation in the Mamu-A*01 epitope CM9 only arises in vivo in the presence of multiple compensatory viral mutations, which might explain why these occur late in the course of infection63,64.
In individuals in which the T242N mutation in the TW10 epitope arises early in the course of infection, compensatory mutations in the virus have been described59,60,65 but these emerge later on. Following the escape mutation in TW10, as mentioned above, the immunodominant T-cell response in chronic infection then targets the HLA-B*57-restricted Gag epitope KAFSPEVIPMF (KF11, Gag 162–172). Recent studies in HLA-B*5703-positive study subjects in KwaZulu-Natal, Durban, South Africa, show that the escape mutation A163G in KF11 confers a fitness cost to the virus that is partially compensated for by the S165N substitution that subsequently arises66. These studies prompt the hypothesis that HLA-B*57 is strongly associated with the control of HIV replication, above all other alleles, because it presents several Gag epitopes. Viral escape from CD8+ T-cell responses against these epitopes reduces viral replicative capacity.
It is of note in this context that the Mamu-A*90120-5-positive Burmese macaques, which control SIV replication so successfully, also show strong T-cell responses to three Gag epitopes that are presented by the same Mamu-A*90120-5 MHC class I allele42,43,44,45. Escape mutations in these three Gag epitopes result in an incremental reduction in viral fitness with the increasing number of mutations. Likewise, control of SIV replication in pigtail macaques is described following early selection of an escape mutation in the Mane-A*10-restricted Gag epitope KP9 that similarly results in a substantial reduction in viral replicative capacity46. A notable exception for this is Mamu-B*08, for which there are few Gag-derived epitopes, and the immunodominant responses in Mamu-B*08-positive animals that control SIV infection successfully are mainly directed towards epitopes in the viral proteins Vif and Nef36. Despite this observation, results from studies of MHC class I molecules associated with control of immunodeficiency virus replication suggest that the targeting of Gag epitopes by CD8+ T cells can result in immune control of viral replication. Subsequent viral escape from these responses may confer a fitness cost to the virus (Fig. 3).

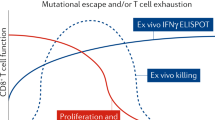
In HIV-infected individuals with HLA class I alleles that are associated with effective control of the virus (such as HLA-B57 and HLA-B27), Gag-specific responses are generated in acute infection. Wild-type virus-specific-cytotoxic T lymphocytes (CTLs) recognize virus-infected cells early in the viral life cycle, before Nef-mediated MHC class I downregulation. Escape mutations that are typically selected in Gag reduce in vitro viral fitness. Remaining wild-type virus-specific CTL responses can contain the infection until new escape variants are selected. However, increasing numbers of costly escape mutations further limit the replicative capacity of the virus, thereby facilitating effective killing of virus-infected cells by remaining CTLs. TCR, T-cell receptor.
Control of HIV by HLA-B–Gag-specific responses
Numerous studies have now been carried out to address the question of whether the protein specificity of the HIV-specific CD8+ T-cell response is related to successful immune control. One of the earliest was a prospective study of 38 asymptomatic HIV-infected subjects that showed that a Gag-specific, but not an Env-specific, CD8+ T-cell response was protective against disease progression67. Eight other cross-sectional studies17,68,69,70,71,72,73,74 showed an association between control of immunodeficiency virus infection and Gag-specific CD8+ T-cell responses. An association between Nef- and Env-specific CD8+ T-cell responses and lack of successful immune control was observed in some studies17,72. An analysis of 578 subjects in KwaZulu-Natal with chronic HIV infection showed that, irrespective of HLA type, an increasing number of Gag-specific responses correlated with decreasing viral load; such a correlation was not observed for non-Gag-specific responses17. A study of the CD8+ T-cell responses in acute HIV infection similarly showed that Gag-specific T-cell activity early in the course of infection was associated with long-term control of HIV replication75. So, a strong consensus that is emerging from these studies is that in HIV infection, Gag-specific but not non-Gag-specific CD8+ T-cell responses in acute and chronic infection are associated with long-term control of HIV infection.
Why are Gag-specific T-cell responses important? The possible reasons for Gag being of such apparent importance have been alluded to earlier, namely, that Gag is highly immunogenic, but also very conserved in sequence (Box 1), and hence mutational escape can often result in a reduction of viral replicative capacity. In support of this hypothesis are the studies (described earlier) of immune control following early escape mutations in Gag. In addition, in a recent analysis of 681 chronically infected study subjects in KwaZulu-Natal, the number of Gag-specific amino-acid polymorphisms associated with each HLA-B allele strongly correlated with the median viral load of individuals expressing that allele117. Importantly, however, this correlation was driven purely by Gag mutations that are likely to revert to the wild-type sequence post-transmission; that is, those that inflict a fitness cost to the virus. These data support an earlier, epitope-specific study76 and suggest an explanation for the well-described association between particular HLA-B molecules and characteristic rates of progression to HIV disease (Table 1). Therefore, it is not just the number of Gag epitopes presented by each allele that is important, but also the ability of the Gag-specific CD8+ T-cell response to drive the selection of escape mutations that have an impact on viral replication. It is noteworthy that this relationship applies predominantly to HLA-B alleles and Gag-specific responses.
A complementary study carried out in a cohort of study subjects with acute HIV infection in Zambia77 showed that reaching the viral setpoint early in the course of infection was strongly related to the number of mutations in HLA-B-restricted Gag epitopes in the transmitted virus. The implication from this and a similar study undertaken in South Africa78 is that an increasing number of Gag mutations decreases viral replicative capacity, which is supported by studies of the SIV-Burmese-macaque model (referred to earlier)42,43,44,45. A recent longitudinal study of HIV-infected individuals showed that decreasing viral replicative capacity over the course of the infection is associated with slow progression to disease, although in this case this was not attributable to epitope mutations in Gag79.
A second reason why Gag may be an important target for CD8+ T cells is that Gag-specific CD8+ T cells may be able to recognize and kill virus-infected cells sooner after infection than non-Gag-specific CD8+ T cells can. Recent studies show that Gag-specific CD8+ T cells recognize SIV-infected targets within 2 hours of infection, whereas Tat-, Env- and Nef-specific CD8+ T cells only recognize virus-infected target cells following de novo synthesis of viral proteins80,81. The significance of this is that Gag-specific CD8+ T cells have a window of approximately 10 hours between expression of Gag on the cell surface and the impact of newly synthesized Nef-mediated downregulation of MHC class I molecules80. It is of interest that the same investigators showed that Pol-specific CD8+ T cells also recognize SIV-infected cells within 2 hours of infection and can eliminate virus-infected cells by 6 hours post-infection, before MHC class I downregulation81. Pol might therefore be an important target for vaccine design, although it seems to be less important in natural infection. A potential explanation for the difference in importance between Gag and Pol in natural infection is the 20-fold higher level of expression of Gag compared to Pol82,83, the large amount of Gag within mature virus particles (1,000–1,500 capsid molecules in each mature virion)84 and the consequent immunodominance of Gag-specific responses in most subjects who show effective control of HIV replication.
Why are HLA-B-restricted T-cell responses important? The explanation for the central role of HLA-B, compared with HLA-A and HLA-C, in the control of HIV replication is even less clear. One possibility relates to the fact that natural killer (NK) cells express killer immunoglobulin-like receptors (KIRs) that recognize MHC class I molecules85. It has been noted that the HLA class I alleles that are most strongly associated with control of HIV are those that contain the Bw4 motif (a ligand for KIRs), and it is becoming increasingly clear that certain combinations of HLA-B alleles (those Bw4-motif-positive alleles that have an isoleucine at residue 80) and KIR allotypes (the activating receptor KIR3DS1 or high expression of the inhibitory receptor KIR3DL1) are associated with slower progression to HIV disease85. However, it is also evident from these analyses that the association of HLA class I alleles, such as HLA-B*57 and HLA-B*27, with control of viraemia is independent of KIRs, although interactions of these and other HLA-B alleles do have important additional consequences for viral setpoint86.
A second possibility is that HLA-B-restricted CD8+ T-cell responses are more polyfunctional (on the basis of cytokine production and proliferative capacity) than HLA-A- or HLA-C-restricted T-cell responses, even when the same peptide is presented87. Why this might be the case remains unclear, and this does not provide an explanation for why HLA-B alleles such as HLA-B*3502 or HLA-B*5802 are associated with high viral setpoints. It is clear that a greater diversity of peptides can be presented by HLA-B compared with HLA-A alleles (Fig. 2), and it could be that, by chance, the alleles presenting the most Gag epitopes are likely to be HLA-B alleles88.
Is HLA-C unimportant? The role of HLA-C-restricted CD8+ T cells remains obscure. In a genome-wide association study of key determinants of host control of HIV-1, the two strongest determinants were a polymorphism (rs2395029) in complete linkage disequilibrium with HLA-B*5701, explaining 9.6% of variation in viral setpoint and a polymorphism (rs9264942) in the region of the HLA-C locus, explaining 6.5% of the variation in viral setpoint3. The HLA-C polymorphism is strongly associated with differences in expression of HLA-C alleles, higher expression being associated with lower viral setpoint. However, it is uncertain whether this HLA-C-related effect is mediated by a gene in linkage disequilibrium with the identified polymorphism, as is the case for HLA-B*5701. The potential role of HLA-C in low viral setpoint has previously been raised following the finding that Nef-mediated downregulation of MHC class I molecules only applies to HLA-A and HLA-B alleles89, suggesting a mechanism by which HIV could evade both CD8+ T-cell responses (by downregulation of HLA-A and HLA-B expression) and NK-cell activity (by lack of downregulation of HLA-C expression). Although HLA-C-restricted CD8+ T-cell responses represent a relatively small minority — approximately 12% — of the total number of HIV-specific CD8+ T-cell responses17, there is evidence that some HLA-C alleles are associated with lower levels of viraemia independent of linkage disequilibrium with HLA-B alleles (A. J. Leslie, unpublished observations), and that, similar to HLA-B alleles, these HLA-C associations with particular viral setpoints are related to the selection of escape mutations that inflict a fitness cost on the virus76.
Polyfunctionality of effective CD8 + T cells
Alternative or additional features of CD8+ T-cell responses that explain distinct rates of HIV disease progression are qualitative differences that are unrelated to T-cell specificity. The crucial role of HIV-specific CD4+ T-cell activity has been highlighted90,91, as has the presence of polyfunctional CD8+ T cells that are capable of producing not only interferon-γ following contact with the cognate-peptide–MHC complex, but also other cytokines, such as interleukin-2, chemokines, such as CC-chemokine ligand 5, which is capable of blocking viral entry through the HIV co-receptor CC-chemokine receptor 5, granzymes and perforin, which are capable of mediating cytotoxicity, and other factors that are capable of inducing proliferative activity92,93. Typically, HIV-specific CD8+ T cells from infected individuals who have low viral loads are polyfunctional and can carry out most or all of these effector functions. Conversely, HIV-infected subjects with high viral loads tend to have HIV-specific CD8+ T cells that are capable of few of these normal effector functions and, at the extreme of the spectrum, their dysfunctionality might be reflected by the expression of PD1 (programmed cell death 1), a marker of T-cell exhaustion94,95,96,97. Similar to CTLA4 (cytotoxic T-lymphocyte antigen 4), PD1 is a member of the B7–CD28 family of immunoregulatory molecules, the expression of which is designed to prevent overactivation of the immune response and consequent autoimmunity. PD1 is expressed at particularly high levels by HIV-specific CD8+ and CD4+ T cells, the level of expression being directly related to viral load and inversely related to absolute CD4+ T-cell numbers. Similarly, CTLA4 is expressed at high levels by HIV-specific CD4+ T cells98. Blockade of the PD1 pathway through the use of antibody against PD1 ligand 1 (PDL1), and blockade of the CTLA4 pathway through the use of a CTLA4-specific antibody, demonstrates the reversibility of these defects and the potential for immunotherapy in HIV-infected subjects96,98. In common with all the studies described earlier, which have identified correlates of protective immunity against HIV disease progression, there is the difficulty in distinguishing cause and effect. However, in mice infected with lymphocytic choriomeningitis virus, in which exhausted CD8+ T cells express PD1, blockade of the PD1–PDL1 pathway restored the ability of the CD8+ T cells to undergo proliferation, secrete cytokines, kill virus-infected cells and decrease viral load94. So, there is a rationale to suggest that reducing PD1 expression could similarly improve CD8+ T-cell function and bring about improved control of viraemia in HIV infection.
Can HIV adapt to HLA-associated control?
One clear attraction of investigating the HLA class I associations with HIV disease outcomes is that the direction of causality is unequivocal. However, an important question is to what extent these associations will persist or whether the virus over time and/or in different localities, might be able to adapt to HLA alleles that were previously associated with control of HIV infection. This is a question of more than just academic interest as, if HIV is capable of adapting to the immune responses that currently mediate control of viraemia, vaccines inducing these immune responses will not be effective.
It is well established that HIV shows rapid within-host adaptation to immune responses including HIV-specific CD8+ T-cell activity, examples of which have been described above. Transmission of viruses with escape mutations has also been documented. Escape mutations in the HLA-B*27-restricted KK10 epitope, which is strongly associated with control of viraemia, abrogates binding of the epitope to the HLA class I molecule34,99. Therefore, transmission of virus encoding the escape mutation to an HLA-B*27-positive recipient would prevent the recipient from making a response to the KK10 epitope, potentially reducing the link between HLA-B*27 and slow progression following HIV infection. A small study of HIV-infected mother–child pairs, in which some children inherited HLA-B*27 from the mother and others inherited it from the father, suggested that this process might have already started99.
Whether HLA is an important driver of HIV evolution at the population level depends to some extent on the likelihood that such transmitted viral escape mutants will revert to wild-type post-transmission, and also on HLA-independent influences such as founder effects. The relative impact of these forces on the evolution of HIV at the population level remains an unresolved issue (Table 2). Some studies suggest that escape mutations within CD8+ T-cell epitopes can be transmitted relatively frequently and persist in HIV-mismatched recipients, thereby accumulating over time to ultimately represent the most prevalent form of the virus100,101,102. Indeed, virus-clade-specific differences, which are otherwise largely unexplained, could result at least in part from the impact of HLA differences between populations living in distinct regions of the world. More recent work has emphasized the role of the founder effect in shaping viral evolution103. As additional large cohort studies from around the world are characterized with respect to the prevailing viral sequences and HLA types, the relative impact of these forces in shaping the nature of the circulating viral sequences will become clearer. However, it is certainly possible that the HLA class I associations with viral control that are described in the published literature might alter with respect to time and place, and that HIV, far from developing a non-pathogenic coexistence with its human host might, over time, adapt to evade the most effective CD8+ T-cell responses currently directed towards it.
Prospects for a T-cell-based HIV vaccine
The prospects for an HIV vaccine seem dim after the recent failure of a CTL-based vaccine to show efficacy in clinical trials led by Merck & Co., Inc.2. Another CTL-based vaccine trial led by researchers at the Vaccine Research Center (VRC), National Institutes of Health, USA, based on priming with a DNA vaccine followed by boosting with a recombinant adenovirus-vector-based vaccine, may proceed in 2008 (see the Vaccine Research Center website). However, it may be argued that neither of these vaccines fully tests the concept that a CTL response induced by a vaccine can control viral replication. The Merck vaccine induced an average of only three epitope-specific responses in the vaccinees104. Furthermore, despite the initially encouraging results against challenge of SHIV89.6P, a challenge virus that in retrospect has been viewed as inadequate105, repeated administration of adenovirus 5 (Ad5) expressing Gag, Pol and Nef failed to control replication of the homologous challenge virus SIVmac239 (D. R. Casimiro, personal communication). The VRC vaccine induces fewer epitope-specific CTL responses than the Merck vaccine did and many of these are directed against epitopes in the Env protein, which is perhaps the most variable of the open reading frames in HIV despite having some conserved regions. So, any challenge virus that the vaccinees might encounter is likely to be different by at least one in three amino acids, or three amino acids in a CTL epitope.
These disappointing Ad5-induced responses in humans are fundamentally different from those induced by Ad5 in macaques for reasons that are as yet unclear. For example, if macaques are vaccinated with Ad5Gag, vaccinated animals make at least five epitope-specific responses (Table 3). By contrast, each Ad5Gag-vaccinated human will make only one epitope-specific response on average. Whether this is related to the fact that most vaccinated humans have previously been exposed to Ad5, thereby inducing an anamnestic CD8+ T-cell response to Ad5 following vaccination, is unknown. This anamnestic Ad5-specific response might 'crowd out' CD8+ T-cell responses against the HIV immunogens in the vaccine. The Ad5-based vaccine is less immunogenic in individuals with Ad5-specific antibody titres of >200, supporting this notion. So, to test the CTL-based vaccine concept, it might be necessary to develop a vaccine that mimics the types of virus-replication-controlling responses seen in humans (or macaques), both in terms of breadth and magnitude. Unfortunately, none of our current vaccines do this.
The challenge to successful HIV vaccine development posed by viral sequence diversity is somewhat distinct from the more technical issue of immunogenicity. The encouraging aspect of studies such as those that show a benefit from targeting multiple Gag epitopes is that the effect is HLA independent. This suggests that individuals receiving a vaccine that can induce broad Gag responses will benefit from it even if they do not express the HLA alleles normally associated with control of viraemia. The sequence conservation of HIV proteins, such as p24 Gag, implies that, if adequate responses can target this region, sequence diversity is to a great extent already addressed: escape mutation will tend to come at a significant cost to the virus, and incoming viral sequence variation in p24 Gag is more likely to be conservative and therefore to be accommodated by epitope cross-recognition.
It seems likely, therefore, that novel approaches to vaccine development will be needed to induce the breadth and nature of CTL responses required to control HIV infection before it can be determined whether a CTL-based vaccine could be successful. Current approaches range from the development of new viral gene delivery vectors that are designed to circumvent the problem of pre-existing vector-specific immunity106 to new peptide immunotherapeutic strategies that have shown promise in the pigtail macaque-SIV model107. However, it is not possible to delineate a clear path forward for an HIV T-cell vaccine, even after more than 25 years of the HIV epidemic. This vaccine will need to induce a sufficiently high magnitude of effective CTL responses against those viral gene products that exhibit limited variability and that have been shown to be the targets of efficacious CD8+ T-cell responses in humans and macaques.

References
HIV vaccine failure prompts Merck to halt trial. [News in Brief] Nature 449, 390 (2007).
Fellay, D. et al. A whole-genome association study of major determinants for host control of HIV-1. Science 317, 944–947 (2007). This study confirms and highlights the central role of HLA in immune control of HIV, in particular that of HLA-B*57, but also suggests an unexpectedly important role for HLA-C.
Rosenberg, E. S. et al. Immune control of HIV-1 after early treatment of acute infection. Nature 407, 523–526 (2000).
Lyles, R. H. et al. Natural history of HIV-1 viremia after seroconversion and proximal to AIDS in a large cohort of homosexual men. J. Infect. Dis. 181, 872–880 (2000).
Koup, R. A. et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68, 4650–4655 (1994).
Borrow, P., Lewicki, H., Hahn, B., Shaw, G. & Oldstone, M. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68, 6103–6110 (1994).
Schmitz, J. E. et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283, 857–860 (1999).
Jin, X. et al. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189, 991–998 (1999).
Matano, T. et al. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J. Virol. 72, 164–169 (1998).
Tsukamoto, T. et al. Induction of CD8+ cells able to suppress CCR5-tropic simian immunodeficiency virus SIVmac239 replication by controlled infection of CXCR4-tropic simian-human immunodeficiency virus in vaccinated rhesus macaques. J. Virol. 81, 11640–11649 (2007).
Friedrich, T. C. et al. Subdominant CD8+ T-cell responses are involved in durable control of AIDS virus replication. J. Virol. 81, 3465–3476 (2007).
Carrington, M. et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283, 1748–1752 (1999).
Tang, J. et al. HLA class I homozygosity accelerates disease progression in HIV-1 infection. AIDS Res. Hum. Retro viruses 15, 317–324 (1999).
Hay, C. M. et al. Lack of viral escape and defective in vivo activation of human immunodeficiency virus type 1-specific cytotoxic T lymphocytes in rapidly progressive infection. J. Virol. 73, 5509–5519 (1999).
Draenert, R. et al. Persistent recognition of autologous virus by high-avidity CD8 T cells in chronic, progressive human immunodeficiency virus type 1 infection. J. Virol. 78, 630–641 (2004).
Kiepiela, P. et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature 432, 769–775 (2004). This analysis of a cohort of more than 700 study subjects in KwaZulu-Natal shows that HIV disease outcome (viral load, CD4+ T-cell count), HIV-specific CD8+ T-cell response and CD8+ T-cell-mediated immune pressure imposed on the virus are all dependent on the particular HLA-B type expressed.
Kiepiela, P. et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nature Med. 13, 46–53 (2007). This analysis of the same cohort as in reference 16 shows that increasing the breadth of the Gag-specific CD8+ T-cell response is associated with decreasing viral load, whereas increasing the breadth of the non-Gag-specific T-cell response is not.
Mungall, A. J. et al. The DNA sequence and analysis of human chromosome 6. Nature 425, 805–811 (2003).
Marsh, S. G. E., Parham, P. & Barber, L. D. The HLA Facts Book. (Academic Press, London, 2000).
Gao, X. et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N. Engl. J. Med. 344, 1668–1675 (2001).
Hill, A. V. S. et al. Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature 360, 434–439 (1992).
Steinle, A. et al. Motif of HLA-B*3503 peptide ligands. Immunogenetics 43, 105–107 (1996).
Smith, K. J. et al. An altered position of the α2 helix of MHC class I is revealed by the crystal structure of HLA-B*3501. Immunity 4, 203–213 (1996).
Jin, X. et al. Human immunodeficiency virus type 1 (HIV-1)-specific CD8+-T-cell responses for groups of HIV-1-infected individuals with different HLA-B*35 genotypes. J. Virol. 76, 12603–12610 (2002).
Leslie, A. et al. Differential selection pressure exerted on HIV by CTL targeting identical epitopes but restricted by distinct HLA alleles from the same HLA supertype. J. Immunol. 177, 4699–4708 (2006).
Kaslow, R. A. et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nature Med. 2, 405–411 (1996).
Hendel, H. et al. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J. Immunol. 162, 6942–6946 (1999).
O'Brien, S. J., Gao, X. & Carrington, M. HLA and AIDS: a cautionary tale. Trends Mol. Med. 7, 379–381 (2001).
Migueles, S. A. et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl Acad. Sci. USA 97, 2709–2714 (2000).
Tang, J. et al. Favorable and unfavorable HLA class I alleles and haplotypes in Zambians predominantly infected with clade C human immunodeficiency virus type 1 in Zambia. J. Virol. 76, 8276–8284 (2002).
Lazaryan, A. et al. Human leukocyte antigen B58 supertype and human immunodeficiency virus type 1 infection in native Africans. J. Virol. 80, 6056–6060 (2006).
Honeyborne, I. et al. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J. Virol. 81, 3667–3672 (2007).
Harrer, E. G. et al. A conserved HLA B13-restricted cytotoxic T lymphocyte epitope in Nef is a dominant epitope in HLA B13-positive HIV-1-infected patients. AIDS 19, 734–735 (2005).
Goulder, P. J. R. et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nature Med. 3, 212–217 (1997).
Ammaranond, P. et al. A new variant cytotoxic T lymphocyte escape mutation in HLA-B27-positive individuals infected with HIV type 1. AIDS Res. Hum. Retroviruses 21, 395–397 (2005).
Loffredo, J. T. et al. CD8+ T cells from SIV elite controller macaques recognize Mamu-B*08-bound epitopes and select for widespread viral variation. PLoS ONE 2, e1152 (2007).
Loffredo, J. T. et al. Patterns of CD8+ immunodominance may influence the ability of Mamu-B*08-positive macaques to naturally control SIVmac239 replication. J. Virol. 82, 1723–1738 (2008).
Giraldo-Vela, J. P. et al. Major histocompatibility complex class II alleles Mamu-DRB1*1003 and -DRB1*0306 are enriched in a cohort of SIV-infected rhesus macaque elite controllers. J. Virol. 82, 859–870 (2008).
Maness, N. J. et al. AIDS virus specific CD8+ T lymphocytes against an immunodominant cryptic epitope select for viral escape. J. Exp. Med. 204, 2505–2512 (2007).
Yant, L. J. et al. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 80, 5074–5077 (2006).
Mothe, B. R. et al. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 77, 2736–2740 (2003).
Tsukamoto, T. et al. Determination of a major histocompatibility complex class I restricting simian immunodeficiency virus Gag241–249 epitope. AIDS 22, 993–994 (2008).
Matano, T. et al. Cytotoxic T lymphocyte-based control of simian immunodeficiency virus replication in a preclinical AIDS vaccine trial. J. Exp. Med. 199, 1709–1718 (2004).
Kobayashi, M., Igarashi, H., Takeda, A., Kato, M. & Matano, T. Reversion in vivo after inoculation of a molecular proviral DNA clone of simian immunodeficiency virus with a cytotoxic-T-lymphocyte escape mutation. J. Virol. 79, 11529–11532 (2005).
Kawada, M. et al. Involvement of multiple epitope-specific cytotoxic T-lymphocyte responses in vaccine-based control of simian immunodeficiency virus replication in rhesus macaques. J. Virol. 80, 1949–1958 (2006).
Fernandez, C. S. et al. Rapid viral escape at an immunodominant simian-human immunodeficiency virus cytotoxic T-lymphocyte epitope exacts a dramatic fitness cost. J. Virol. 79, 5721–5731 (2005).
Fernandez, C. S. et al. Vaccine-induced T cells control reversion of AIDS virus immune escape mutants. J. Virol. 81, 4137–4144 (2007).
Herberts, C. et al. Cutting Edge: HLA-B27 acquires many N-terminal dibasic peptides: coupling cytosolic peptide stability to antigen presentation. J. Immunol. 176, 2697–2701 (2006).
Nixon D. F. et al. HIV-1 gag-specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature 336, 484–487 (1988).
Feeney, M. E. et al. Immune escape precedes breakthrough HIV-1 viremia and broadening of the CTL response in a HLA-B27-positive long-term nonprogressing child. J. Virol. 78, 8927–8930 (2004).
Casimiro, D. R. et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag. J. Virol. 79, 15547–15555 (2005).
Allen, T. M. et al. Characterization of the peptide binding motif of a rhesus MHC class I molecule (Mamu-A*01) that binds an immunodominant CTL epitope from simian immunodeficiency virus. J. Immunol. 160, 6062–6071 (1998).
Allen, T. M. et al. CD8+ lymphocytes from simian immunodeficiency virus-infected rhesus macaques recognize 14 different epitopes bound by the major histocompatibility complex class I molecule Mamu-A*01: implications for vaccine design and testing. J. Virol. 75, 738–749 (2001).
Pal, R. et al. ALVAC-SIV-gag-pol-env-based vaccination and macaque major histocompatibility complex class I (A*01) delay simian immunodeficiency virus SIVmac-induced immunodeficiency. J. Virol. 76, 292–302 (2002).
Zhang, Z. Q. et al. Mamu-A*01 allele-mediated attenuation of disease progression in simian-human immunodeficiency virus infection. J. Virol. 76, 12845–12854 (2002).
Barouch, D. H. et al. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415, 335–339 (2002).
Barouch, D. H. et al. Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J. Virol. 77, 7367–7375 (2003).
McDermott, A. B. et al. Cytotoxic T-lymphocyte escape does not always explain the transient control of simian immunodeficiency virus SIVmac239 viremia in adenovirus-boosted and DNA-primed Mamu-A*01-positive rhesus macaques. J. Virol. 79, 15556–15566 (2005).
Leslie, A. J. et al. HIV evolution: CTL escape mutation and reversion after transmission. Nature Med. 10, 282–289 (2004). This is the first observation of in vivo reversion following transmission of a viral escape mutant in the dominant epitope targeted by CTLs in HLA-B*57- and HLA-B*5801-positive subjects.
Martinez-Picado, J. et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80, 3617–3623 (2006).
Friedrich, T. C. et al. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nature Med. 10, 275–281 (2004). This is the first observation of in vivo reversion following transmission in the SIV-macaque model, also associated with immune control.
Friedrich, T. C. et al. Extraepitopic compensatory substitutions partially restore fitness to simian immunodeficiency virus variants that escape from an immunodominant cytotoxic-T-lymphocyte response. J. Virol. 78, 2581–2585 (2004).
Kelleher, A. D. et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193, 375–386 (2001).
Schneidewind, A. et al. Late escape from a dominant Gag-specific CTL response in HLA-B27+ subjects is associated with a dramatic cost to HIV-1 fitness. J. Virol. 81, 12382–12393 (2007).
Brockman, M. A. et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 81, 12608–12618 (2007).
Crawford H. et al. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81, 8346–8351 (2007).
Riviere, Y. et al. Gag-specific cytotoxic responses to HIV type 1 are associated with a decreased risk of progression to AIDS-related complex or AIDS. AIDS Res. Hum. Retroviruses 11, 903–907 (1995).
Klein, M. R. et al. Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infection: a longitudinal analysis of rapid progressors and long-term asymptomatics. J. Exp. Med. 181, 1365–1372 (1995).
Ogg, G. S. et al. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279, 2103–2106 (1998).
Edwards, B. H. et al. Magnitude of functional CD8+ T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J. Virol. 76, 2298–2305 (2002).
Novitsky, V. et al. Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J. Virol. 77, 882–890 (2003).
Masemola, A. et al. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: correlation with viral load. J. Virol. 78, 3233–3243 (2004).
Zuniga, R. et al. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 80, 3122–3125 (2006).
Geldmacher, C. et al. CD8 T-cell recognition of multiple epitopes within specific gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 81, 2440–2448 (2007).
Streeck, H. et al. Recognition of a defined region within p24 gag by CD8+ T cells during primary human immunodeficiency virus type 1 infection in individuals expressing protective HLA class I alleles. J. Virol. 81, 7725–7731 (2007).
Frater, A. J. et al. Effective T-cell responses select human immunodeficiency virus mutants and slow disease progression. J. Virol. 81, 6742–6751 (2007).
Goepfert, P. A. et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 205, 1009–1017 (2008). This study shows that the generation of HLA-B-associated Gag escape mutations as a result of the CD8+ T-cell response is of benefit not only to the primary infected host, but also to a secondary recipient following transmission of the virus, as long as the escape mutations do not diminish the ability of the recipient to make an effective HLA-B-restricted Gag-specific response against the virus.
Chopera, R. S. et al. Transmission of HIV-1 escape variants provides HLA-mismatched recipients with a survival advantage. PLoS Pathog. 4, e1000033 (2008).
Navis, M. et al. Viral replication capacity as a correlate of HLA B57/B5801-associated nonprogressive HIV-1 infection. J. Immunol. 179, 3133–3143 (2007).
Sacha, J. B. et al. Gag-specific CD8+ T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. J. Immunol. 178, 2746–2754 (2007). This study shows that Gag-specific CD8+ T cells can recognize SIV-infected target cells within 2 hours of infection.
Sacha, J. B. et al. Pol-specific CD8+ T cells recognize simian immunodeficiency virus-infected cells prior to Nef-mediated major histocompatibility complex class I downregulation. J. Virol. 81, 11703–11712 (2007).
Shehu-Xhilaga, M., Crowe, S. M. & Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 75, 1834–1841 (2001).
Tsomides, T. J. et al. Naturally processed viral peptides recognized by cytotoxic T lymphocytes on cells chronically infected by human immunodeficiency virus type 1. J. Exp. Med. 180, 1283–1293 (1994).
Briggs, J. A. et al. The stoichiometry of Gag protein in HIV-1. Nature Struct. Mol. Biol. 11, 672–675 (2004).
Altfeld, M. & Goulder, P. J. R. Unleashed natural killers hinder HIV. Nature Genet. 39, 708–710 (2007).
Martin, M. P. et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nature Genet. 39, 733–740 (2007).
Harari, A. et al. Skewed association of polyfunctional antigen-specific CD8 T cell populations with HLA-B genotype. Proc. Natl Acad. Sci. USA 104, 16233–16238 (2007).
Borghans, J. A., Mølgaard, A., de Boer, R. J. & Kesmir, C. HLA alleles associated with slow progression to AIDS truly prefer to present HIV-1 p24. PLoS ONE 2, e920 (2007).
Cohen, G. B. et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10, 661–671 (1999).
Rosenberg, E. S. et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science 278, 1447–1450 (1997).
Kalams, S. A. & Walker, B. D. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 188, 2199–2204 (1998).
Betts, M. R. et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107, 4781–4789 (2006).
Freeman, G. J., Wherry, E. J., Ahmed, R. & Sharpe, A. H. Reinvigorating exhausted HIV-specific T cells via PD-1–PD-1 ligand blockade. J. Exp. Med. 203, 2223–2227 (2006).
Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006).
Petrovas, C. et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 203, 2281–2292 (2006).
Day, C. L. et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354 (2006).
Trautmann, L. et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nature Med. 12, 1198–1202 (2006).
Kaufmann, D. E. et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nature Immunol. 8, 1246–1254 (2007).
Goulder, P. J. et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412, 334–338 (2001).
Draenert, R. et al. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199, 905–915 (2004).
Leslie, A. et al. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J. Exp. Med. 201, 891–902 (2005).
Moore, C. B. et al. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296, 1439–1443 (2002).
Bhattacharya, T. et al. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 315, 1583–1586 (2007).
Robertson, M. et al. Immunological characterization of subjects from the STEP study: a phase IIB test of concept trial of the MRKAd5HIV-1 Gag/Pol/Nef trivalent vaccine. CROI 2008, Abstract 89LB.
Feinberg, M. B. & Moore, J. P. AIDS vaccine models: challenging challenge viruses. Nature Med. 8, 207–210 (2002).
Roberts, D. M. et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature 441, 239–243 (2006).
DeRose, R. et al. Control of viremia and prevention of AIDS following immunotherapy of SIV-infected macaques with peptide-pulsed blood. PLoS Pathog. 4, e1000055 (2008).
Tomiyama, H., Fujiwara, M., Oka, S. & Takiguchi, M. Epitope-dependent effect of Nef-mediated HLA class I down-regulation on ability of HIV-1-specific CTLs to suppress HIV-1 replication. J. Immunol. 174, 36–40 (2005).
Goulder, P. J. et al. Novel, cross-restricted, conserved and immunodominant cytotoxic T lymphocyte epitopes in slow progressors in HIV-1 infection. AIDS Res. Hum. Retroviruses 12, 1691–1698 (1996).
Klein, M. R. et al. Characterisation of HLA-B57-restricted HIV-1 Gag- and RT-specific cytotoxic T lymphocyte responses. J. Gen. Virol. 79, 2191–2201 (1998).
Ngumbela, K. C. et al. Positive association between targeting of an epitope presented by HLA-B*5802 and markers of HIV disease progression. AIDS Res. Hum. Retroviruses 24, 72–82 (2008).
O'Connor, D. H. et al. Major histocompatibility complex class I alleles associated with slow simian immunodeficiency virus disease progression bind epitopes recognized by dominant acute-phase cytotoxic-T-lymphocyte responses. J. Virol. 77, 9029–9040 (2003).
Allen, T. M. et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407, 386–390 (2000).
Loffredo, J. T. et al. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J. Virol. 81, 8827–8832 (2007).
Feeney, M. E. et al. HIV-1 viral escape in infancy followed by emergence of a variant-specific CTL response. J. Immunol. 174, 7524–7530 (2005).
Wilson, N. A. et al. Vaccine-induced cellular immune responses reduce plasma viral load concentrations after repeated low-dose challenge with pathogenic SIVmac239. J. Virol. 80, 5875–5885 (2006).
Matthews, P. et al. Central role of reverting mutations in HLA associations with HIV viral setpoint. J. Virol. (in the press).
Acknowledgements
P.J.R.G is supported by the Wellcome Trust, the US National Institutes of Health (grant RO1 AI46995) and the Phillip and Susan Ragon Foundation. D.I.W. and his laboratory are supported by US National Institutes of Health (grants RO1 AI049120, RO1 AI052056, RO1 AI076114, R24 RR015371, R24 RR016038, R21 AI077472, contract HHSN266200400088C) and grants from the International AIDS Vaccine Initiative.
Author information
Authors and Affiliations
Corresponding author
Related links
Glossary
- Viral setpoint
-
This is the time at which plasma viraemia settles to a relatively stable level (within approximately 3–6 months of the onset of HIV infection). Viral setpoint is strongly predictive of both how quickly HIV infection will progress and the risk of HIV transmission.
- MHC class I tetramers
-
Soluble tetrahedral complexes artificially generated by using a fluorochrome-coupled avidin to join four biotinylated MHC class I molecules with a peptide of interest and β2-microglobulin. The resulting MHC class I tetramers can be used as reagents to identify antigen-specific CD8+ T-cell populations.
- HLA B7 supertype
-
A group of HLA alleles (HLA-B*0702, HLA-B*3910, HLA-B*4201, HLA-B*4202 and HLA-B*8101) that have very similar peptide-binding motifs and that can frequently bind the same peptides.
- SHIV89.6P
-
A virus that has the backbone of SIVmac239 with the envelope of HIV and which has been used to test T-cell-based vaccines. It is relatively easy to protect against.
- SIVsmE660
-
A swarm virus that is heterologous to the SIVmac239 virus and has been used as a heterologous challenge in macaques vaccinated with SIVmac239-based sequences.
- Replicative capacity
-
A measure of the ability of a virus to replicate under standard conditions, such as in an in vitro-assay system that compares, for example, the virus under study with a laboratory-adapted strain.
- Viral fitness
-
Viral fitness is context specific, and refers to the relative ability of a virus to replicate under particular conditions, such as those that occur when the immune response or antiretroviral therapy is acting on the virus.
- In vitro competition assays
-
Comparison of the replicative capacity of two different HIV viruses is achieved most effectively by infection of target cells by both viruses simultaneously and determination of the relative proportions of the two competing viruses over time.
- SIVmac239
-
A cloned virus used for pathogenesis and challenge studies. It is a rigorous challenge that is difficult to protect against.
- Bw4 motif
-
The amino-acid sequences at positions 77–83 in the α1 domain of the HLA class I heavy chain. There are four Bw4 motif sequences, which differ only at positions 77 (asparagine, aspartate or serine) and 80 (isoleucine or threonine). The Bw4 motif is involved in the recognition of certain HLA alleles by killer immunoglobulin-like receptors.
- Linkage disequilibrium
-
The non-random association of alleles from two independent loci, owing to their close physical proximity and a lack of recombination between them.
- Founder effects
-
Gene mutations that are observed in high frequency in a specific colony owing to their presence in a few members of the original population.
- Virus clade
-
HIV is subdivided, based on degree of sequence divergence, into three major groups, M, N and O; group M is subdivided into 10 subtypes or clades, of which clade C is the predominant subtype worldwide (prevalent in subSaharan Africa and India) and clade B is the most studied subtype (prevalent in North America and Eastern Europe).
Rights and permissions
About this article
Cite this article
Goulder, P., Watkins, D. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol 8, 619–630 (2008). https://doi.org/10.1038/nri2357
Issue Date:
DOI: https://doi.org/10.1038/nri2357
This article is cited by
-
Expression of type I interferon-associated genes at antiretroviral therapy interruption predicts HIV virological rebound
Scientific Reports (2022)
-
Haplotype structures and polymorphisms of dog leukocyte antigen (DLA) class I loci shaped by intralocus and interlocus recombination events
Immunogenetics (2022)
-
Long-term protective immunity induced by an adjuvant-containing live-attenuated AIDS virus
npj Vaccines (2021)
-
Neutrophil subtypes shape HIV-specific CD8 T-cell responses after vaccinia virus infection
npj Vaccines (2021)
-
SIV-induced terminally differentiated adaptive NK cells in lymph nodes associated with enhanced MHC-E restricted activity
Nature Communications (2021)
