
Abstract
Organ-specific differences in epidermal growth factor receptor (EGFR) mutational spectra and frequencies were found in lung cancer and sporadic and BRCA 1/2-related breast cancers. Additionally, we found a high frequency of EGFR mutations in the tumour stroma of these invasive breast carcinomas. Those organ-specific mutational spectra and potential targets in the cancer-associated stroma might influence the efficacy of TKI therapy.
Similar content being viewed by others

Main
The most common cancers worldwide are lung and breast cancer, accounting for over 2 million new cases each year. Approximately 3 years ago, molecular targeted therapy, starting with the introduction of imatinib, which targets the tyrosine kinases (TK) BCR–ABL, KIT and PDGFR, for the treatment of chronic myeloid leukaemia, was anticipated to provide a new approach to fight malignancies. In clinical trials, the response to epidermal growth factor receptor (EGFR) inhibitors, such as gefitinib, has varied widely, ranging from rare in breast cancers to 10–30% in metastatic non-small-cell lung carcinoma (NSCLC) (Dancey and Freidlin, 2003). Recently, somatic mutations in the TK domain of the EGFR gene have been identified in the NSCLC of the patients who showed increased response to gefitinib, suggesting clinical utility as a predictive factor (Lynch et al, 2004; Paez et al, 2004). Despite the promising response in a subset of NSCLC, other solid tumours, such as breast cancer, do not share this. We hypothesised that differences in EGFR somatic mutational spectra and frequencies among the different solid tumours may result in different responsiveness. Thus, we sought to determine and compare the mutational frequencies and spectra in NSCLC and breast carcinomas. In addition, regarding the growing understanding of the tumour–stroma interaction and the possible role of cancer-associated mesenchyme as a novel target for anticancer therapy, we also analysed the stroma of invasive breast adenocarcinomas for EGFR mutations.
Materials and methods
The TK domain of EGFR, encoded by exons 18–21, was directly sequenced in 60 NSCLC samples, 48 samples of sporadic breast carcinoma and 24 samples from hereditary breast cancers (17 with BRCA1 mutations and seven with BRCA2 mutations, respectively). All samples were obtained as anonymised archival material under approval from the respective Institutional Review Boards.
DNA extraction and mutation analysis
In the breast cancer samples, tumour epithelial and stromal components were collected separately with laser-capture microdissection (LCM). For NSCLC, tumour-enriched paraffin-embedded samples have been used. Genomic DNA was extracted by proteinase K digestion as described by us previously (Fukino et al, 2004). We directly sequenced exons 18–21 of the TK domain in the EGFR gene with the primers listed below. PCR consisted of 40 cycles using an annealing temperature of 55°C in a 15 μl reaction mixture containing 7.5 μl HotStar MasterMix, 1.5 μl 5xQ-Solution (Invitrogen, Carlsbad, CA, USA) and 0.25 μl of each primer. PCR products were then sequenced using Big Dye v3.1 terminator technology and the ABI 3730 analyzer (Applied Biosystems, Perkin-Elmer Corp., Norwalk, CT, USA) according to the manufacturer's recommendation for mutation analysis (supplements 1 and 2).
We used two-sided Fisher exact test to evaluate differences between groups.
Primers for mutation analysis were as follows: exon 18 – GCTGAGGTGACCCTTGTCTC (sense), ACAGCTTGCAAGGACTCTGG (antisense); exon 19 – CATGTGGCACCATCTCACA (sense), CAGCTGCCAGACATGAGAAA (antisense); exon 20 – CACACTGACGTGGCCTCTCC (sense), TATCTCCCCTCCCCGTATCT (antisense); exon 21 – CCTCACAGCAGGGTCTTCTC (sense), CCTGGTGTCAGGAAAATGCT (antisense).
Results
In two (3.3%) of the 60 NSCLC samples, somatic in-frame deletions were detected in exon 19 (Table 1, Figure 1). Both samples showed a heterozygous in-frame deletion of five amino acids (E, L, R, E, A) (delE746-A750) through loss of nucleotides 2235–2249 and 2236–2250, respectively. These two deletions coincided with those reported in the previous reports, suggesting gefitinib responsiveness (Lynch et al, 2004; Paez et al, 2004).

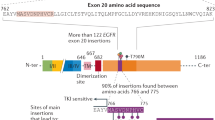
Spectra of somatic EGFR mutations. Location of somatic mutations found in 60 NSCLC, 48 sporadic and 24 hereditary (BRCA1/2 mutation positive) breast cancers. Exons are shown as bars with intronic regions as lines. For breast cancer samples, labels above each bar/line indicate mutations in the stroma, and labels below denote mutations found in neoplastic epithelium. Green labels indicate silent variants.
In total, seven somatic missense mutations were detected in seven (14.6%) of 48 sporadic breast cancer samples. No correlation was detected between tumour grade and mutation status. We identified 14 missense mutations in 11 (45.8%) of 24 breast cancers from BRCA1/2 mutation carriers (Figures 1 and 2, Table 1). Thus, the frequency of EGFR mutations was significantly higher in BRCA1/2-related breast cancers compared to that in sporadic ones (P=0.0079). In addition, three silent mutations that did not alter the amino acid were identified in three hereditary breast cancer samples, of which two also harboured other missense mutations. There was no difference in the frequency of EGFR mutations between BRCA1- (eight out of 17, 47%) and BRCA2- (three out of seven, 43%) related breast cancers (P=1.0). It is noteworthy that, among the 11 BRCA1/2-related breast cancers with EGFR somatic mutations, eight (72.7%) were located exclusively in the stroma (Table 2, Figure 1). Similarly, of the seven sporadic breast cancers with somatic EGFR mutations, four (57.1%) had mutations only in the stroma (Table 2, Figures 1 and 2). Furthermore, 57% (eight out of 14 hereditary, four out of seven sporadic) of all mutations were located in exon 20. In addition, we identified 10 somatic intronic single-nucleotide variants (ISNV) in seven of 24 (29.2%) hereditary breast cancers and nine ISNV in seven out of 48 (14.6%) sporadic breast cancers. Finally, nonsense mutations were identified in one hereditary breast cancer and two sporadic breast cancers. No in-frame deletions as reported for NSCLC were identified in either hereditary or sporadic breast cancer samples.

Somatic EGFR mutations in the epithelium or stroma of sporadic and hereditary breast carcinomas. Each of the four columns (A–D, E–H, I–L and M–P) represents one EGFR mutation-positive sample and the corresponding images taken during the LCM process. The sample codes corresponding to Table 1 are indicated below each set of chromatograms. Each set of chromatograms (A, E, I and M) shows the control (wild-type) sequence in the top row, followed by the sequence of the mutation-negative compartment. The heterozygous mutation and surrounding sequences are shown in forward (f) and reverse (r) directions in the bottom two rows. The first column shows sample H2, harbouring a somatic D761N mutation in the tumour epithelium (A, f and r) but not tumour stroma (mut. neg. in A). Image (B) shows an overview of this tumour (H&E, × 100 and × 200) and images (C) and (D) confirm that we accurately captured stroma (C) and epithelium (D). The second column shows the chromatograms (E) and tissue image (F) of sample H9, harbouring the somatic Q791R mutation in the stroma (f and r in (E)) but not epithelium (E, mut. neg.). The corresponding images (G) and (H) depict the captured epithelium (G) and the tissue image after extraction of the epithelial component by LCM (H). The third column represents the sporadic breast adenocarcinoma sample S1 (J) with a somatic T693A mutation in the stromal compartment (I, f and r) but not epithelium (mut. neg. in (I)). Again, images (K) and (L) verify the separation of tumour epithelium (L) and stroma (K). The last column shows sample S13 harbouring a W817X mutation in the tumour epithelium (M, f and r) but not stroma (M, mut. neg.). The neoplastic epithelium is microdissected (O) out of the whole tumour section (N), leaving the stromal compartment (P).
Discussion
Our data show that EGFR mutations occur at a significantly higher frequency in hereditary breast cancer compared to sporadic breast cancer (P=0.0079). This may not be surprising given the functional effect of BRCA1/2 mutations: defects in BRCA1 and BRCA2 have been shown to disrupt the DNA repair mechanism, which leads to genomic instability. Despite the difference in mutation frequencies between sporadic and hereditary breast cancers, it is obvious that sporadic and heritable breast cancers both have a similarly high frequency of EGFR mutations in tumour stroma, and that the majority of missense mutations lie in exon 20, in contrast to those in NSCLC.
The mutations reported in gefitinib-sensitive NSCLC were located in the TK domain encoded by exons 18–21, and have been shown to increase growth factor signalling and confer susceptibility to gefitinib in vitro (Lynch et al, 2004; Paez et al, 2004). These data suggest that the clinical outcome after molecular targeted therapy strongly depends on acquired alterations in target genes, and, by extrapolation, perhaps germline alterations (host risk) and/or functional status of the molecular target as well. At this point, we can only speculate on the functional properties that the mutations described in our breast cancers will have on EGFR receptor signalling. The majority of variants identified lie in close proximity to the highly conserved amino-acid residues within the TK domain I to VI (Table 1). Extrapolating from other reports, it seams likely that these could affect the ATP-binding pocket and result in gain of function (Huang et al, 2004). Truncating mutations such as the nonsense mutations found in breast cancers are predicted to result in loss of function, and, by extrapolation from a recent report by Huang et al (2004) lack of responsiveness to EGFR-TKI's. Since the samples have been obtained as anonymised material, we are not able to link our results to the outcome of possible EGFR-directed therapy. Her2-neu expression is reported to be associated with responsiveness to therapy, especially in tamoxifen-resistant cases and it is suggested that the heterodimerisation of EGFR/Her2-neu might contribute. Based on our data, we did not find any evidence for a correlation between EGFR mutation status and Her2-neu expression (Table 1).
What the effect of EGFR mutations as possible targets for anticancer therapy, in the stroma on responsiveness to EGFR-TKI's, is unknown. Given our and other existing data on the tumour microenvironment (Dancey and Freidlin, 2003; Fukino et al, 2004), it may be postulated that the high frequencies of stromal EGFR mutations in sporadic and hereditary breast cancers could confound responsiveness to EGFR-TKI and may help explain interpatient variation. Thus, we suggest that future clinical trials employing molecular targeted therapy evaluate these genetic factors, not only in the traditional epithelial neoplasm but also in the surrounding tumour stroma in order to establish their role in and predictive value for interindividual variation in responses.
In summary, we have shown that EGFR mutations are found in a distinct, organ-specific pattern, and suggest that mutational spectra may be only one basis for prediction of response to EGFR-TKI's. Furthermore, we have demonstrated that the tumour stroma was rich in EGFR gene alterations compared to the epithelium. We previously reported on a model of tumour–microenviroment interaction in multistep breast carcinogenesis and pointed out the importance of mutations found exclusively in the tumour stroma (Kurose et al, 2001, 2002). It is acknowledged that the stroma can modulate the neoplastic epithelium by mediating invasion and progression. Therefore, it is possible that any EGFR-TK-directed therapy should consider anticancer targets in the tumour stroma as well as neoplastic epithelium, and, indeed, perhaps such TK-directed therapy in breast cancer will predominantly affect this tumour–microenvironment interaction by inhibiting invasion and progression rather than influence tumour mass.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Dancey JE, Freidlin B (2003) Targeting epidermal growth factor receptor – are we missing the mark? Lancet 362: 62–64
Fukino K, Shen L, Matsumoto S, Morrison CD, Mutter GL, Eng C (2004) Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res 64: 7231–7236
Huang SF, Liu HP, Li LH, Ku YC, Fu YN, Tsai HY, Chen YT, Lin YF, Chang WC, Kuo HP, Wu YC, Chen YR, Tsai SF (2004) High frequency of epidermal growth factor receptor mutations with complex patterns in non-small cell lung cancers related to gefitinib responsiveness in Taiwan. Clin Cancer Res 10: 8195–8203
Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C (2002) Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet 32: 355–357
Kurose K, Hoshaw-Woodard S, Adeyinka A, Lemeshow S, Watson PH, Eng C (2001) Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: clues to tumour-microenvironment interactions. Hum Mol Genet 10: 1907–1913
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129–2139
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304: 1497–1500
Acknowledgements
Funding source: US Department of Defense Breast Cancer Research Program (DAMD17-02-1-0528 to CE); National Cancer Institute, Bethesda, USA (P01CA97189 and P50CA113001 to CE; P30CA16058 to The Ohio State University Comprehensive Cancer Center); CE is a recipient of the Doris Duke Distinguished Clinical Scientist Award. The Tissue Procurement Shared Resource and the Human Cancer Genetics Program Genotyping-Sequencing Core Facility of the Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio is in part supported by grant P30 CA16059, National Cancer Institute, Bethesda, MD. The sponsors mentioned had no role in study design, collection and analysis of data, writing of the report and decision to submit the paper for publication. No author had any financial and personal relationships with other people or organisations that could inappropriately influence or bias this work.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Weber, F., Fukino, K., Sawada, T. et al. Variability in organ-specific EGFR mutational spectra in tumour epithelium and stroma may be the biological basis for differential responses to tyrosine kinase inhibitors. Br J Cancer 92, 1922–1926 (2005). https://doi.org/10.1038/sj.bjc.6602557
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6602557
Keywords
This article is cited by
-
Role of EGFR and FASN in breast cancer progression
Journal of Cell Communication and Signaling (2023)
-
Detection of IKKε by immunohistochemistry in primary breast cancer: association with EGFR expression and absence of lymph node metastasis
BMC Cancer (2017)
-
Rational design of non-resistant targeted cancer therapies
Scientific Reports (2017)
-
EGFR protein overexpression correlates with chromosome 7 polysomy and poor prognostic parameters in clear cell renal cell carcinoma
Journal of Biomedical Science (2012)
-
Role of epidermal growth factor receptor in breast cancer
Breast Cancer Research and Treatment (2012)
